
Identification of SARS-CoV-2 Main Protease Inhibitors Using Chemical Similarity Analysis Combined with Machine Learning

 ,
,  ,
,  ,
, 
 and
and 
Abstract
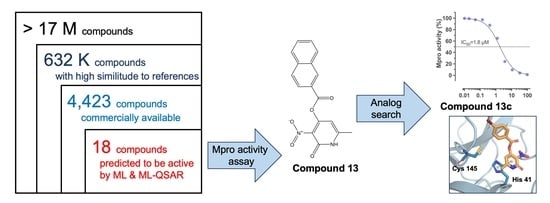
1. Introduction
2. Results and Discussion
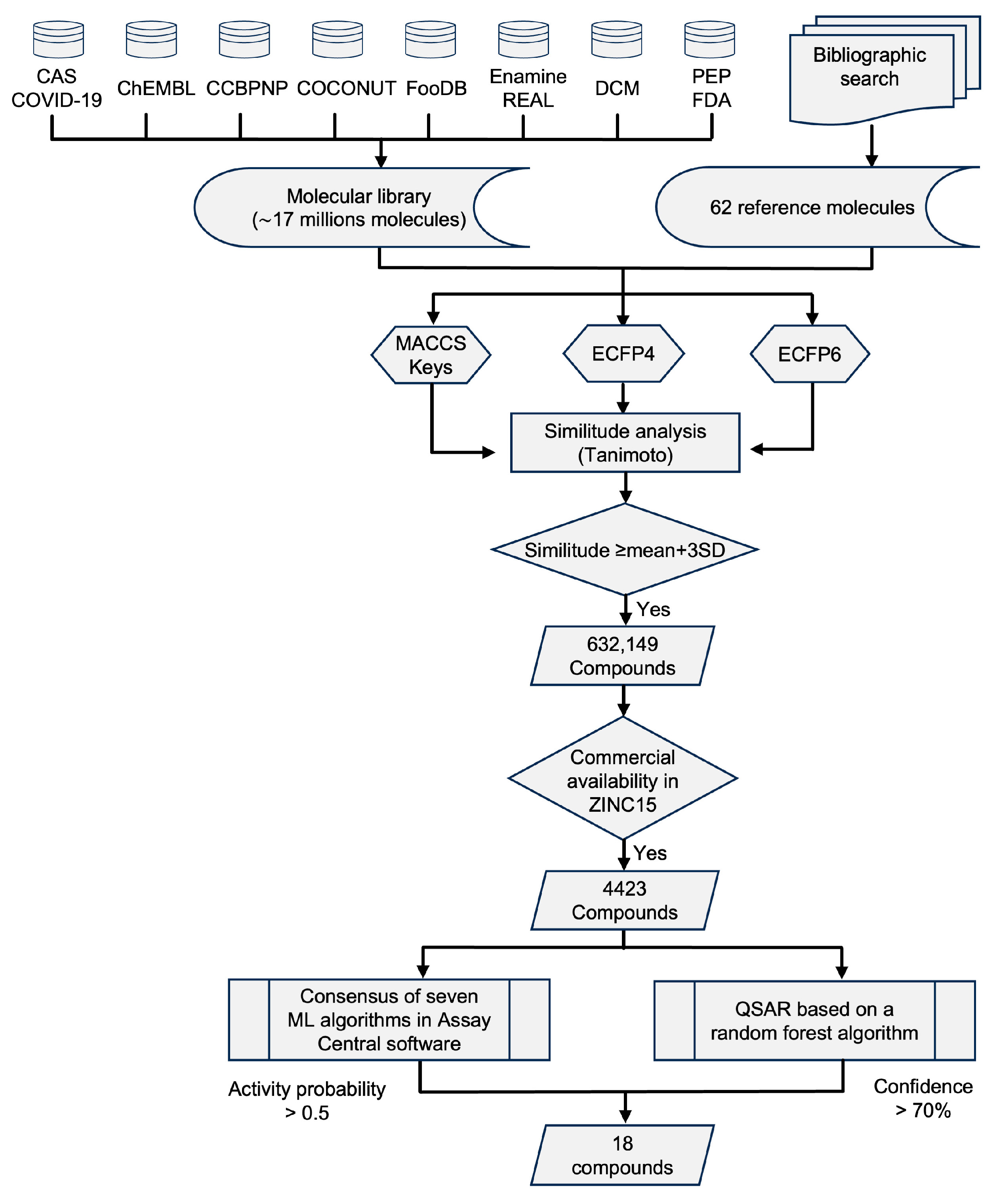
2.1. In Silico Identification of Potential Mpro Inhibitors
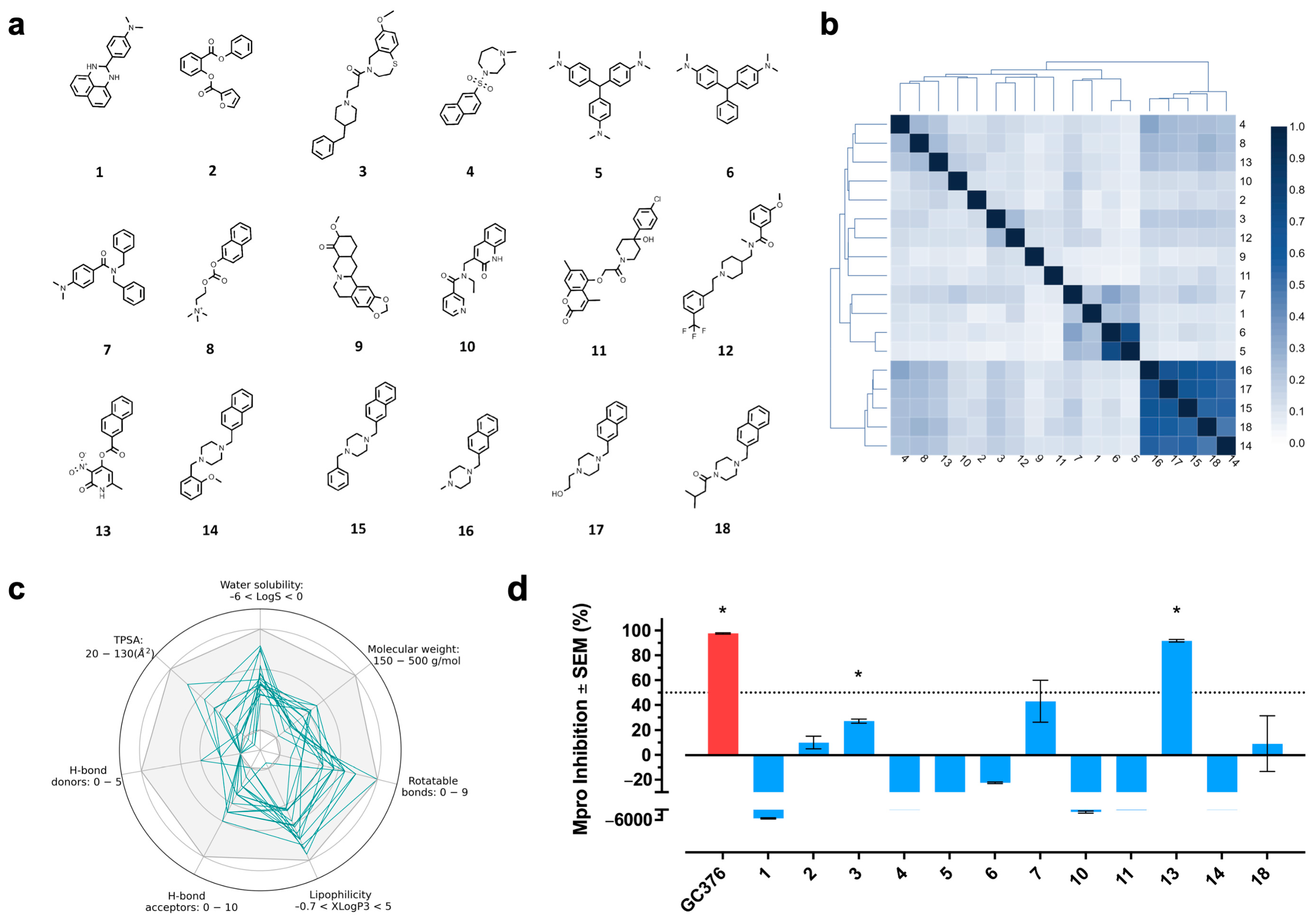
2.2. Property and Diversity Profiling of Computational Hits and Experimental Validation
2.3. Identification of New Mpro Inhibitors by Searching Analogs of Compound 13
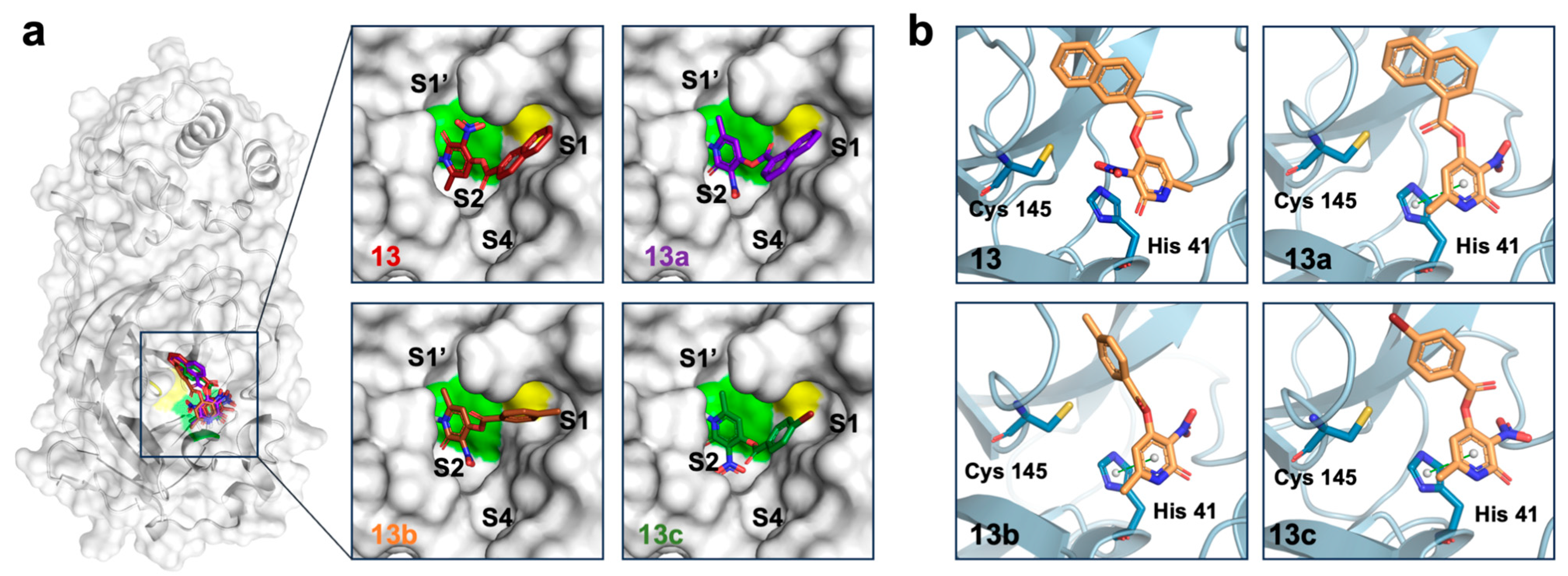
2.4. In Silico Characterization of Compound Binding to SARS-CoV-2 Mpro
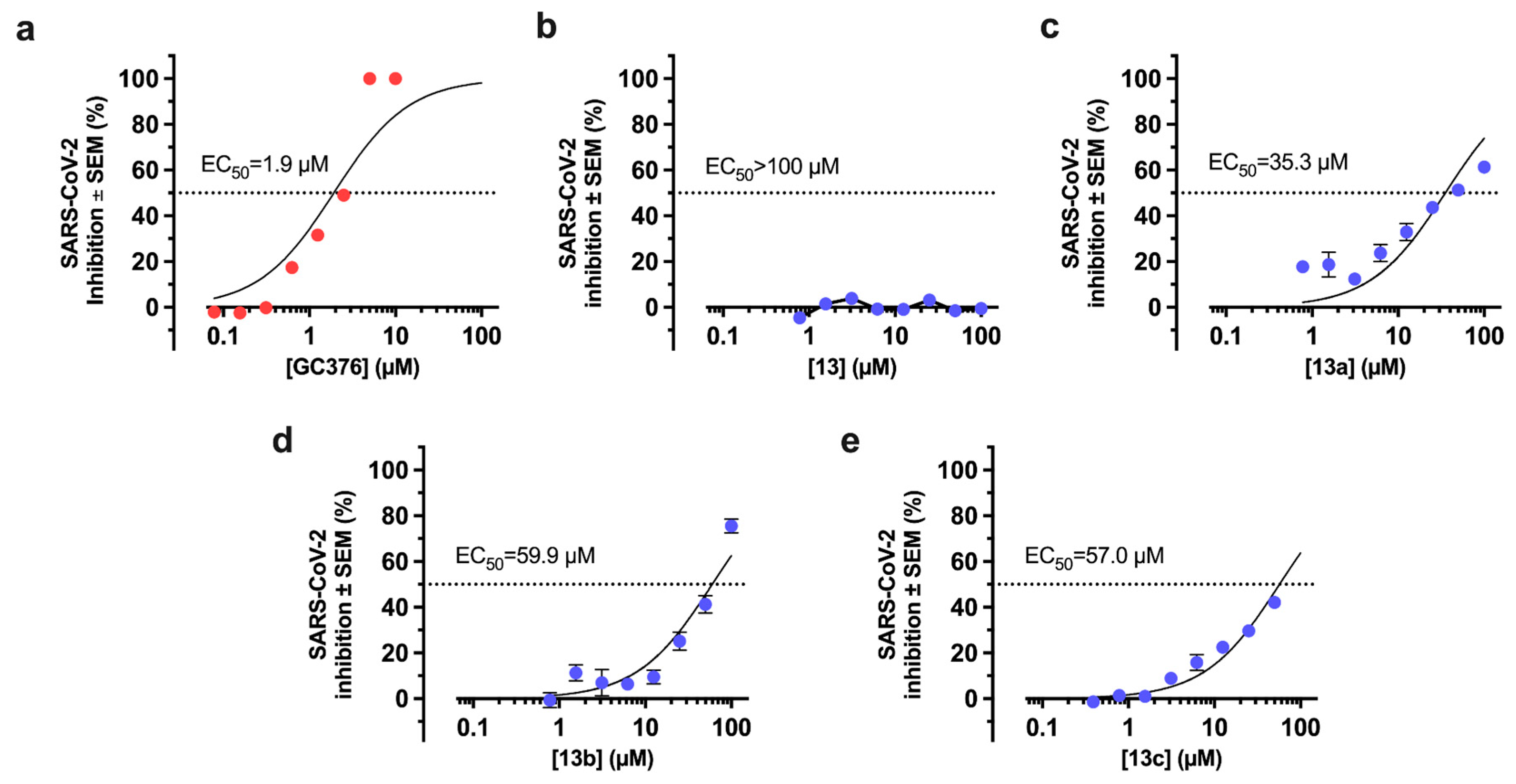
2.5. Evaluation of Antiviral Activity in Cell Culture
3. Materials and Methods
3.1. Preparation of the Screening Chemical Library and the Reference Set of Active Compounds
3.2. VS
3.3. Analysis of Structural Diversity and Physicochemical Properties of Computational Hits
3.4. Compounds
3.5. Mpro Enzymatic Activity Assays
3.6. Molecular Docking
3.7. Molecular Dynamics Simulations
3.8. Cytotoxicity Assessment
3.9. Evaluation of Antiviral Activity by Plaque Reduction Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Helmy, Y.A.; Fawzy, M.; Elaswad, A.; Sobieh, A.; Kenney, S.P.; Shehata, A.A. The COVID-19 Pandemic: A Comprehensive Review of Taxonomy, Genetics, Epidemiology, Diagnosis, Treatment, and Control. J. Clin. Med. 2020, 9, 1225. [Google Scholar] [CrossRef] [PubMed]
- Naseer, S.; Khalid, S.; Parveen, S.; Abbass, K.; Song, H.; Achim, M.V. COVID-19 Outbreak: Impact on Global Economy. Front. Public Health 2023, 10, 1009393. [Google Scholar] [CrossRef] [PubMed]
- WHO WHO Coronavirus Disease (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 2 September 2023).
- Zhang, Y.-Z.; Holmes, E.C. A Genomic Perspective on the Origin and Emergence of SARS-CoV-2. Cell 2020, 181, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell Entry Mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef] [PubMed]
- Suryawanshi, R.K.; Koganti, R.; Agelidis, A.; Patil, C.D.; Shukla, D. Dysregulation of Cell Signaling by SARS-CoV-2. Trends Microbiol. 2021, 29, 224–237. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of Therapeutic Targets for SARS-CoV-2 and Discovery of Potential Drugs by Computational Methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef] [PubMed]
- Cortese, M.; Lee, J.-Y.; Cerikan, B.; Neufeldt, C.J.; Oorschot, V.M.J.; Köhrer, S.; Hennies, J.; Schieber, N.L.; Ronchi, P.; Mizzon, G.; et al. Integrative Imaging Reveals SARS-CoV-2-Induced Reshaping of Subcellular Morphologies. Cell Host Microbe 2020, 28, 853–866.e5. [Google Scholar] [CrossRef]
- Twu, W.-I.; Lee, J.-Y.; Kim, H.; Prasad, V.; Cerikan, B.; Haselmann, U.; Tabata, K.; Bartenschlager, R. Contribution of Autophagy Machinery Factors to HCV and SARS-CoV-2 Replication Organelle Formation. Cell Rep. 2021, 37, 110049. [Google Scholar] [CrossRef]
- Shahid, M.; Shahzad-ul-Hussan, S. Structural Insights of Key Enzymes into Therapeutic Intervention against SARS-CoV-2. J. Struct. Biol. 2021, 213, 107690. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal Structure of SARS-CoV-2 Main Protease Provides a Basis for Design of Improved α-Ketoamide Inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef]
- de Vries, M.; Mohamed, A.S.; Prescott, R.A.; Valero-Jimenez, A.M.; Desvignes, L.; O’Connor, R.; Steppan, C.; Devlin, J.C.; Ivanova, E.; Herrera, A.; et al. A Comparative Analysis of SARS-CoV-2 Antivirals Characterizes 3CL pro Inhibitor PF-00835231 as a Potential New Treatment for COVID-19. J. Virol. 2021, 95, 1128. [Google Scholar] [CrossRef] [PubMed]
- Cannalire, R.; Cerchia, C.; Beccari, A.R.; Di Leva, F.S.; Summa, V. Targeting SARS-CoV-2 Proteases and Polymerase for COVID-19 Treatment: State of the Art and Future Opportunities. J. Med. Chem. 2022, 65, 2716–2746. [Google Scholar] [CrossRef]
- Kuzikov, M.; Costanzi, E.; Reinshagen, J.; Esposito, F.; Vangeel, L.; Wolf, M.; Ellinger, B.; Claussen, C.; Geisslinger, G.; Corona, A.; et al. Identification of Inhibitors of SARS-CoV-2 3CL-Pro Enzymatic Activity Using a Small Molecule in Vitro Repurposing Screen. ACS Pharmacol. Transl. Sci. 2021, 4, 1096–1110. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhu, Y.; Liu, X.; Jin, Z.; Duan, Y.; Zhang, Q.; Wu, C.; Feng, L.; Du, X.; Zhao, J.; et al. Structural Basis for Replicase Polyprotein Cleavage and Substrate Specificity of Main Protease from SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2022, 15, e2117142119. [Google Scholar] [CrossRef] [PubMed]
- Nextstrain Latest Global SARS-CoV-2 Analysis (GISAID Data). Available online: https://nextstrain.org/ncov/gisaid/global/6m (accessed on 20 December 2023).
- Oerlemans, R.; Ruiz-Moreno, A.J.; Cong, Y.; Dinesh Kumar, N.; Velasco-Velazquez, M.A.; Neochoritis, C.G.; Smith, J.; Reggiori, F.; Groves, M.R.; Dömling, A. Repurposing the HCV NS3–4A Protease Drug Boceprevir as COVID-19 Therapeutics. RSC Med. Chem. 2021, 3, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Vuong, W.; Khan, M.B.; Fischer, C.; Arutyunova, E.; Lamer, T.; Shields, J.; Saffran, H.A.; McKay, R.T.; van Belkum, M.J.; Joyce, M.A.; et al. Feline Coronavirus Drug Inhibits the Main Protease of SARS-CoV-2 and Blocks Virus Replication. Nat. Commun. 2020, 11, 4282. [Google Scholar] [CrossRef]
- Huang, C.; Shuai, H.; Qiao, J.; Hou, Y.; Zeng, R.; Xia, A.; Xie, L.; Fang, Z.; Li, Y.; Yoon, C.; et al. A New Generation Mpro Inhibitor with Potent Activity against SARS-CoV-2 Omicron Variants. Signal Transduct. Target. Ther. 2023, 8, 128. [Google Scholar] [CrossRef]
- Haritha, C.V.; Sharun, K.; Jose, B. Ebselen, a New Candidate Therapeutic against SARS-CoV-2. Int. J. Surg. 2020, 84, 53–56. [Google Scholar] [CrossRef]
- Harrach, S.; Haag, J.; Steinbüchel, M.; Schröter, R.; Neugebauer, U.; Bertrand, J.; Ciarimboli, G. Interaction of Masitinib with Organic Cation Transporters. Int. J. Mol. Sci. 2022, 23, 14189. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, Q.; Yao, P.; Yao, Q.; Zhang, J. Multidimensional Virtual Screening Approaches Combined with Drug Repurposing to Identify Potential Covalent Inhibitors of SARS-CoV-2 3CL Protease. J. Biomol. Struct. Dyn. 2023, 41, 15262–15285. [Google Scholar] [CrossRef]
- Mukae, H.; Yotsuyanagi, H.; Ohmagari, N.; Doi, Y.; Sakaguchi, H.; Sonoyama, T.; Ichihashi, G.; Sanaki, T.; Baba, K.; Tsuge, Y.; et al. Efficacy and Safety of Ensitrelvir in Patients With Mild-to-Moderate Coronavirus Disease 2019: The Phase 2b Part of a Randomized, Placebo-Controlled, Phase 2/3 Study. Clin. Infect. Dis. 2023, 76, 1403–1411. [Google Scholar] [CrossRef] [PubMed]
- Food Drug Administration Paxlovid EUA Letter of Authorization. Available online: https://www.fda.gov/media/155049/download (accessed on 20 December 2023).
- Chakraborty, C.; Bhattacharya, M.; Alshammari, A.; Alharbi, M.; Albekairi, T.H.; Zheng, C. Exploring the Structural and Molecular Interaction Landscape of Nirmatrelvir and Mpro Complex: The Study Might Assist in Designing More Potent Antivirals Targeting SARS-CoV-2 and Other Viruses. J. Infect. Public Health 2023, 16, 1961–1970. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Rani, C.; Pant, P.; Vijayan, V.; Vikram, N.; Kaur, P.; Singh, T.P.; Sharma, S.; Sharma, P. Structure-Based Virtual Screening and Biochemical Validation to Discover a Potential Inhibitor of the SARS-CoV-2 Main Protease. ACS Omega 2020, 5, 33151–33161. [Google Scholar] [CrossRef]
- Menendez, C.A.; Mohamed, A.; Perez-Lemus, G.R.; Weiss, A.M.; Rawe, B.W.; Liu, G.; Crolais, A.E.; Kenna, E.; Byléhn, F.; Alvarado, W.; et al. Development of Masitinib Derivatives with Enhanced Mpro Ligand Efficiency and Reduced Cytotoxicity. Molecules 2023, 28, 6643. [Google Scholar] [CrossRef] [PubMed]
- Menéndez, C.A.; Byléhn, F.; Perez-Lemus, G.R.; Alvarado, W.; de Pablo, J.J. Molecular Characterization of Ebselen Binding Activity to SARS-CoV-2 Main Protease. Sci. Adv. 2020, 6, eabd0345. [Google Scholar] [CrossRef] [PubMed]
- Reymond, J.-L. The Chemical Space Project. Acc. Chem. Res. 2015, 48, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Restrepo, G. Chemical Space: Limits, Evolution and Modelling of an Object Bigger than Our Universal Library. Digit. Discov. 2022, 1, 568–585. [Google Scholar] [CrossRef]
- Medina-Franco, J.L. Grand Challenges of Computer-Aided Drug Design: The Road Ahead. Front. Drug Discov. 2021, 1, 728551. [Google Scholar] [CrossRef]
- Hassan Baig, M.; Ahmad, K.; Roy, S.; Mohammad Ashraf, J.; Adil, M.; Haris Siddiqui, M.; Khan, S.; Amjad Kamal, M.; Provazník, I.; Choi, I. Computer Aided Drug Design: Success and Limitations. Curr. Pharm. Des. 2016, 22, 572–581. [Google Scholar] [CrossRef]
- Muratov, E.N.; Amaro, R.; Andrade, C.H.; Brown, N.; Ekins, S.; Fourches, D.; Isayev, O.; Kozakov, D.; Medina-Franco, J.L.; Merz, K.M.; et al. A Critical Overview of Computational Approaches Employed for COVID-19 Drug Discovery. Chem. Soc. Rev. 2021, 50, 9121–9151. [Google Scholar] [CrossRef]
- Ton, A.; Gentile, F.; Hsing, M.; Ban, F.; Cherkasov, A. Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds. Mol. Inform. 2020, 39, 2000028. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.M.; Agarwal, R.; Vermaas, J.V.; Smith, M.D.; Rajeshwar, R.T.; Cooper, C.; Sedova, A.; Boehm, S.; Baker, M.; Glaser, J.; et al. SARS-CoV2 Billion-Compound Docking. Sci. Data 2023, 10, 173. [Google Scholar] [CrossRef] [PubMed]
- Khanfar, M.A.; Salaas, N.; Abumostafa, R. Discovery of Natural-derived M pro Inhibitors as Therapeutic Candidates for COVID-19: Structure-based Pharmacophore Screening Combined with QSAR Analysis. Mol. Inform. 2023, 42, 202200198. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Kogej, T.; Engkvist, O. Cheminformatics in Drug Discovery, an Industrial Perspective. Mol. Inform. 2018, 37, 1800041. [Google Scholar] [CrossRef] [PubMed]
- Keyvanpour, M.R.; Shirzad, M.B. An Analysis of QSAR Research Based on Machine Learning Concepts. Curr. Drug Discov. Technol. 2021, 18, 17–30. [Google Scholar] [CrossRef]
- Macip, G.; Garcia-Segura, P.; Mestres-Truyol, J.; Saldivar-Espinoza, B.; Ojeda-Montes, M.J.; Gimeno, A.; Cereto-Massagué, A.; Garcia-Vallvé, S.; Pujadas, G. Haste Makes Waste: A Critical Review of Docking-based Virtual Screening in Drug Repurposing for SARS-CoV-2 Main Protease (M-pro) Inhibition. Med. Res. Rev. 2022, 42, 744–769. [Google Scholar] [CrossRef]
- Negru, P.A.; Miculas, D.C.; Behl, T.; Bungau, A.F.; Marin, R.-C.; Bungau, S.G. Virtual Screening of Substances Used in the Treatment of SARS-CoV-2 Infection and Analysis of Compounds with Known Action on Structurally Similar Proteins from Other Viruses. Biomed. Pharmacother. 2022, 153, 113432. [Google Scholar] [CrossRef]
- Castillo-Garit, J.A.; Cañizares-Carmenate, Y.; Pham-The, H.; Pérez-Doñate, V.; Torrens, F.; Pérez-Giménez, F. A Review of Computational Approaches Targeting SARS-CoV-2 Main Protease to the Discovery of New Potential Antiviral Compounds. Curr. Top. Med. Chem. 2023, 23, 3–16. [Google Scholar] [CrossRef]
- Mignani, S.; Rodrigues, J.; Tomas, H.; Jalal, R.; Singh, P.P.; Majoral, J.-P.; Vishwakarma, R.A. Present Drug-Likeness Filters in Medicinal Chemistry during the Hit and Lead Optimization Process: How Far Can They Be Simplified? Drug Discov. Today 2018, 23, 605–615. [Google Scholar] [CrossRef]
- Ren, H.-C.; Sai, Y.; Chen, T. Evaluation of Generic Methods to Predict Human Pharmacokinetics Using Physiologically Based Pharmacokinetic Model for Early Drug Discovery of Tyrosine Kinase Inhibitors. Eur. J. Drug Metab. Pharmacokinet. 2019, 44, 121–132. [Google Scholar] [CrossRef]
- Gawriljuk, V.O.; Zin, P.P.K.; Puhl, A.C.; Zorn, K.M.; Foil, D.H.; Lane, T.R.; Hurst, B.; Tavella, T.A.; Costa, F.T.M.; Lakshmanane, P.; et al. Machine Learning Models Identify Inhibitors of SARS-CoV-2. J. Chem. Inf. Model. 2021, 61, 4224–4235. [Google Scholar] [CrossRef] [PubMed]
- Puhl, A.C.; Gomes, G.F.; Damasceno, S.; Godoy, A.S.; Noske, G.D.; Nakamura, A.M.; Gawriljuk, V.O.; Fernandes, R.S.; Monakhova, N.; Riabova, O.; et al. Pyronaridine Protects against SARS-CoV-2 Infection in Mouse. ACS Infect. Dis. 2022, 8, 1147–1160. [Google Scholar] [CrossRef] [PubMed]
- Puhl, A.C.; Gomes, G.F.; Damasceno, S.; Fritch, E.J.; Levi, J.A.; Johnson, N.J.; Scholle, F.; Premkumar, L.; Hurst, B.L.; Lee-Montiel, F.; et al. Vandetanib Blocks the Cytokine Storm in SARS-CoV-2-Infected Mice. ACS Omega 2022, 7, 31935–31944. [Google Scholar] [CrossRef] [PubMed]
- Alves, V.M.; Bobrowski, T.; Melo-Filho, C.C.; Korn, D.; Auerbach, S.; Schmitt, C.; Muratov, E.N.; Tropsha, A. QSAR Modeling of SARS-CoV M pro Inhibitors Identifies Sufugolix, Cenicriviroc, Proglumetacin, and Other Drugs as Candidates for Repurposing against SARS-CoV-2. Mol. Inform. 2021, 40, 2000113. [Google Scholar] [CrossRef] [PubMed]
- Jukič, M.; Janežič, D.; Bren, U. Ensemble Docking Coupled to Linear Interaction Energy Calculations for Identification of Coronavirus Main Protease (3CLpro) Non-Covalent Small-Molecule Inhibitors. Molecules 2020, 25, 5808. [Google Scholar] [CrossRef] [PubMed]
- Jukič, M.; Škrlj, B.; Tomšič, G.; Pleško, S.; Podlipnik, Č.; Bren, U. Prioritisation of Compounds for 3CLpro Inhibitor Development on SARS-CoV-2 Variants. Molecules 2021, 26, 3003. [Google Scholar] [CrossRef]
- Chen, F.; Chan, K.H.; Jiang, Y.; Kao, R.Y.T.; Lu, H.T.; Fan, K.W.; Cheng, V.C.C.; Tsui, W.H.W.; Hung, I.F.N.; Lee, T.S.W. In Vitro Susceptibility of 10 Clinical Isolates of SARS Coronavirus to Selected Antiviral Compounds. J. Clin. Virol. 2004, 31, 69–75. [Google Scholar] [CrossRef]
- De Clercq, E. Potential Antivirals and Antiviral Strategies against SARS Coronavirus Infections. Expert Rev. Anti Infect. Ther. 2006, 4, 291–302. [Google Scholar] [CrossRef]
- Dyall, J.; Coleman, C.M.; Hart, B.J.; Venkataraman, T.; Holbrook, M.R.; Kindrachuk, J.; Johnson, R.F.; Olinger, G.G.; Jahrling, P.B.; Laidlaw, M.; et al. Repurposing of Clinically Developed Drugs for Treatment of Middle East Respiratory Syndrome Coronavirus Infection. Antimicrob. Agents Chemother. 2014, 58, 4885–4893. [Google Scholar] [CrossRef]
- Cong, Y.; Hart, B.J.; Gross, R.; Zhou, H.; Frieman, M.; Bollinger, L.; Wada, J.; Hensley, L.E.; Jahrling, P.B.; Dyall, J.; et al. MERS-CoV Pathogenesis and Antiviral Efficacy of Licensed Drugs in Human Monocyte-Derived Antigen-Presenting Cells. PLoS ONE 2018, 13, e0194868. [Google Scholar] [CrossRef]
- Liang, R.; Wang, L.; Zhang, N.; Deng, X.; Su, M.; Su, Y.; Hu, L.; He, C.; Ying, T.; Jiang, S.; et al. Development of Small-Molecule MERS-CoV Inhibitors. Viruses 2018, 10, 721. [Google Scholar] [CrossRef]
- Richardson, P.; Griffin, I.; Tucker, C.; Smith, D.; Oechsle, O.; Phelan, A.; Rawling, M.; Savory, E.; Stebbing, J. Baricitinib as Potential Treatment for 2019-nCoV Acute Respiratory Disease. Lancet 2020, 395, e30–e31. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and Chloroquine Effectively Inhibit the Recently Emerged Novel Coronavirus (2019-nCoV) In Vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Meenakshisundaram, S.; Manickam, M. Recent Discovery and Development of Inhibitors Targeting Coronaviruses. Drug Discov. Today 2020, 25, 668–688. [Google Scholar] [CrossRef]
- Caly, L.; Druce, J.D.; Catton, M.G.; Jans, D.A.; Wagstaff, K.M. The FDA-Approved Drug Ivermectin Inhibits the Replication of SARS-CoV-2 in Vitro. Antiviral Res. 2020, 178, 104787. [Google Scholar] [CrossRef]
- Yu, P.-C.; Huang, C.-H.; Kuo, C.-J.; Liang, P.-H.; Wang, L.H.-C.; Pan, M.Y.-C.; Chang, S.-Y.; Chao, T.-L.; Ieong, S.-M.; Fang, J.-T.; et al. Drug Repurposing for the Identification of Compounds with Anti-SARS-CoV-2 Capability via Multiple Targets. Pharmaceutics 2022, 14, 176. [Google Scholar] [CrossRef]
- Mody, V.; Ho, J.; Wills, S.; Mawri, A.; Lawson, L.; Ebert, M.C.C.J.C.; Fortin, G.M.; Rayalam, S.; Taval, S. Identification of 3-Chymotrypsin like Protease (3CLPro) Inhibitors as Potential Anti-SARS-CoV-2 Agents. Commun. Biol. 2021, 4, 93. [Google Scholar] [CrossRef]
- van de Sand, L.; Bormann, M.; Alt, M.; Schipper, L.; Heilingloh, C.S.; Steinmann, E.; Todt, D.; Dittmer, U.; Elsner, C.; Witzke, O.; et al. Glycyrrhizin Effectively Inhibits SARS-CoV-2 Replication by Inhibiting the Viral Main Protease. Viruses 2021, 13, 609. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and Discovery of Its Inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Anton, D.B.; Galvez Bulhões Pedreira, J.; Zvirtes, M.L.; Laufer, S.A.; Ducati, R.G.; Goettert, M.; Saraiva Macedo Timmers, L.F. Targeting SARS-CoV-2 Main Protease (MPro) with Kinase Inhibitors: A Promising Approach for Discovering Antiviral and Anti-Inflammatory Molecules against SARS-CoV-2. J. Chem. Inf. Model. 2023, 63, 4138–4146. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Yao, S.; Zhao, W.; Li, M.; Liu, J.; Shang, W.; Xie, H.; Ke, C.; Hu, H.; Gao, M.; et al. Anti-SARS-CoV-2 Activities in Vitro of Shuanghuanglian Preparations and Bioactive Ingredients. Acta Pharmacol. Sin. 2020, 41, 1167–1177. [Google Scholar] [CrossRef]
- Kuo, C.-J.; Chao, T.-L.; Kao, H.-C.; Tsai, Y.-M.; Liu, Y.-K.; Wang, L.H.-C.; Hsieh, M.-C.; Chang, S.-Y.; Liang, P.-H. Kinetic Characterization and Inhibitor Screening for the Proteases Leading to Identification of Drugs against SARS-CoV-2. Antimicrob. Agents Chemother. 2021, 65, e02577-20. [Google Scholar] [CrossRef]
- Soleimani Asl, S.; Roozbahani, M.H. A Novel Robust Inhibitor of Papain-like Protease (PLpro) as a COVID-19 Drug. J. Biomol. Struct. Dyn. 2023, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ribaudo, G.; Yun, X.; Ongaro, A.; Oselladore, E.; Ng, J.P.L.; Haynes, R.K.; Law, B.Y.K.; Memo, M.; Wong, V.K.W.; Coghi, P.; et al. Combining Computational and Experimental Evidence on the Activity of Antimalarial Drugs on papain-like Protease of SARS-CoV-2: A Repurposing Study. Chem. Biol. Drug Des. 2023, 101, 809–818. [Google Scholar] [CrossRef]
- Chou, C.-Y.; Chien, C.-H.; Han, Y.-S.; Prebanda, M.T.; Hsieh, H.-P.; Turk, B.; Chang, G.-G.; Chen, X. Thiopurine Analogues Inhibit Papain-like Protease of Severe Acute Respiratory Syndrome Coronavirus. Biochem. Pharmacol. 2008, 75, 1601–1609. [Google Scholar] [CrossRef]
- Lin, C.; Tsai, F.-J.; Hsu, Y.-M.; Ho, T.-J.; Wang, G.-K.; Chiu, Y.-J.; Ha, H.-A.; Yang, J.-S. Study of Baicalin toward COVID-19 Treatment: In Silico Target Analysis and in Vitro Inhibitory Effects on SARS-CoV-2 Proteases. Biomed. Hub 2021, 6, 122–137. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Hu, Y.; Townsend, J.A.; Lagarias, P.I.; Marty, M.T.; Kolocouris, A.; Wang, J. Ebselen, Disulfiram, Carmofur, PX-12, Tideglusib, and Shikonin Are Nonspecific Promiscuous SARS-CoV-2 Main Protease Inhibitors. ACS Pharmacol. Transl. Sci. 2020, 3, 1265–1277. [Google Scholar] [CrossRef]
- Medina-Franco, J.L.; Giulianotti, M.A.; Yu, Y.; Shen, L.; Yao, L.; Singh, N. Discovery of a Novel Protein Kinase B Inhibitor by Structure-Based Virtual Screening. Bioorg. Med. Chem. Lett. 2009, 19, 4634–4638. [Google Scholar] [CrossRef] [PubMed]
- Prado-Romero, D.L.; Gómez-García, A.; Cedillo-González, R.; Villegas-Quintero, H.; Avellaneda-Tamayo, J.F.; López-López, E.; Saldívar-González, F.I.; Chávez-Hernández, A.L.; Medina-Franco, J.L. Consensus Docking Aid to Model the Activity of an Inhibitor of DNA Methyltransferase 1 Inspired by de Novo Design. Front. Drug Discov. 2023, 3, 1261094. [Google Scholar] [CrossRef]
- Kim, B.-K.; Ko, H.; Jeon, E.-S.; Ju, E.-S.; Jeong, L.S.; Kim, Y.-C. 2,3,4-Trihydroxybenzyl-Hydrazide Analogues as Novel Potent Coxsackievirus B3 3C Protease Inhibitors. Eur. J. Med. Chem. 2016, 120, 202–216. [Google Scholar] [CrossRef]
- Shaqra, A.M.; Zvornicanin, S.N.; Huang, Q.Y.J.; Lockbaum, G.J.; Knapp, M.; Tandeske, L.; Bakan, D.T.; Flynn, J.; Bolon, D.N.A.; Moquin, S.; et al. Defining the Substrate Envelope of SARS-CoV-2 Main Protease to Predict and Avoid Drug Resistance. Nat. Commun. 2022, 13, 3556. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T.; Yang, Q.; Gribenko, A.; Perrin, B.S.; Zhu, Y.; Cardin, R.; Liberator, P.A.; Anderson, A.S.; Hao, L. Genetic Surveillance of SARS-CoV-2 M pro Reveals High Sequence and Structural Conservation Prior to the Introduction of Protease Inhibitor Paxlovid. mBio 2022, 13, e00869-22. [Google Scholar] [CrossRef]
- Ip, J.D.; Wing-Ho Chu, A.; Chan, W.-M.; Cheuk-Ying Leung, R.; Umer Abdullah, S.M.; Sun, Y.; To, K.K.-W. Global Prevalence of SARS-CoV-2 3CL Protease Mutations Associated with Nirmatrelvir or Ensitrelvir Resistance. eBioMedicine 2023, 91, 104559. [Google Scholar] [CrossRef]
- von Delft, A.; Hall, M.D.; Kwong, A.D.; Purcell, L.A.; Saikatendu, K.S.; Schmitz, U.; Tallarico, J.A.; Lee, A.A. Accelerating Antiviral Drug Discovery: Lessons from COVID-19. Nat. Rev. Drug Discov. 2023, 22, 585–603. [Google Scholar] [CrossRef]
- Zev, S.; Raz, K.; Schwartz, R.; Tarabeh, R.; Gupta, P.K.; Major, D.T. Benchmarking the Ability of Common Docking Programs to Correctly Reproduce and Score Binding Modes in SARS-CoV-2 Protease Mpro. J. Chem. Inf. Model. 2021, 61, 2957–2966. [Google Scholar] [CrossRef] [PubMed]
- Department of Health & Human Services; Food and Drug Administration; Center for Drug Evaluation and Research Guidance for Industry Antiviral Product Development- Conducting and Submitting Virology Studies to the Agency. Available online: https://www.fda.gov/media/71223/download (accessed on 19 December 2023).
- Hung, H.-C.; Ke, Y.-Y.; Huang, S.Y.; Huang, P.-N.; Kung, Y.-A.; Chang, T.-Y.; Yen, K.-J.; Peng, T.-T.; Chang, S.-E.; Huang, C.-T.; et al. Discovery of M Protease Inhibitors Encoded by SARS-CoV-2. Antimicrob. Agents Chemother. 2020, 64, 1128. [Google Scholar] [CrossRef] [PubMed]
- Chávez-Hernández, A.L.; Sánchez-Cruz, N.; Medina-Franco, J.L. A Fragment Library of Natural Products and Its Comparative Chemoinformatic Characterization. Mol. Inform. 2020, 39, 202000050. [Google Scholar] [CrossRef]
- Weininger, D.; Weininger, A.; Weininger, J.L. SMILES. 2. Algorithm for Generation of Unique SMILES Notation. J. Chem. Inf. Comput. Sci. 1989, 29, 97–101. [Google Scholar] [CrossRef]
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL Keys for Use in Drug Discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. [Google Scholar] [CrossRef]
- Jaccard, P. Étude Comparative de La Distribution Florale Dans Une Portion Des Alpes et Du Jura. Bull. Société Vaudoise Sci. Nat. 1901, 37, 547–579. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.S.; Ma, X.R.; Ma, Y.; Alugubelli, Y.R.; Scott, D.A.; Vatansever, E.C.; Drelich, A.K.; Sankaran, B.; Geng, Z.Z.; Blankenship, L.R.; et al. A Speedy Route to Multiple Highly Potent SARS-CoV-2 Main Protease Inhibitors. bioRxiv2020, 2020–07.
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Cheng, X.; Lee, J.; Kim, S.; Park, S.; Patel, D.S.; Beaven, A.H.; Lee, K.I.; Rui, H.; Park, S.; et al. CHARMM-GUI 10 Years for Biomolecular Modeling and Simulation. J. Comput. Chem. 2017, 38, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Vásquez-Bochm, L.X.; Velázquez-Paniagua, M.; Castro-Vázquez, S.S.; Guerrero-Rodríguez, S.L.; Mondragon-Peralta, A.; De La Fuente-Granada, M.; Pérez-Tapia, S.M.; González-Arenas, A.; Velasco-Velázquez, M.A. Transcriptome-Based Identification of Lovastatin as a Breast Cancer Stem Cell-Targeting Drug. Pharmacol. Rep. 2019, 71, 535–544. [Google Scholar] [CrossRef] [PubMed]
- González-González, E.; Carballo-Uicab, G.; Salinas-Trujano, J.; Cortés-Paniagua, M.I.; Vázquez-Leyva, S.; Vallejo-Castillo, L.; Mendoza-Salazar, I.; Gómez-Castellano, K.; Pérez-Tapia, S.M.; Almagro, J.C. In Vitro and In Vivo Characterization of a Broadly Neutralizing Anti-SARS-CoV-2 Antibody Isolated from a Semi-Immune Phage Display Library. Antibodies 2022, 11, 57. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Descriptor | Compound 13 | Compound 13a | Compound 13b | Compound 13c |
|---|---|---|---|---|
| Intestinal absorption (%) | 89.21 | 93.79 | 86.80 | 87.54 |
| Lipinski violations | None | None | None | None |
| Verber violations | None | None | None | None |
| BBB permeability | No | No | No | No |
| P-glycoprotein substrate | No | Yes | Yes | No |
| P-glycoprotein inhibitor | Yes | No | No | No |
| CYP1A2 inhibitor | Yes | Yes | No | No |
| CYP2C19 inhibitor | Yes | Yes | No | Yes |
| CYP2C9 inhibitor | Yes | Yes | No | No |
| CYP2D6 inhibitor | No | No | No | No |
| CYP3A4 inhibitor | No | No | No | No |
| Total clearance (log mL/min/kg) | 0.75 | 0.75 | 0.79 | -0.16 |
| AMES toxicity | Yes | Yes | Yes | Yes |
| Hepatotoxicity | Yes | Yes | No | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juárez-Mercado, K.E.; Gómez-Hernández, M.A.; Salinas-Trujano, J.; Córdova-Bahena, L.; Espitia, C.; Pérez-Tapia, S.M.; Medina-Franco, J.L.; Velasco-Velázquez, M.A. Identification of SARS-CoV-2 Main Protease Inhibitors Using Chemical Similarity Analysis Combined with Machine Learning. Pharmaceuticals 2024, 17, 240. https://doi.org/10.3390/ph17020240
Juárez-Mercado KE, Gómez-Hernández MA, Salinas-Trujano J, Córdova-Bahena L, Espitia C, Pérez-Tapia SM, Medina-Franco JL, Velasco-Velázquez MA. Identification of SARS-CoV-2 Main Protease Inhibitors Using Chemical Similarity Analysis Combined with Machine Learning. Pharmaceuticals. 2024; 17(2):240. https://doi.org/10.3390/ph17020240
Chicago/Turabian StyleJuárez-Mercado, Karina Eurídice, Milton Abraham Gómez-Hernández, Juana Salinas-Trujano, Luis Córdova-Bahena, Clara Espitia, Sonia Mayra Pérez-Tapia, José L. Medina-Franco, and Marco A. Velasco-Velázquez. 2024. "Identification of SARS-CoV-2 Main Protease Inhibitors Using Chemical Similarity Analysis Combined with Machine Learning" Pharmaceuticals 17, no. 2: 240. https://doi.org/10.3390/ph17020240
APA StyleJuárez-Mercado, K. E., Gómez-Hernández, M. A., Salinas-Trujano, J., Córdova-Bahena, L., Espitia, C., Pérez-Tapia, S. M., Medina-Franco, J. L., & Velasco-Velázquez, M. A. (2024). Identification of SARS-CoV-2 Main Protease Inhibitors Using Chemical Similarity Analysis Combined with Machine Learning. Pharmaceuticals, 17(2), 240. https://doi.org/10.3390/ph17020240