
Machine Learning Application for Medicinal Chemistry: Colchicine Case, New Structures, and Anticancer Activity Prediction



Abstract
1. Introduction
- 1.
- A549—adenocarcinomic human alveolar basal epithelial cells, lung cancer related [15];
- 2.
- BALB/3T3—detection of the carcinogenic potential of chemicals [16];
- 3.
- LoVo/DX—human colon adenocarcinoma doxorubicin-resistant cell line [17];
- 4.
- LoVo—human colon adenocarcinoma cell line, colorectal cancer related [18];
- 5.
- MCF-7—breast-cancer-related cell line [19].
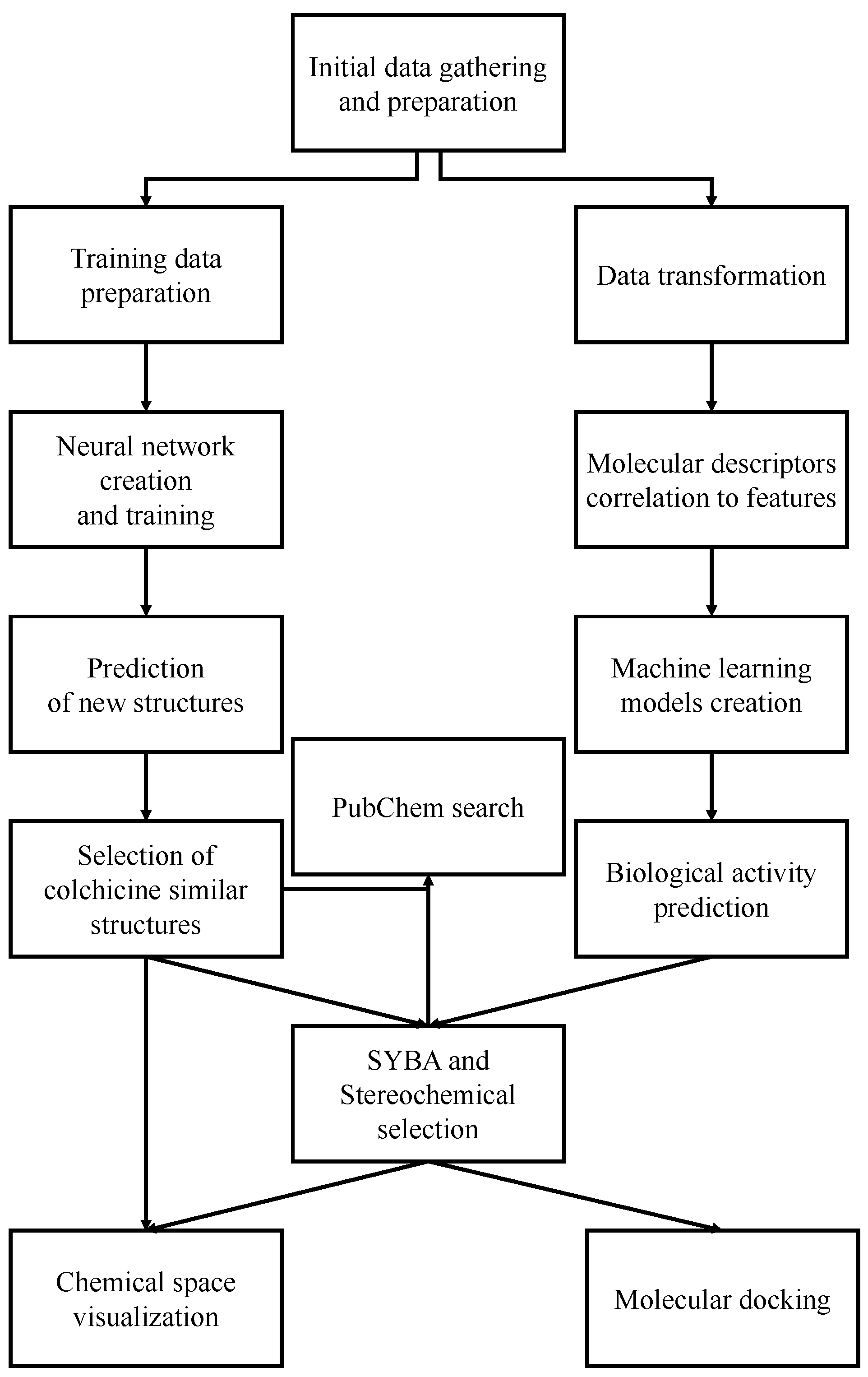
2. Results and Discussion
2.1. Training Data
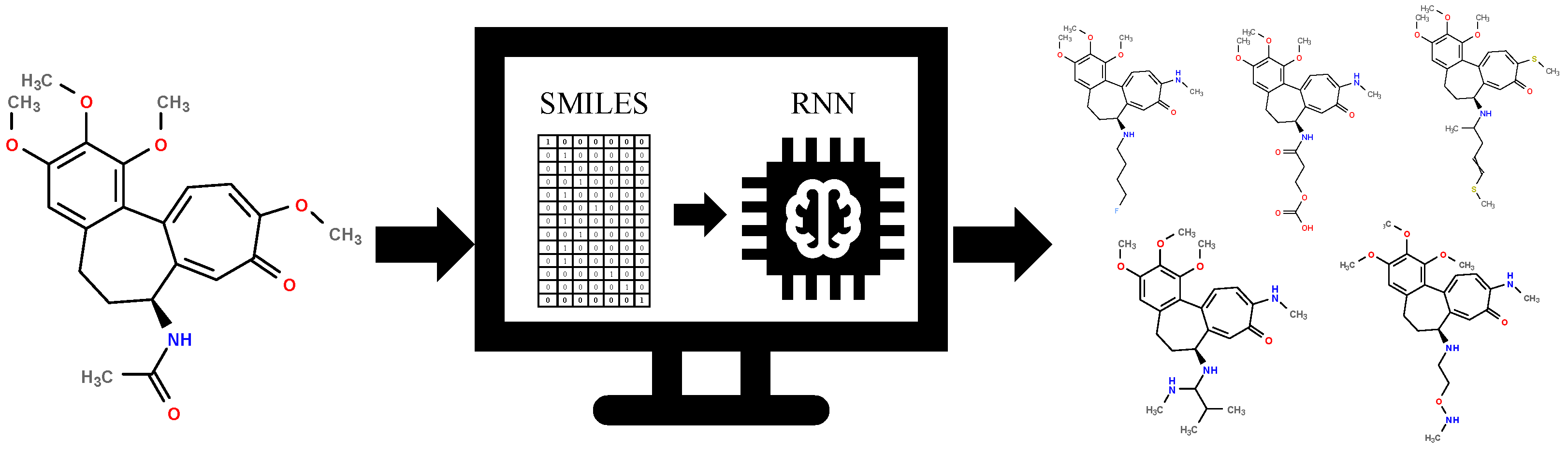
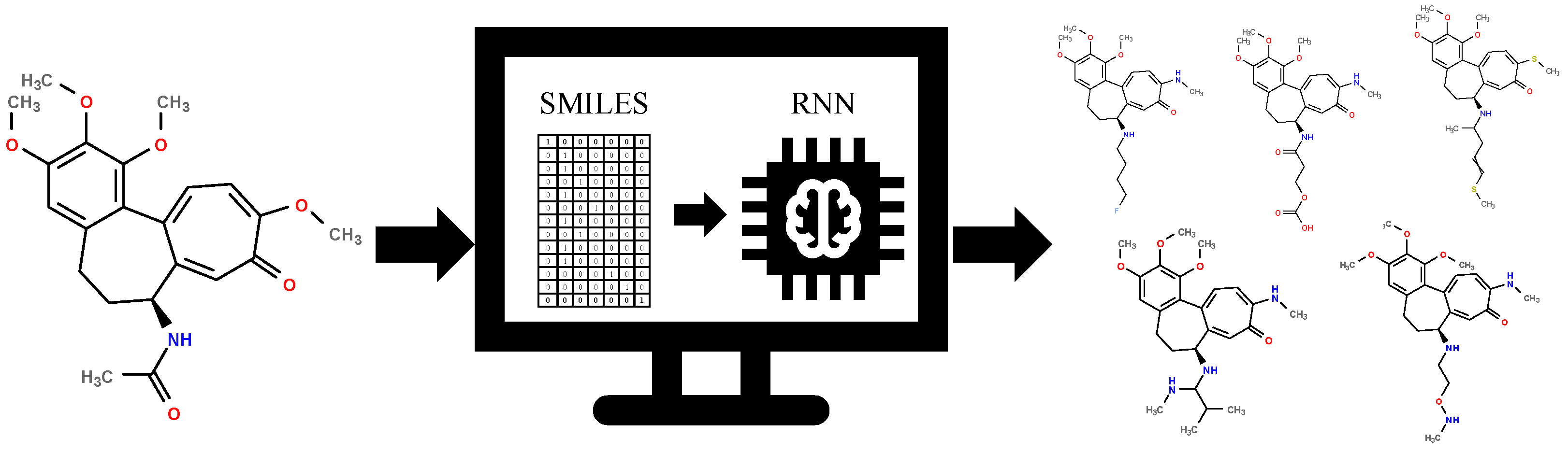
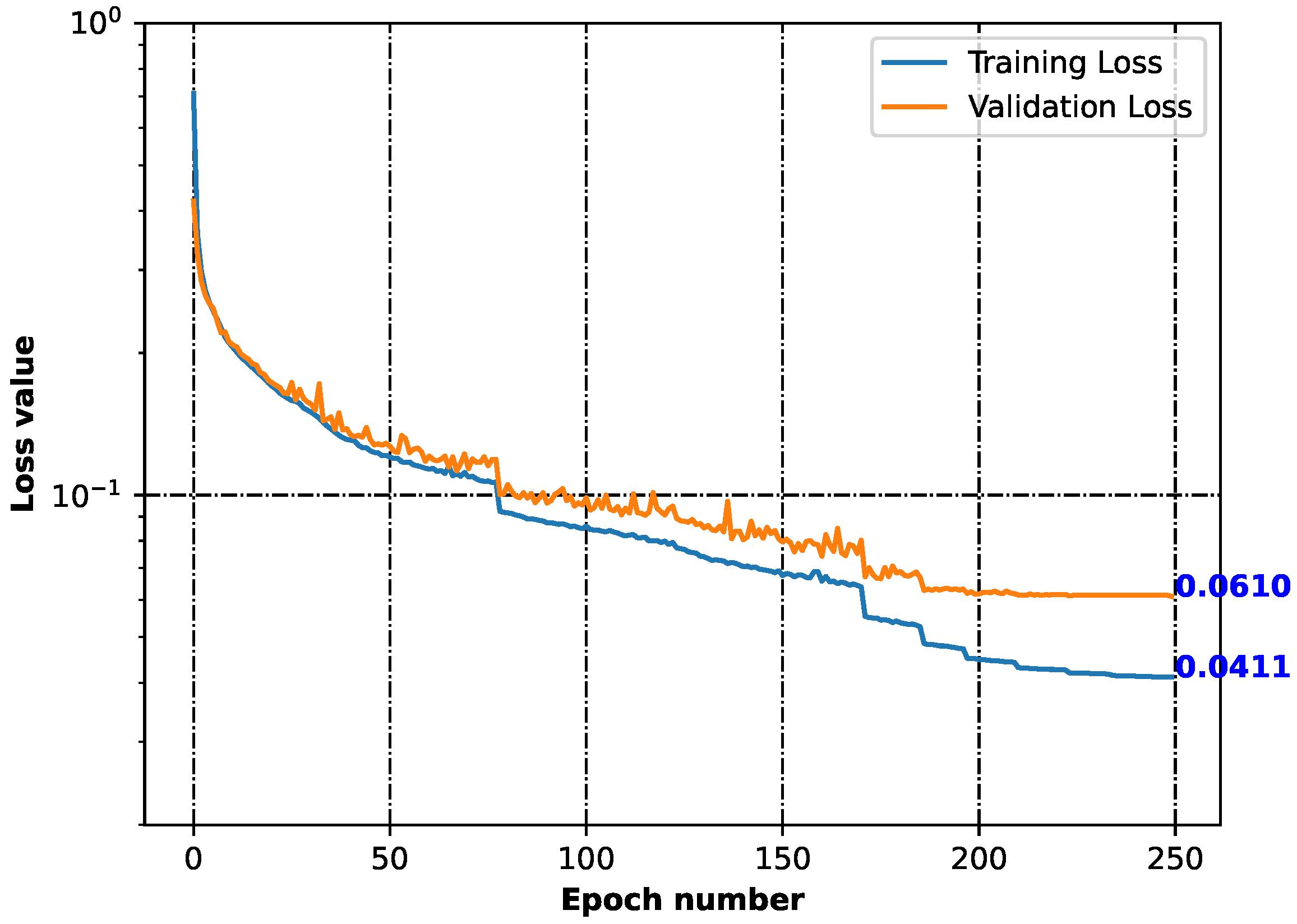
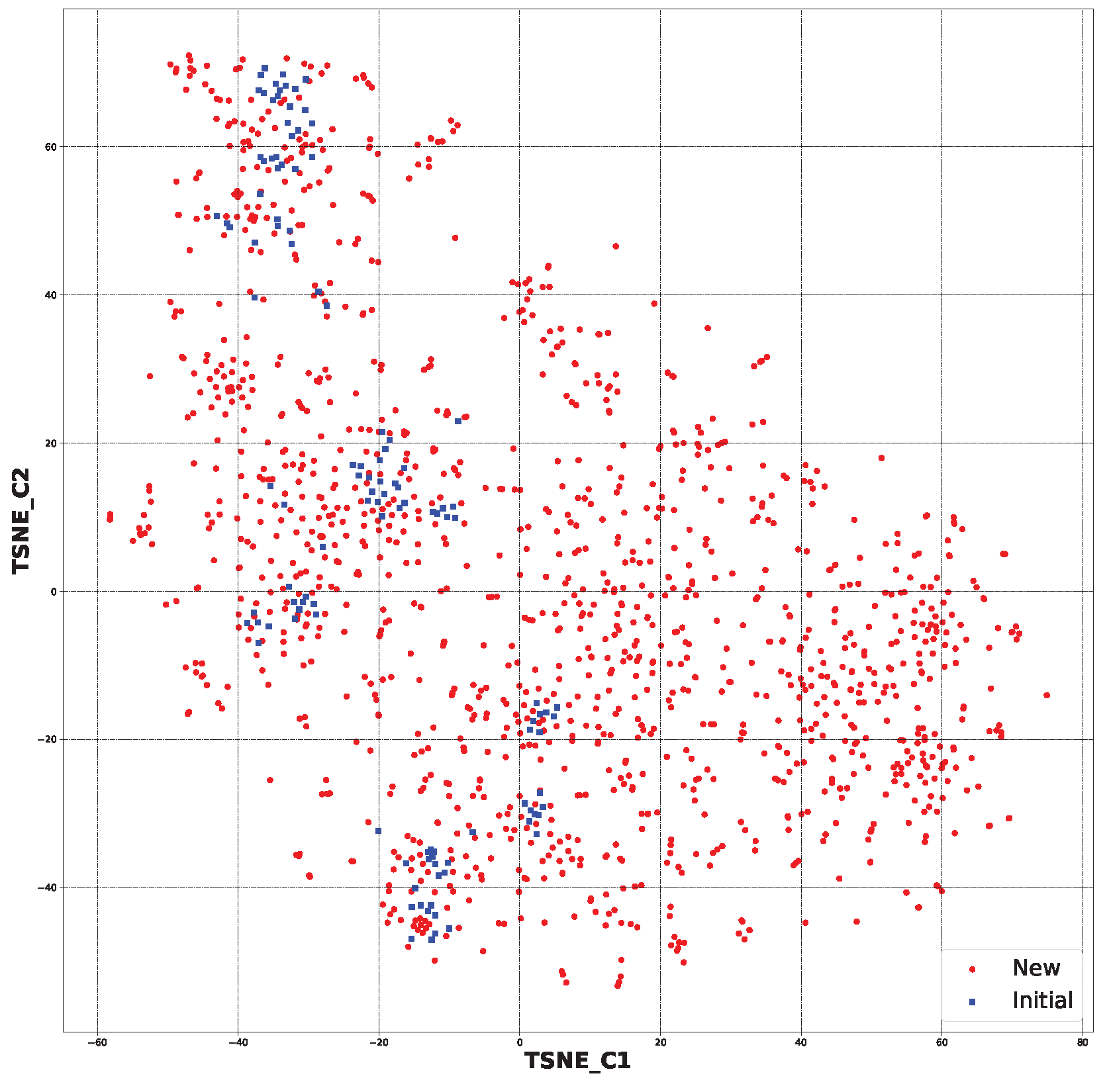
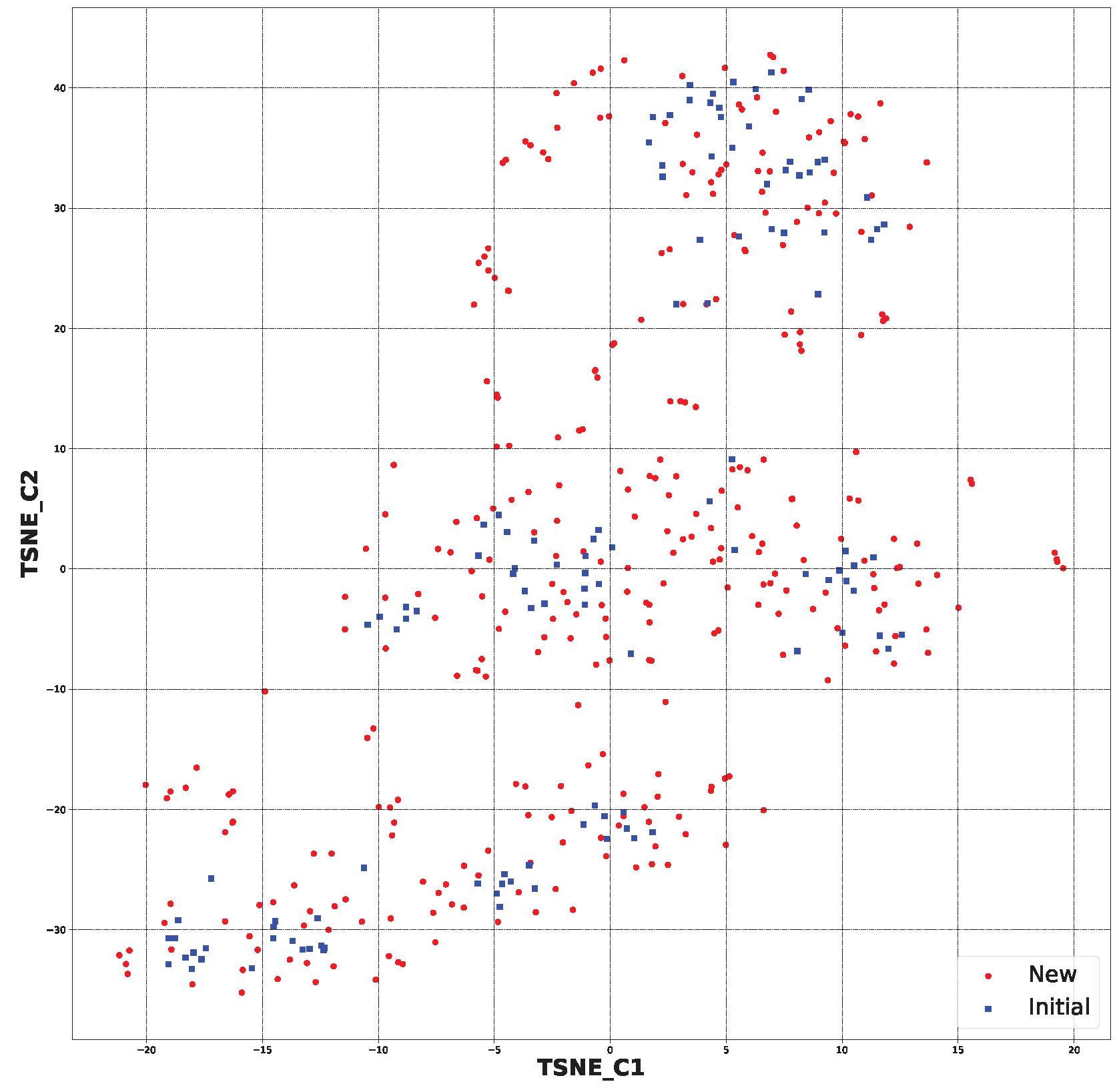
2.2. Generative Neural Network
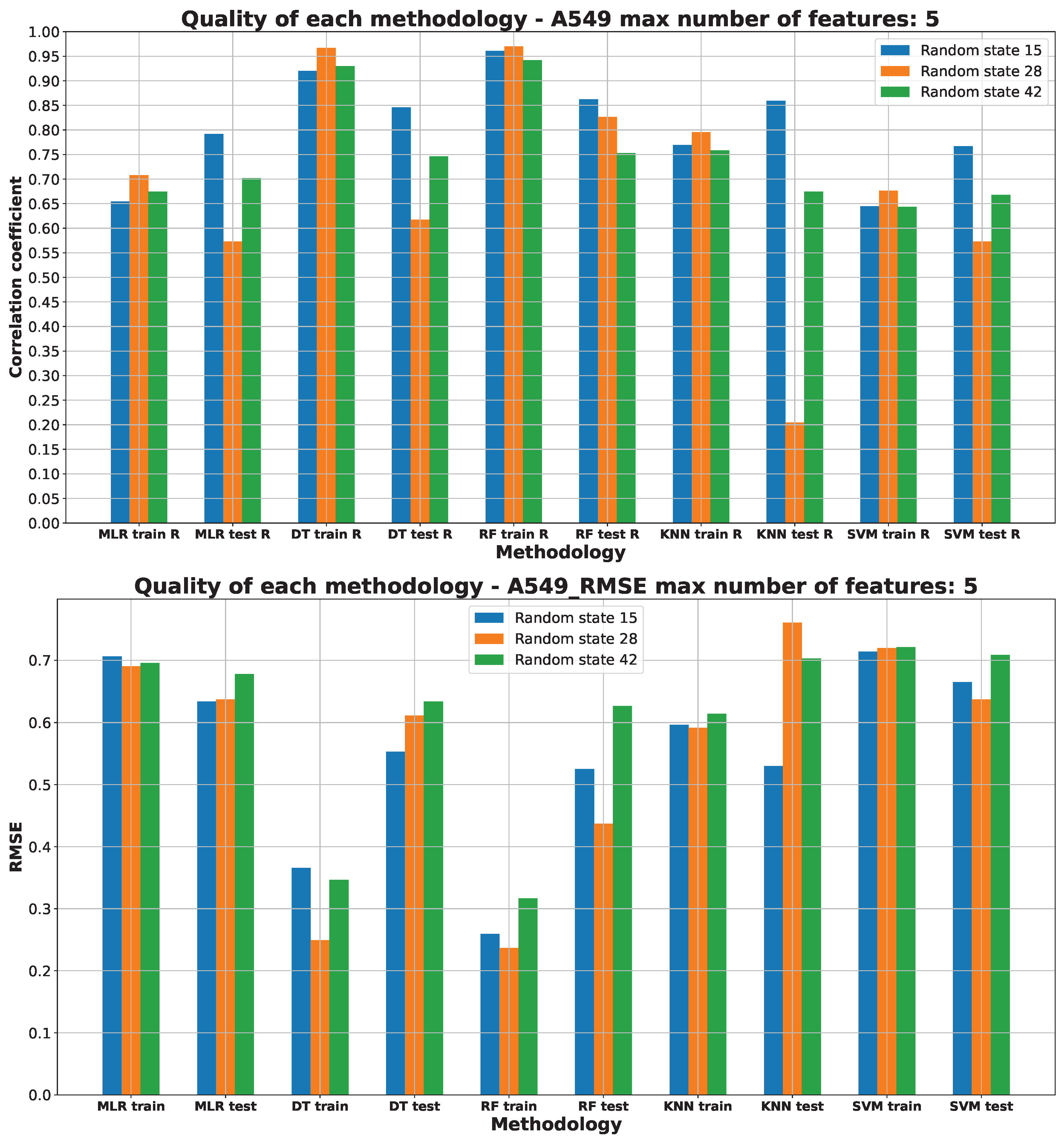
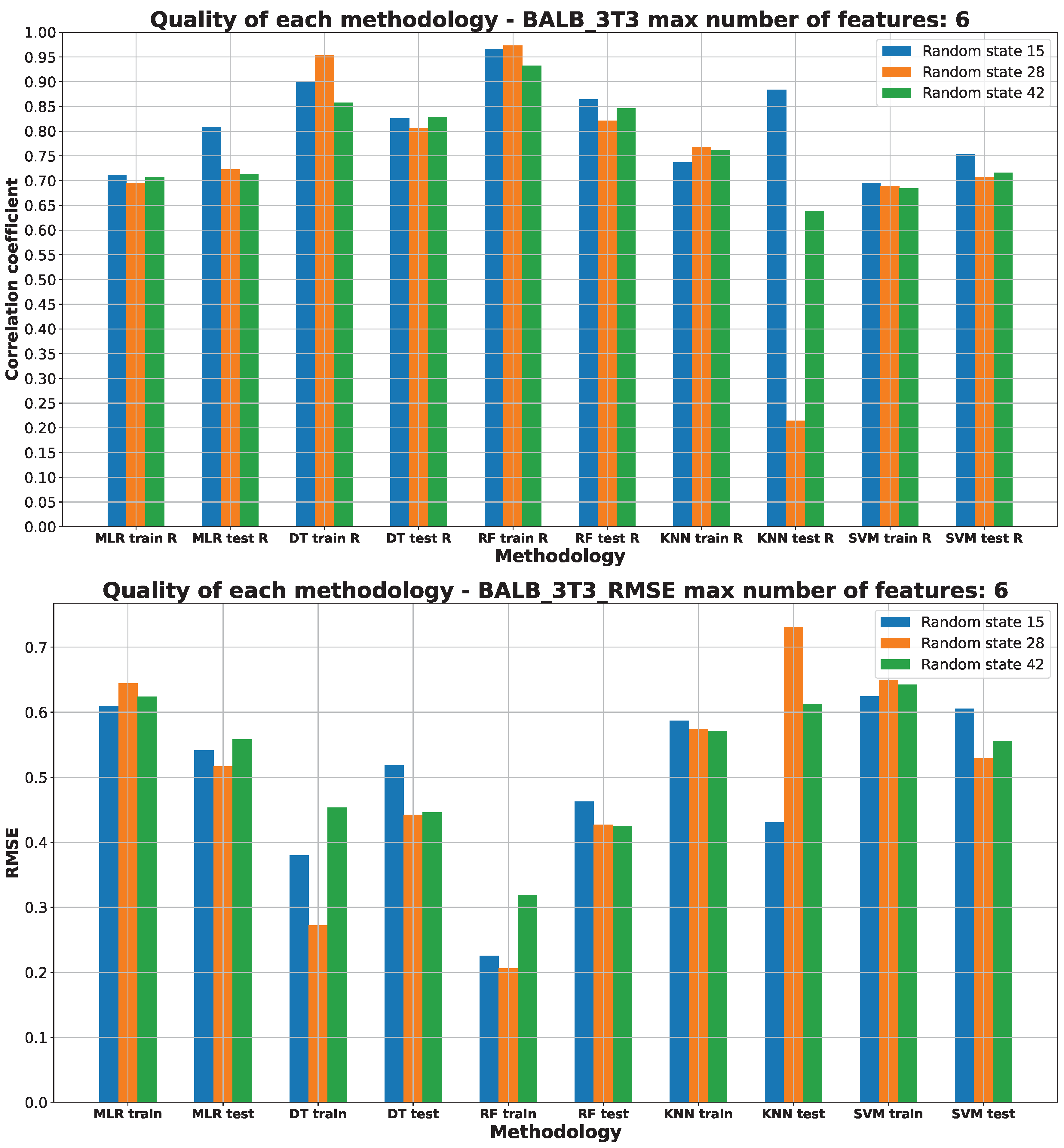
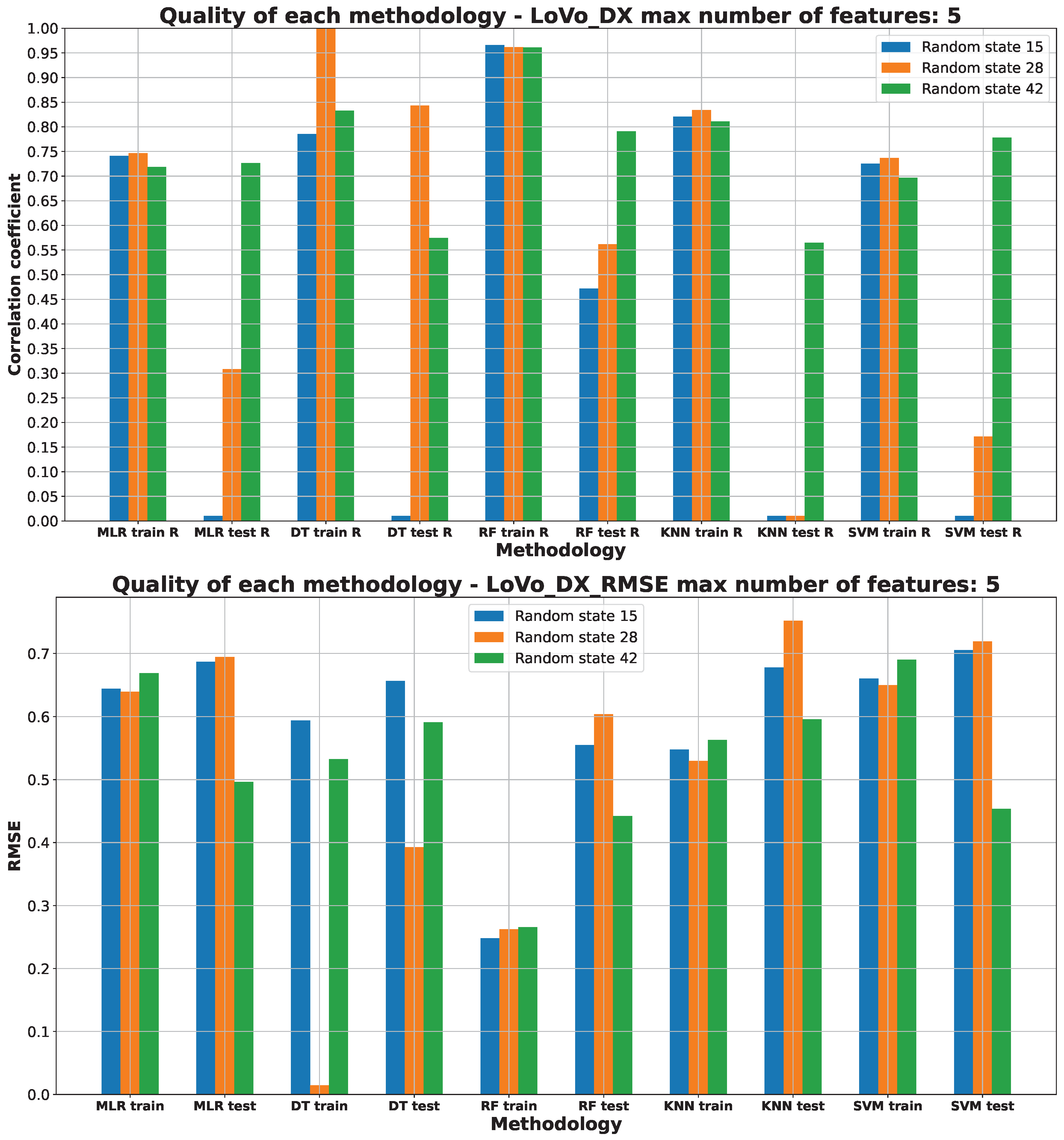
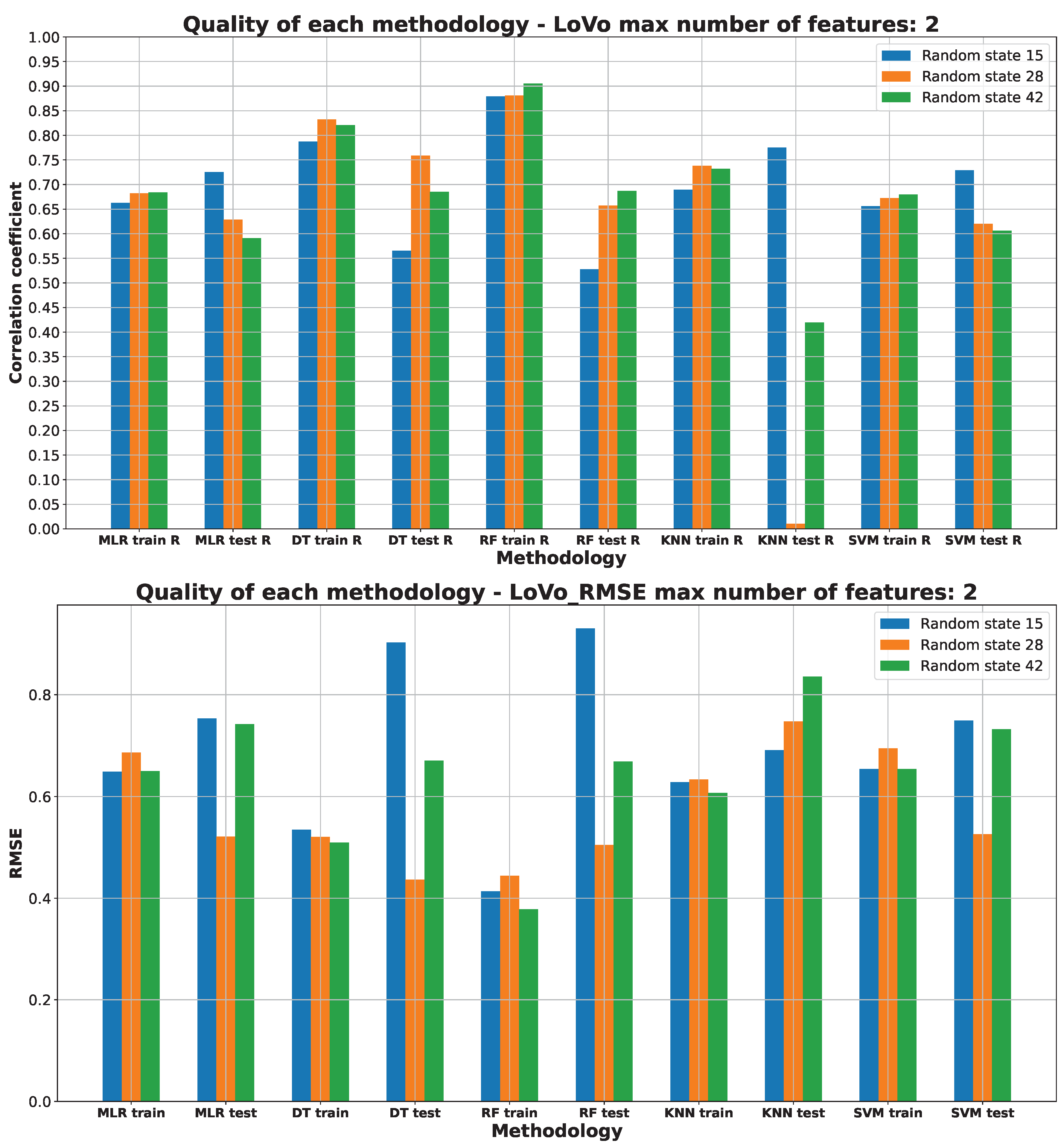
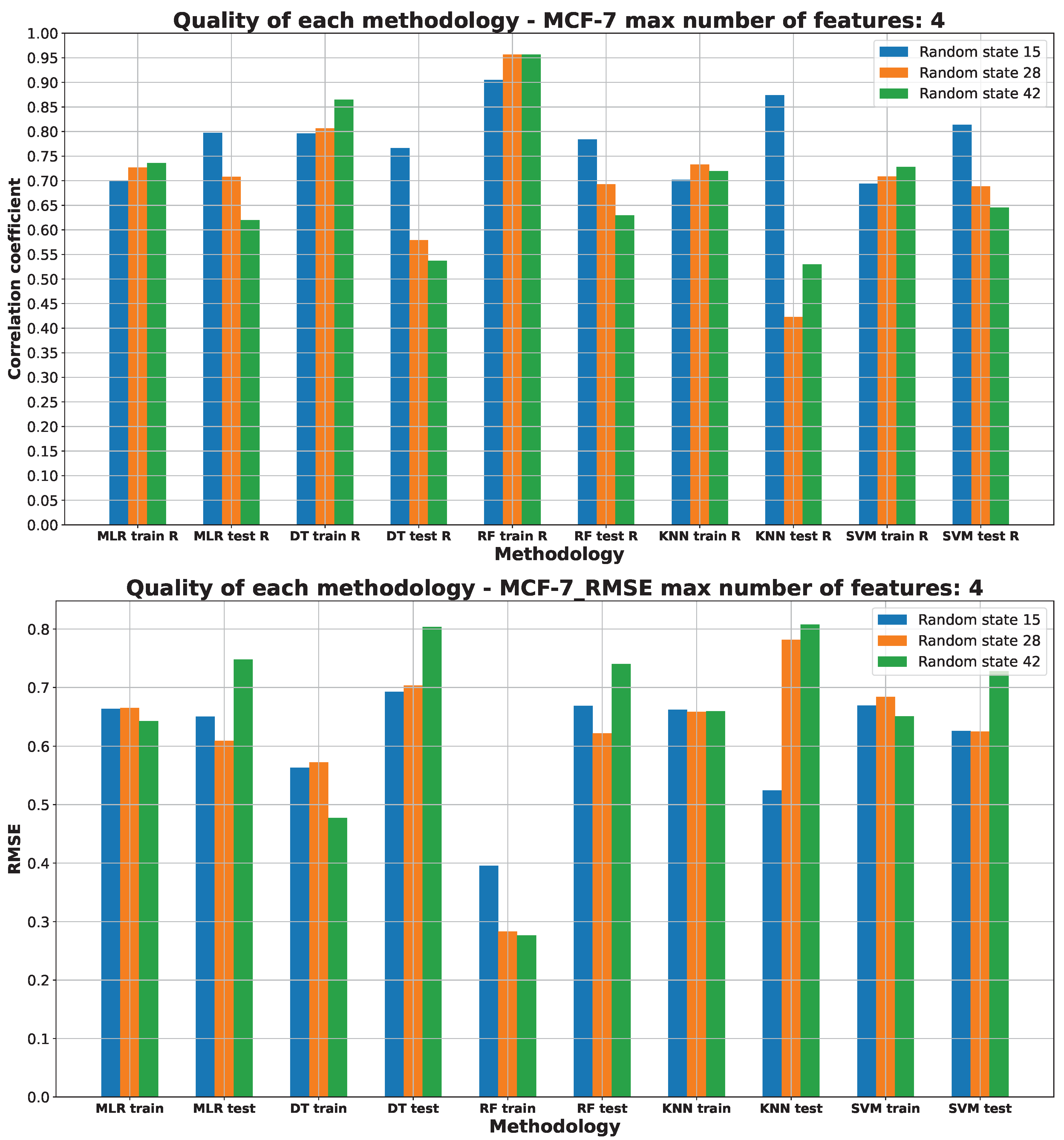
2.3. Machine Learning Models for Anticancer Activity





2.4. Data Selection
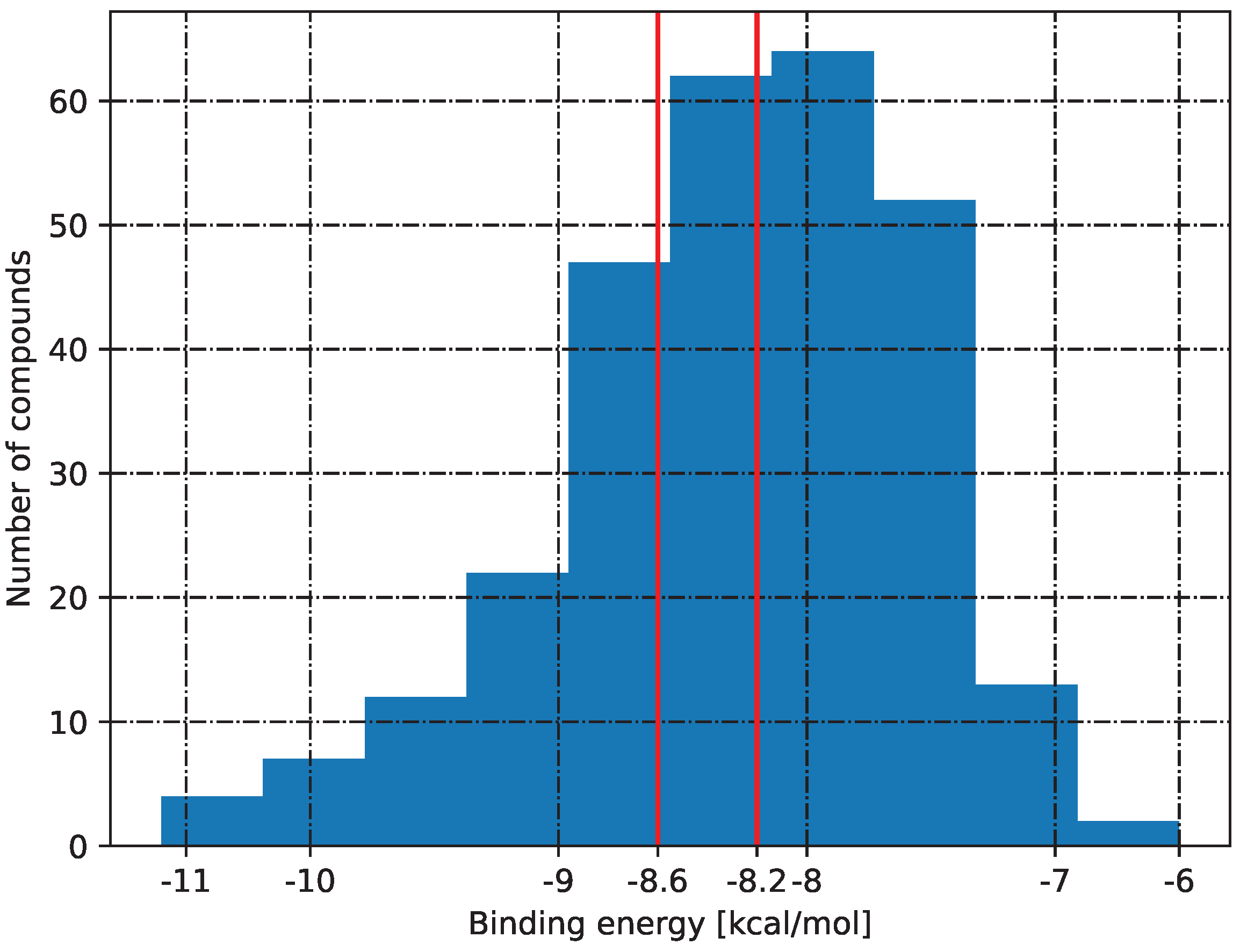
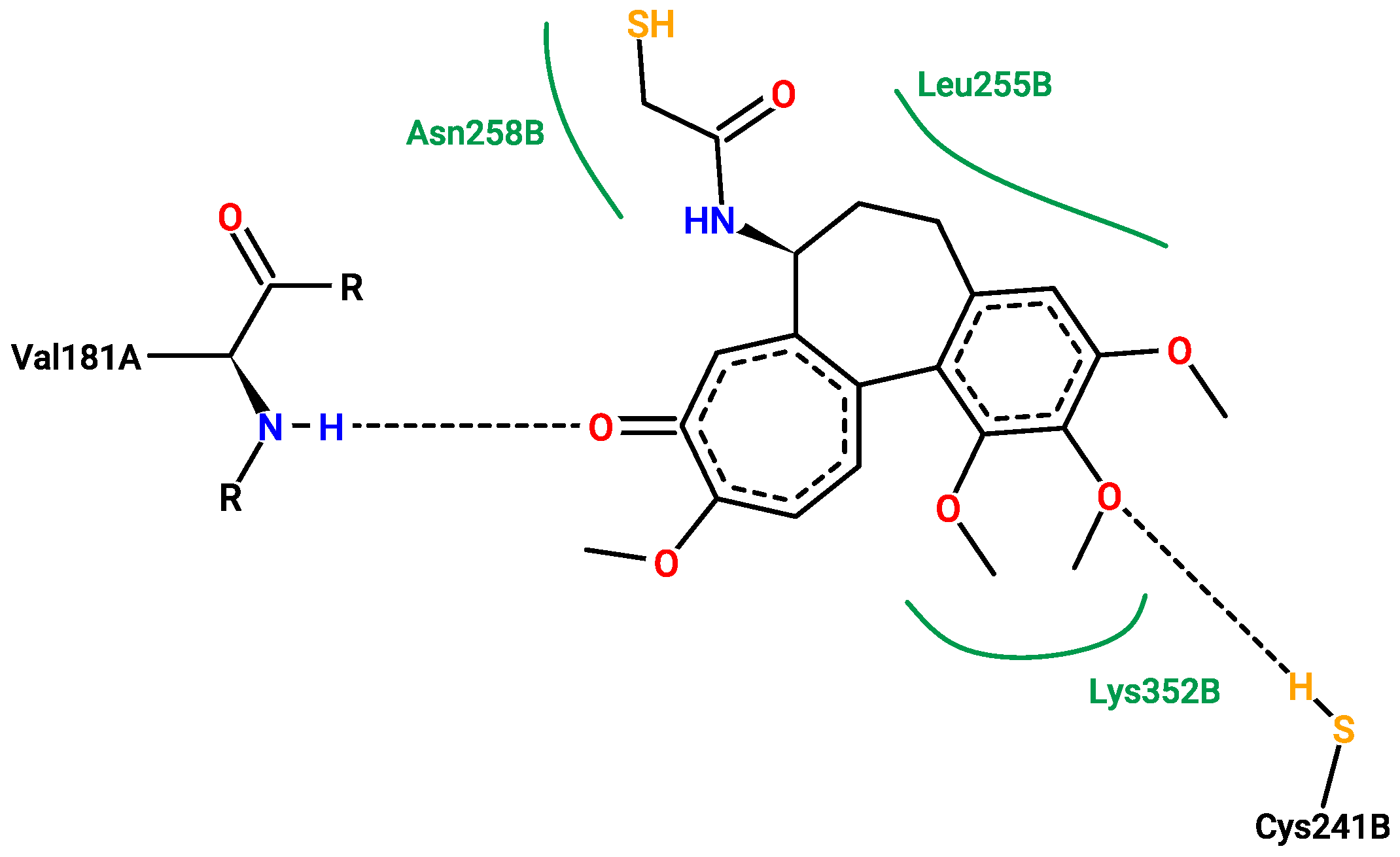
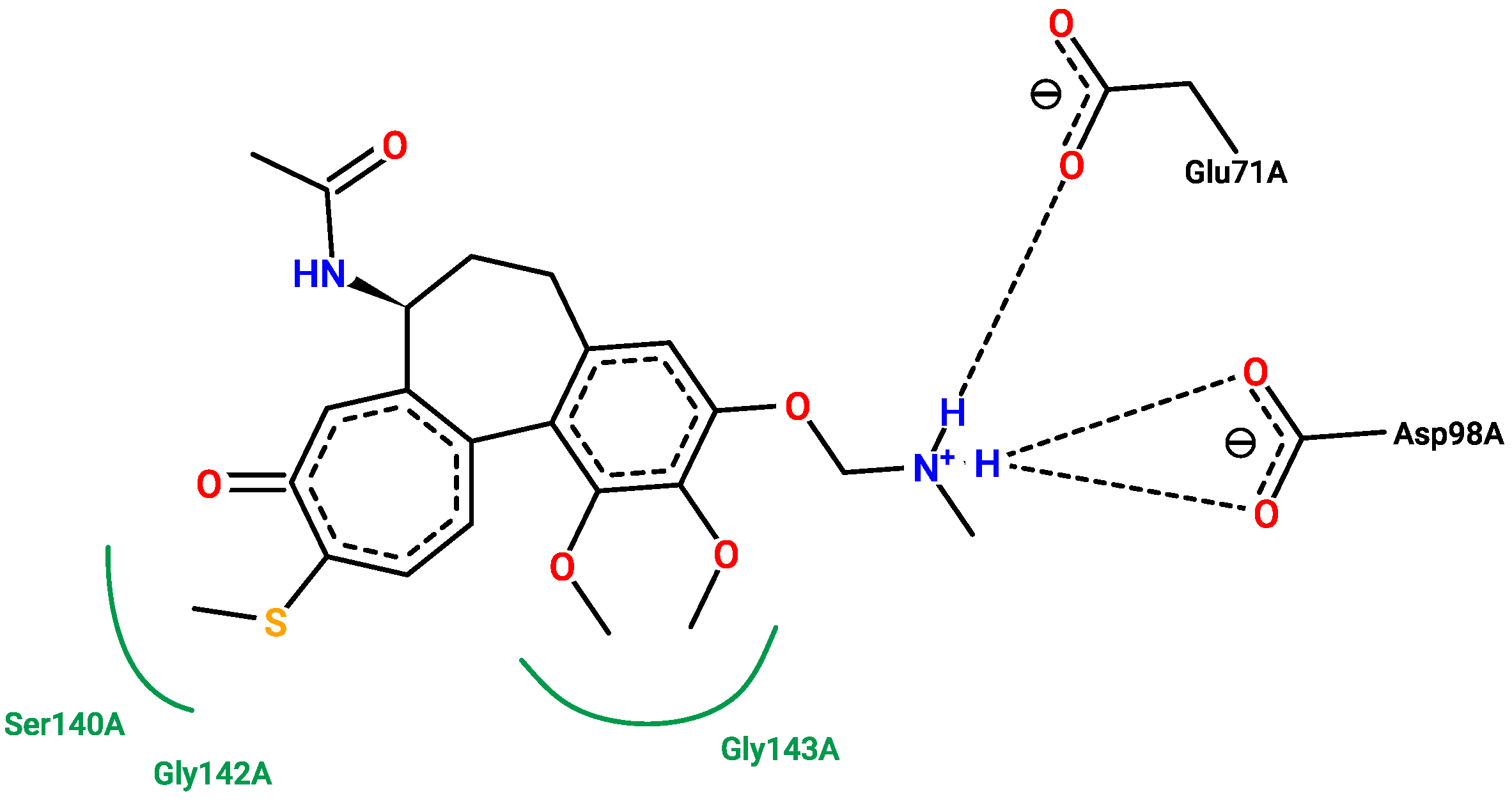
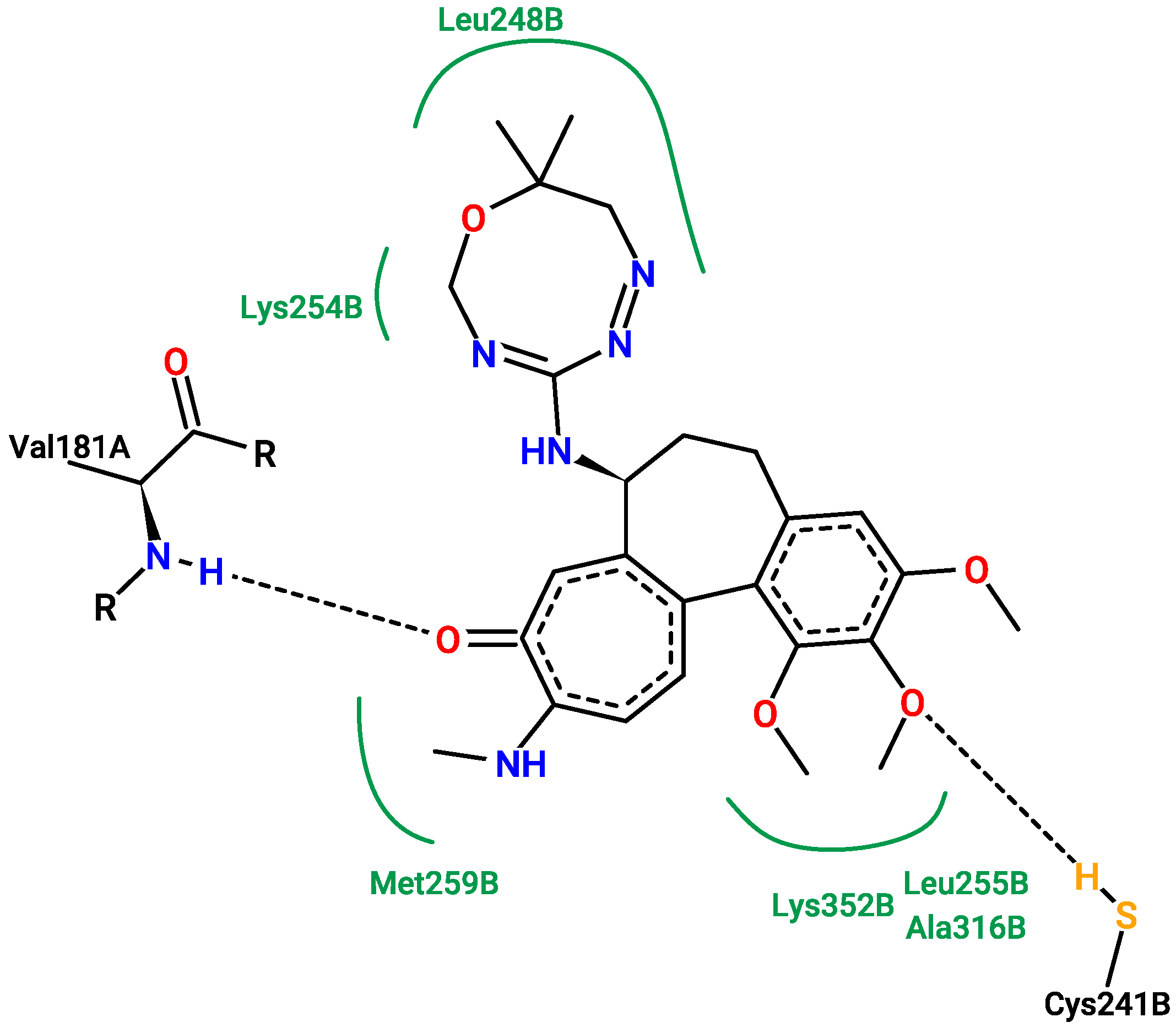
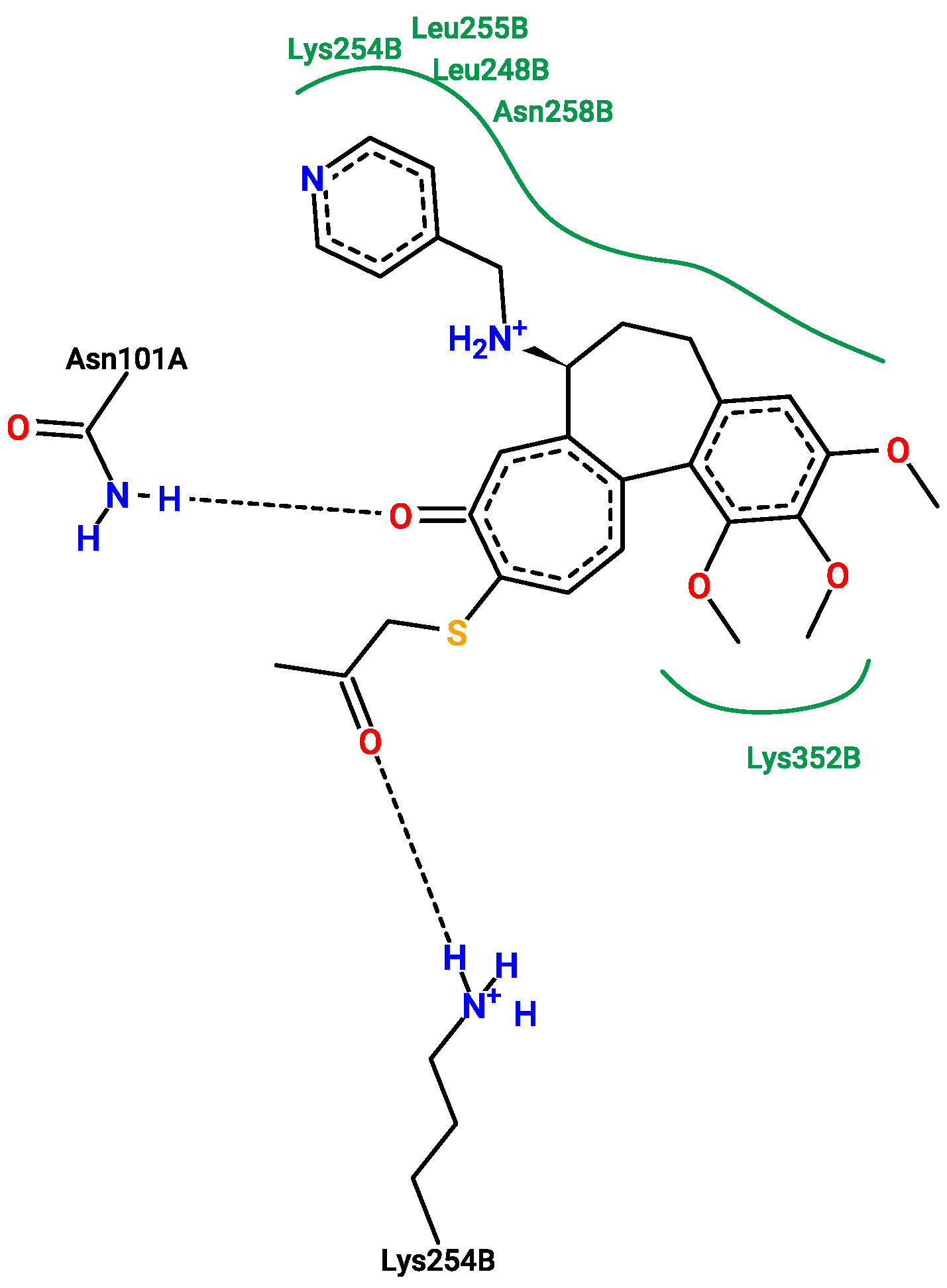
2.5. Molecular Docking
3. Materials and Methods
3.1. Training Data
3.2. Generative Neural Network
3.3. Machine Learning Models for Anticancer Activity
- 1.
- A549 (Supplementary Files S15–S17);
- 2.
- BALB/3T3 (Supplementary Files S18–S20);
- 3.
- LoVo/DX (Supplementary Files S21–S23);
- 4.
- LoVo (Supplementary Files S24–S26);
- 5.
- MCF-7 (Supplementary Files S27–S29).
3.4. Data Selection
- 1.
- Preservation of structures that are highly similar to the colchicine core. The Tanimoto similarity [43,56,57] threshold value was set to the lowest similarity found among the starting structures to the colchicine core, namely, 0.257 (Supplementary Files S37–S39).
- 2.
- The SYBA selection process, as documented in the study by Vorsilak et al. [42], this stage serves the purpose of eliminating structures that could pose challenges during the synthesis process. The SYBA algorithm yields a numerical SYBA score, wherein higher values indicate greater feasibility for molecular synthesis. The algorithm computes SYBA scores for the initial set of structures, and the lowest recorded score, set at 19.48 (documented in Supplementary File S40), is subsequently employed as the threshold for evaluating newly generated structures. The results of this analysis are stored in Supplementary File S41.
- 3.
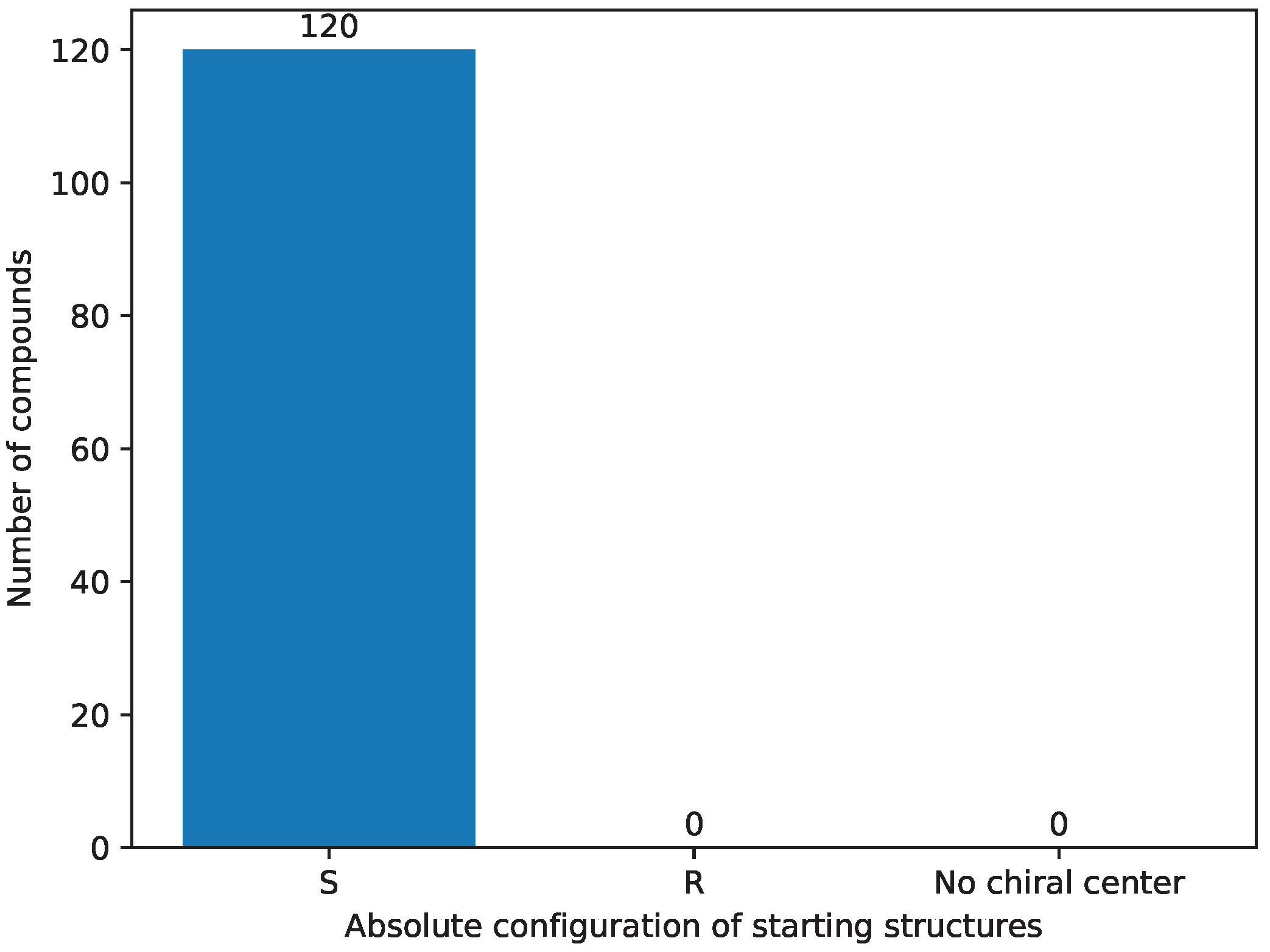
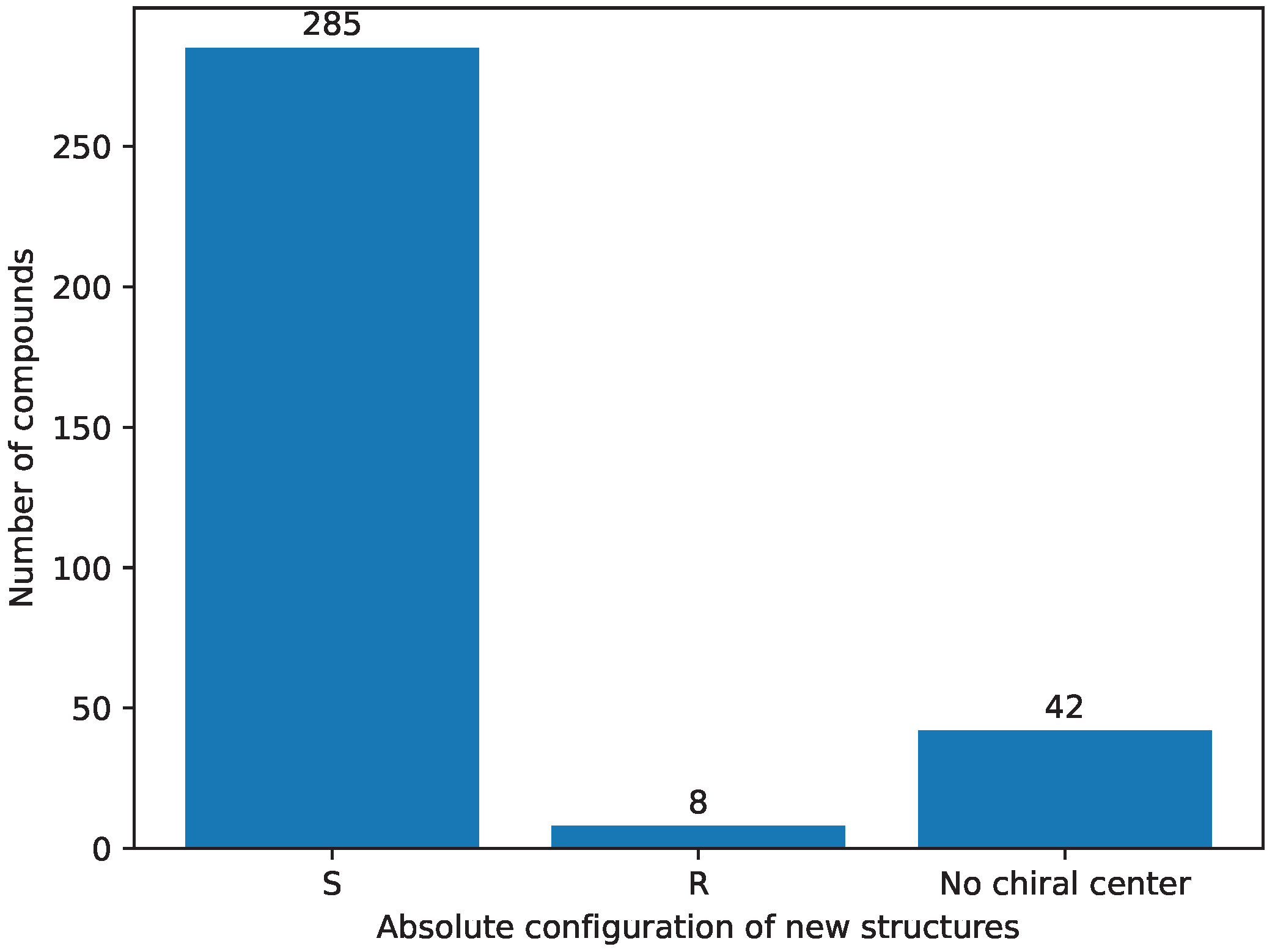
- Stereochemistry [9] selection was performed as the third step; it assumes compounds have the `S’ absolute configuration on the seventh carbon [58]. The `R’ absolute configuration structures do not tend to be biologically active [22,23,24,25,26], thus they are removed from further consideration. The results of this step are stored in Supplementary File S42.
- 4.
- The process of RI and SI selection is conducted subsequent to the prediction of values, as described in Section 3.3. This pivotal step enables the identification and retention of AI-generated colchicine-based structures that satisfy the prerequisites concerning drug resistance (RI) and specificity towards cancer cell lines (SI). The resultant indices are stored within Supplementary File S54.
3.5. Molecular Docking
- 1.
- Raw 1SA0.pdb structure was downloaded from the PDB;
- 2.
- Native ligand present inside the pocket was saved separately;
- 3.
- 3D structure of raw colchicine was prepared (Supplementary File S48);
- 4.
- All the selected new colchicine-based structures were transformed into 3D objects and prepared for molecular docking procedures (Supplementary Files S49 and S50);
- 5.
- Molecular docking was conducted (Supplementary File S51), and the results are saved in Supplementary File S52. The visualization of the results is in Supplementary File S53. The final results are stored in Supplementary File S54.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AI | Artificial intelligence |
| ML | Machine learning |
| RNN | Recurrent neural network |
| SMILES | Simplified molecular-input line-entry system |
| SELFIES | Self-referencing embedded strings |
| MLR | Multiple linear regression |
| DT | Decision tree |
| RF | Random forest |
| KNN | K-nearest neighbors |
| SVM | Support vector machines |
| R | Correlation coefficient |
| MSE | Mean squared error |
| MAE | Mean absolute error |
| RMSE | Root mean square error |
| PDB | Protein Data Bank |
| RI | Resistance index |
| SI | Selectivity index |
References
- Bjerrum, E.J.; Threlfall, R. Molecular Generation with Recurrent Neural Networks (RNNs). arXiv 2017, arXiv:1705.04612. [Google Scholar] [CrossRef]
- Kotsias, P.C.; Arús-Pous, J.; Chen, H.; Engkvist, O.; Tyrchan, C.; Bjerrum, E.J. Direct steering of de novo molecular generation with descriptor conditional recurrent neural networks. Nat. Mach. Intell. 2020, 2, 254–265. [Google Scholar] [CrossRef]
- Segler, M.H.S.; Kogej, T.; Tyrchan, C.; Waller, M.P. Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks. ACS Cent. Sci. 2018, 4, 120–131. [Google Scholar] [CrossRef]
- Nowak, D.; Bachorz, R.A.; Hoffmann, M. Neural Networks in the Design of Molecules with Affinity to Selected Protein Domains. Int. J. Mol. Sci. 2023, 24, 1762. [Google Scholar] [CrossRef]
- Ranjan, A.; Shukla, S.; Datta, D.; Misra, R. Generating novel molecule for target protein (SARS-CoV-2) using drug–target interaction based on graph neural network. Netw. Model Anal. Health Inform. Bioinform. 2022, 11, 6. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, Z.; Wang, L.; Wang, J.; Li, P.; Cao, D.; Zeng, X.; Ye, X.; Sakurai, T. Deep generative model for drug design from protein target sequence. J. Cheminform. 2023, 15, 38. [Google Scholar] [CrossRef]
- Li, J.N.; Yang, G.; Zhao, P.C.; Wei, X.X.; Shi, J.Y. CProMG: Controllable protein-oriented molecule generation with desired binding affinity and drug-like properties. Bioinformatics 2023, 39, i326–i336. [Google Scholar] [CrossRef]
- Bachorz, R.A.; Pastwińska, J.; Nowak, D.; Karaś, K.; Karwaciak, I.; Ratajewski, M. The application of machine learning methods to the prediction of novel ligands for RORγ/RORγT receptors. Comput. Struct. Biotechnol. J. 2023, 21, P5491–P5505. [Google Scholar] [CrossRef]
- Landrum, G. RDKit: Open-Source Cheminformatics Software. 2016. Available online: https://zenodo.org/records/7415128 (accessed on 16 August 2023).
- Moriwaki, H.; Tian, Y.S.; Kawashita, N.; Takagi, T. Mordred: A molecular descriptor calculator. J. Cheminform. 2018, 10, 4. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Model. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Nowak, D.; Babijczuk, K.; Jaya, L.O.I.; Bachorz, R.A.; Mrówczyńska, L.; Jasiewicz, B.; Hoffmann, M. Artificial Intelligence in Decrypting Cytoprotective Activity under Oxidative Stress from Molecular Structure. Int. J. Mol. Sci. 2023, 24, 11349. [Google Scholar] [CrossRef]
- Kumar, A.; Sharma, P.R.; Mondhe, D.M. Potential anticancer role of colchicine-based derivatives: An overview. Anti-Cancer Drugs 2017, 28, 250–262. [Google Scholar] [CrossRef]
- Aykul, S.; Martinez-Hackert, E. Determination of half-maximal inhibitory concentration using biosensor-based protein interaction analysis. Anal. Biochem. 2016, 508, 97–103. [Google Scholar] [CrossRef]
- Foster, K.A.; Oster, C.G.; Mayer, M.M.; Avery, M.L.; Audus, K.L. Characterization of the A549 Cell Line as a Type II Pulmonary Epithelial Cell Model for Drug Metabolism. Exp. Cell Res. 1998, 243, 359–366. [Google Scholar] [CrossRef]
- Aaronson, S.A.; Todaro, G.J. Development of 3T3-like lines from Balb-c mouse embryo cultures: Transformation susceptibility to SV40. J. Cell Physiol. 1968, 72, 141–148. [Google Scholar] [CrossRef]
- Grandi, M.; Geroni, C.; Giuliani, F.C. Isolation and characterization of a human colon adenocarcinoma cell line resistant to doxorubicin. Br. J. Cancer 1986, 54, 515–518. [Google Scholar] [CrossRef]
- Hsu, H.H.; Chen, M.C.; Day, C.H.; Lin, Y.M.; Li, S.Y.; Tu, C.C.; Padma, V.V.; Shih, H.N.; Kuo, W.W.; Huang, C.Y. Thymoquinone suppresses migration of LoVo human colon cancer cells by reducing prostaglandin E2 induced COX-2 activation. World J. Gastroenterol. 2017, 23, 1171–1179. [Google Scholar] [CrossRef]
- Lee, A.V.; Oesterreich, S.; Davidson, N.E. MCF-7 Cells—Changing the Course of Breast Cancer Research and Care for 45 Years. JNCI J. Natl. Cancer Inst. 2015, 107, djv073. [Google Scholar] [CrossRef]
- Klejborowska, G.; Urbaniak, A.; Maj, E.; Wietrzyk, J.; Moshari, M.; Preto, J.; Tuszynski, J.A.; Chambers, T.C.; Huczyński, A. Synthesis, anticancer activity and molecular docking studies of N-deacetylthiocolchicine and 4-iodo-N-deacetylthiocolchicine derivatives. Bioorg. Med. Chem. 2021, 32, 116014. [Google Scholar] [CrossRef]
- Huczyński, A.; Rutkowski, J.; Popiel, K.; Maj, E.; Wietrzyk, J.; Stefańska, J.; Majcher, U.; Bartl, F. Synthesis, antiproliferative and antibacterial evaluation of C-ring modified colchicine analogues. Eur. J. Med. Chem. 2015, 90, 296–301. [Google Scholar] [CrossRef]
- Czerwonka, D.; Sobczak, S.; Maj, E.; Wietrzyk, J.; Katrusiak, A.; Huczyński, A. Synthesis and Antiproliferative Screening of Novel Analogs of Regioselectively Demethylated Colchicine and Thiocolchicine. Molecules 2020, 25, 1180. [Google Scholar] [CrossRef] [PubMed]
- Krzywik, J.; Aminpour, M.; Maj, E.; Mozga, W.; Wietrzyk, J.; Tuszyński, J.A.; Huczyński, A. New Series of Double-Modified Colchicine Derivatives: Synthesis, Cytotoxic Effect and Molecular Docking. Molecules 2020, 25, 3540. [Google Scholar] [CrossRef]
- Czerwonka, D.; Maj, E.; Wietrzyk, J.; Huczyński, A. Synthesis of thiocolchicine amine derivatives and evaluation of their antiproliferative activity. Bioorg. Med. Chem. Lett. 2021, 52, 128382. [Google Scholar] [CrossRef]
- Krzywik, J.; Nasulewicz-Goldeman, A.; Mozga, W.; Wietrzyk, J.; Huczyński, A. Novel Double-Modified Colchicine Derivatives Bearing 1,2,3-Triazole: Design, Synthesis, and Biological Activity Evaluation. ACS Omega 2021, 6, 26583–26600. [Google Scholar] [CrossRef]
- Krzywik, J.; Maj, E.; Nasulewicz-Goldeman, A.; Mozga, W.; Wietrzyk, J.; Huczyński, A. Synthesis and antiproliferative screening of novel doubly modified colchicines containing urea, thiourea and guanidine moieties. Bioorg. Med. Chem. Lett. 2021, 47, 128197. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 update. Nucleic Acids Res. 2023, 51, D1373–D1380. [Google Scholar] [CrossRef]
- Galton, F. Regression Towards Mediocrity in Hereditary Stature. J. Anthropol. Inst. Great Br. Irel. 1886, 15, 246. [Google Scholar] [CrossRef]
- Jobson, J.D. Multiple Linear Regression. In Applied Multivariate Data Analysis; Springer Texts in Statistics; Springer: New York, NY, USA, 1991; pp. 219–398. [Google Scholar] [CrossRef]
- von Winterfeldt, D.; Edwards, W. Decision Analysis and Behavioral Research; Cambridge University Press: Cambridge, UK, 1986. [Google Scholar]
- Ho, T.K. Random decision forests. In Proceedings of the 3rd International Conference on Document Analysis and Recognition, San José, CA, USA, 21–26 August 1995; Volume 1, pp. 278–282. [Google Scholar]
- Fix, E.; Hodges, J.L. Discriminatory Analysis. Nonparametric Discrimination: Consistency Properties. Int. Stat. Rev. Rev. Int. Stat. 1989, 57, 238. [Google Scholar] [CrossRef]
- Awad, M.; Khanna, R. Support Vector Regression. In Efficient Learning Machines; Apress: New York, NY, USA, 2015; pp. 67–80. [Google Scholar] [CrossRef]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar] [CrossRef]
- Choi, J.; Park, S.; Ahn, J. RefDNN: A reference drug based neural network for more accurate prediction of anticancer drug resistance. Sci. Rep. 2020, 10, 1861. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Lai, X.; Pan, Y.; Zheng, H.; Vera, J.; Liu, S.; Deng, S.; Zhang, L. Artificial intelligence in cancer target identification and drug discovery. Sig. Transduct. Target Ther. 2022, 7, 156. [Google Scholar] [CrossRef]
- Everitt, B.; Skrondal, A. The Cambridge Dictionary of Statistics, 4th ed.; Cambridge University Press: Cambridge, UK, 2010. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Nguyen, N.T.; Nguyen, T.H.; Pham, T.N.H.; Huy, N.T.; Bay, M.V.; Pham, M.Q.; Nam, P.C.; Vu, V.V.; Ngo, S.T. Autodock Vina Adopts More Accurate Binding Poses but Autodock4 Forms Better Binding Affinity. J. Chem. Inf. Model. 2020, 60, 204–211. [Google Scholar] [CrossRef]
- Plewczynski, D.; Łaźniewski, M.; Augustyniak, R.; Ginalski, K. Can we trust docking results? Evaluation of seven commonly used programs on PDBbind database. J. Comput. Chem. 2011, 32, 742–755. [Google Scholar] [CrossRef]
- Ravelli, R.B.; Gigant, B.; Curmi, P.A.; Jourdain, I.; Lachkar, S.; Sobel, A.; Knossow, M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 2004, 428, 198–202. [Google Scholar] [CrossRef]
- Voršilák, M.; Kolář, M.; Čmelo, I.; Svozil, D. SYBA: Bayesian estimation of synthetic accessibility of organic compounds. J. Cheminform. 2020, 12, 35. [Google Scholar] [CrossRef]
- Maggiora, G.; Vogt, M.; Stumpfe, D.; Bajorath, J. Molecular Similarity in Medicinal Chemistry: Miniperspective. J. Med. Chem. 2014, 57, 3186–3204. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Open Babel Development Team. Open Babel. Version: 3.1.1. 2020. Available online: http://openbabel.org/wiki/Main_Page (accessed on 14 October 2023).
- Mauri, A.; Consonni, V.; Todeschini, R. Molecular Descriptors. In Handbook of Computational Chemistry; Leszczynski, J., Ed.; Springer: Dordrecht, The Netherlands, 2016; pp. 1–29. [Google Scholar] [CrossRef]
- Müller, A.C.; Guido, S. Introduction to Machine Learning with Python: A Guide for Data Scientists, 1st ed.; O’Reilly Media, Inc.: Sebastopol, CA, USA, 2016; ISBN 978-1-4493-6941-5. [Google Scholar]
- Freedman, D.; Pisani, R.; Purves, R. Statistics (International Student Edition), 4th ed.; WW Norton & Company: New York, NY, USA, 2007. [Google Scholar]
- Chai, T.; Draxler, R.R. Root mean square error (RMSE) or mean absolute error (MAE)?— Arguments against avoiding RMSE in the literature. Geosci. Model Dev. 2014, 7, 1247–1250. [Google Scholar] [CrossRef]
- Chicco, D.; Warrens, M.J.; Jurman, G. The coefficient of determination R-squared is more informative than SMAPE, MAE, MAPE, MSE and RMSE in regression analysis evaluation. PeerJ Comput. Sci. 2021, 7, e623. [Google Scholar] [CrossRef] [PubMed]
- Lučić, B.; Batista, J.; Bojović, V.; Lovrić, M.; Sović Kržić, A.; Bešlo, D.; Nadramija, D.; Vikić-Topić, D. Estimation of Random Accuracy and its Use in Validation of Predictive Quality of Classification Models within Predictive Challenges. Croat. Chem. Acta 2019, 92, 379–391. [Google Scholar] [CrossRef]
- Hellström, T.; Dignum, V.; Bensch, S. Bias in Machine Learning—What is it Good for? arXiv 2020, arXiv:2004.00686. [Google Scholar] [CrossRef]
- Mordred Descriptor List. Available online: https://mordred-descriptor.github.io/documentation/master/descriptors.html (accessed on 7 December 2023).
- Schluchter, M.D. Mean Square Error. In Wiley StatsRef: Statistics Reference Online, 1st ed.; Balakrishnan, N., Colton, T., Everitt, B., Piegorsch, W., Ruggeri, F., Teugels, J.L., Eds.; Wiley: Hoboken, NJ, USA, 2014. [Google Scholar] [CrossRef]
- Fürnkranz, J.; Chan, P.K.; Craw, S.; Sammut, C.; Uther, W.; Ratnaparkhi, A.; Jin, X.; Han, J.; Yang, Y.; Morik, K.; et al. Mean Absolute Error. In Encyclopedia of Machine Learning; Sammut, C., Webb, G.I., Eds.; Springer: New York, NY, USA, 2011; p. 652. [Google Scholar] [CrossRef]
- Cereto-Massagué, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Garcia-Vallvé, S.; Pujadas, G. Molecular fingerprint similarity search in virtual screening. Methods 2015, 71, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Bajorath, J. Selected Concepts and Investigations in Compound Classification, Molecular Descriptor Analysis, and Virtual Screening. J. Chem. Inf. Comput. Sci. 2001, 41, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Gao, R.; Gu, X.; Yu, B.; Wu, Y.; Li, Q.; Xiang, P.; Xu, H. A New Insight into Toxicity of Colchicine Analogues by Molecular Docking Analysis Based on Intestinal Tight Junction Protein ZO-1. Molecules 2022, 27, 1797. [Google Scholar] [CrossRef] [PubMed]
- Berdzik, N.; Jasiewicz, B.; Ostrowski, K.; Sierakowska, A.; Szlaużys, M.; Nowak, D.; Mrówczyńska, L. Novel gramine-based bioconjugates obtained by click chemistry as cytoprotective compounds and potent antibacterial and antifungal agents. Nat. Prod. Res. 2023, 1–7. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Jurafsky, D.; Martin, J.H. Speech and Language Processing: An Introduction to Natural Language Processing, Computational Linguistics, and Speech Recognition; Prentice Hall Series in Artificial Intelligence; Prentice Hall: Upper Saddle River, NJ, USA, 2000. [Google Scholar]
- Krenn, M.; Häse, F.; Nigam, A.; Friederich, P.; Aspuru-Guzik, A. Self-Referencing Embedded Strings (SELFIES): A 100% robust molecular string representation. arXiv 2019, arXiv:1905.13741. [Google Scholar] [CrossRef]
- Murphy, K.P. Probabilistic Machine Learning: An Introduction; MIT Press: Cambridge, MA, USA, 2022. [Google Scholar]
- van der Maaten, L.; Hinton, G. Visualizing Data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Swain, M. PuBChemPy. 2017. Available online: https://github.com/mcs07/PubChemPy/ (accessed on 4 August 2023).
- RCBS Protein Data Bank. 1971. Available online: https://www.rcsb.org/ (accessed on 9 August 2023).
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- ProteinsPlus Development Team. ProteinsPlus. Available online: https://proteins.plus (accessed on 14 October 2023).
- Fährrolfes, R.; Bietz, S.; Flachsenberg, F.; Meyder, A.; Nittinger, E.; Otto, T.; Volkamer, A.; Rarey, M. ProteinsPlus: A web portal for structure analysis of macromolecules. Nucleic Acids Res. 2017, 45, W337–W343. [Google Scholar] [CrossRef]
- Schöning-Stierand, K.; Diedrich, K.; Fährrolfes, R.; Flachsenberg, F.; Meyder, A.; Nittinger, E.; Steinegger, R.; Rarey, M. ProteinsPlus: Interactive analysis of protein–ligand binding interfaces. Nucleic Acids Res. 2020, 48, W48–W53. [Google Scholar] [CrossRef] [PubMed]
- Schöning-Stierand, K.; Diedrich, K.; Ehrt, C.; Flachsenberg, F.; Graef, J.; Sieg, J.; Penner, P.; Poppinga, M.; Ungethüm, A.; Rarey, M. Proteins Plus: A comprehensive collection of web-based molecular modeling tools. Nucleic Acids Res. 2022, 50, W611–W615. [Google Scholar] [CrossRef]
- Stierand, K.; Rarey, M. Drawing the PDB: Protein–Ligand Complexes in Two Dimensions. ACS Med. Chem. Lett. 2010, 1, 540–545. [Google Scholar] [CrossRef]
- Stierand, K.; Rarey, M. From Modeling to Medicinal Chemistry: Automatic Generation of Two-Dimensional Complex Diagrams. ChemMedChem 2007, 2, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Stierand, K.; Maaß, P.C.; Rarey, M. Molecular complexes at a glance: Automated generation of two-dimensional complex diagrams. Bioinformatics 2006, 22, 1710–1716. [Google Scholar] [CrossRef] [PubMed]
- Fricker, P.C.; Gastreich, M.; Rarey, M. Automated Drawing of Structural Molecular Formulas under Constraints. J. Chem. Inf. Comput. Sci. 2004, 44, 1065–1078. [Google Scholar] [CrossRef]
- Diedrich, K.; Krause, B.; Berg, O.; Rarey, M. PoseEdit: Enhanced ligand binding mode communication by interactive 2D diagrams. J. Comput. Aided Mol. Des. 2023, 37, 491–503. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Starting structures 1 | |
 (nM): A549: 10.8 ± 1.4, MCF-7: 10.3 ± 0.4, LoVo: 6.5 ± 1.9, LoVo/DX: 54.9 ± 22.0, BALB/3T3: 10.2 ± 1.9 SMILES: COc2c3C1=CC=C(SC)C(=O)C=C1[C@H](CCc3cc(OC)c2OC)NCC |  (nM): A549: 9.6 ± 1.3, MCF-7: 9.7 ± 1.5, LoVo: 7.8 ± 1.0, LoVo/DX: 8.5 ± 1.1, BALB/3T3: 7.5 ± 1.5 SMILES: CC(CC)N[C@H]2CCc3cc(OC)c(OC)c(OC)c3C1=CC=C(NC)C(=O)C=C12 |
| Newly proposed structures 1 | |
 Tanimoto similarity: 0.844 (nM): A549: 8.3, MCF-7: 8.4, LoVo: 6.2, LoVo/DX: 94.3, BALB/3T3: 10.9 SMILES: COC1=C2C3=CC=C(NC)C(=O)C=C3[C@H1](CCC2=CC(OC)=C1OC)NCCONC |  Tanimoto similarity: 0.931 (nM): A549: 11.3, MCF-7: 7.9, LoVo: 9.6, LoVo/DX: 129.6, BALB/3T3: 21.3 SMILES: CNC(C(C)C)N[C@@H1]1C=2C(C3=C(C=C(OC)C(OC)=C3OC)CC1)=CC=C(C(=O)C=2)NC |
 Tanimoto similarity: 0.953 (nM): A549: 11.2, MCF-7: 12.7, LoVo: 14.5, LoVo/DX: 71.1, BALB/3T3: 10.2 SMILES: C1=2[C@H1](CCC3=CC(OC)=C(OC)C(OC)=C3C1=CC=C(SC)C(=O)C=2)NC(C)CC=CSC |  Tanimoto similarity: 0.978 (nM): A549: 15.9, MCF-7: 8.6, LoVo: 9.6, LoVo/DX: 21.6, BALB/3T3: 23.5 SMILES: N([C@H1]1CCC2=CC(OC)=C(OC)C(OC)=C2C3=CC=C(NC)C(C=C31)=O)CCCCF |
 Tanimoto similarity: 0.983 (nM): A549: 10.6, MCF-7: 12.2, LoVo: 14.5, LoVo/DX: 18.1, BALB/3T3: 11.5 SMILES: COC1=C2C3=CC=C(SC)C(=O)C=C3[C@H1](CCC2=CC(OC)=C1OC)NCCCCl |  Tanimoto similarity: 0.931 (nM): A549: 900, MCF-7: 4259, LoVo: 345, LoVo/DX: 9490, BALB/3T3: 835 SMILES: O=C(N[C@H1]1CCC2=CC(OC)=C(OC)C(OC)=C2C3=CC=C(NC)C(=O)C=C31)CCOC(=O)O |
| Target | Methodology | Random State | Number of Features | Correlation Threshold | Overall R Score | MSE | MAE | RMSE |
|---|---|---|---|---|---|---|---|---|
| A549 | RF | 15 | 5 | 0.51 | 0.944 | 0.099 | 0.205 | 0.314 |
| BALB/3T3 | RF | 15 | 6 | 0.51 | 0.950 | 0.075 | 0.183 | 0.274 |
| LoVo/DX | RF | 42 | 5 | 0.63 | 0.950 | 0.089 | 0.217 | 0.299 |
| LoVo | RF | 28 | 2 | 0.54 | 0.865 | 0.206 | 0.320 | 0.453 |
| MCF-7 | RF | 15 | 4 | 0.51 | 0.883 | 0.200 | 0.258 | 0.447 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nowak, D.; Huczyński, A.; Bachorz, R.A.; Hoffmann, M. Machine Learning Application for Medicinal Chemistry: Colchicine Case, New Structures, and Anticancer Activity Prediction. Pharmaceuticals 2024, 17, 173. https://doi.org/10.3390/ph17020173
Nowak D, Huczyński A, Bachorz RA, Hoffmann M. Machine Learning Application for Medicinal Chemistry: Colchicine Case, New Structures, and Anticancer Activity Prediction. Pharmaceuticals. 2024; 17(2):173. https://doi.org/10.3390/ph17020173
Chicago/Turabian StyleNowak, Damian, Adam Huczyński, Rafał Adam Bachorz, and Marcin Hoffmann. 2024. "Machine Learning Application for Medicinal Chemistry: Colchicine Case, New Structures, and Anticancer Activity Prediction" Pharmaceuticals 17, no. 2: 173. https://doi.org/10.3390/ph17020173
APA StyleNowak, D., Huczyński, A., Bachorz, R. A., & Hoffmann, M. (2024). Machine Learning Application for Medicinal Chemistry: Colchicine Case, New Structures, and Anticancer Activity Prediction. Pharmaceuticals, 17(2), 173. https://doi.org/10.3390/ph17020173