

Present Scenario and Future Landscape of Payloads for ADCs: Focus on DNA-Interacting Agents

Abstract
1. Introduction
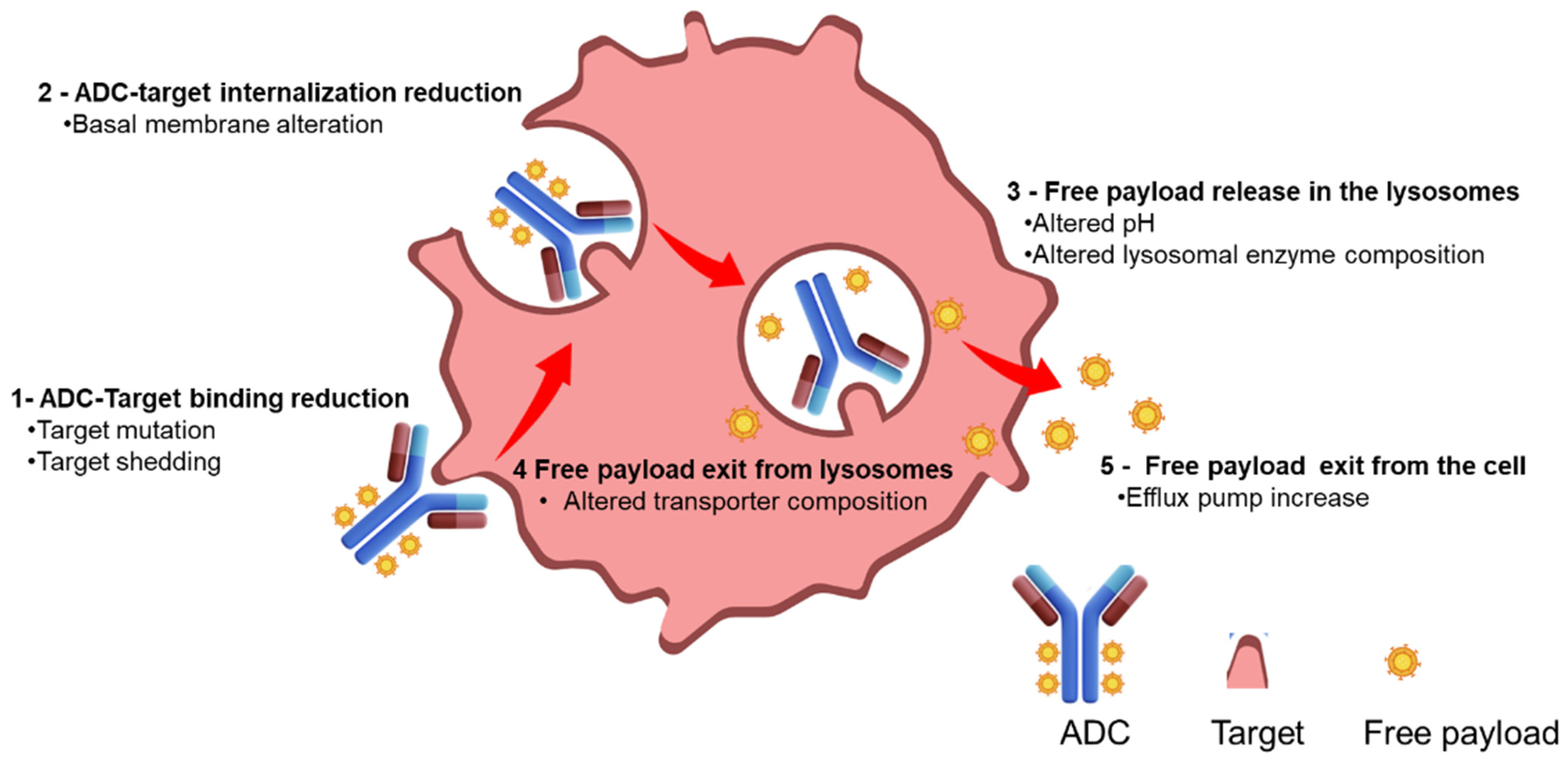
2. Mechanism of Resistance
3. Current Status of ADCs Containing DNA-Binding Payloads
4. DNA-Binding Payload Background and Clinical Results
4.1. Calicheamicin
4.2. Pyrrolobenzodiazepine Dimers (PBD)
4.3. Indolino Benzodiazepine Dimers (IGN)
4.4. Anthracycline
4.5. Duocarmycins
5. Comparative Evaluation of DNA Interacting Payload Features
6. Conclusions and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- ADC Beacon [Database]. 2024. Available online: https://beacon-intelligence.com (accessed on 21 September 2024).
- Hamann, P.R.; Hinman, L.M.; Hollander, I.; Beyer, C.F.; Lindh, D.; Holcomb, R.; Hallett, W.; Tsou, H.-R.; Upeslacis, J.; Shochat, D.; et al. Gemtuzumab ozogamicin, a potent and selective anti-CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Bioconjug. Chem. 2002, 13, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.; Chari, R.V.J. Ado-trastuzumab Emtansine (T-DM1): An Antibody–Drug Conjugate (ADC) for HER2-Positive Breast Cancer. J. Med. Chem. 2014, 57, 6949–6964. [Google Scholar] [CrossRef] [PubMed]
- Wynne, J.; Wright, D.; Stock, W. Inotuzumab: From preclinical development to success in B-cell acute lymphoblastic leukemia. Blood Adv. 2019, 3, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Smith, S.W.; Ghone, S.; Tomczuk, B. Current ADC Linker Chemistry. Pharm. Res. 2015, 32, 3526–3540. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.-B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Burki, T.K. Sacituzumab govitecan activity in advanced breast cancer. Lancet Oncol. 2017, 18, e246. [Google Scholar] [CrossRef]
- Panowski, S.; Bhakta, S.; Raab, H.; Polakis, P.; Junutula, J.R. Site-specific antibody drug conjugates for cancer therapy. MAbs 2014, 6, 34–45. [Google Scholar] [CrossRef]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable linkers in antibody-drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef]
- Giugliano, F.; Corti, C.; Tarantino, P.; Michelini, F.; Curigliano, G. Bystander effect of antibody-drug conjugates: Fact or fiction? Curr. Oncol. Rep. 2022, 24, 809–817. [Google Scholar] [CrossRef]
- Modi, S.; Jacot, W.; Yamashita, T.; Sohn, J.; Vidal, M.; Tokunaga, E.; Tsurutani, J.; Ueno, N.T.; Prat, A.; Chae, Y.S.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. N. Engl. J. Med. 2022, 387, 9–20. [Google Scholar] [CrossRef]
- D’Arienzo, A.; Verrazzo, A.; Pagliuca, M.; Napolitano, F.; Parola, S.; Viggiani, M.; Caputo, R.; Puglisi, F.; Giuliano, M.; Del Mastro, L.; et al. Toxicity profile of antibody-drug conjugates in breast cancer: Practical considerations. EClinicalMedicine 2023, 62, 102113. [Google Scholar] [CrossRef] [PubMed]
- García-Alonso, S.; Ocaña, A.; Pandiella, A. Resistance to Antibody–Drug Conjugates. Cancer Res. 2018, 78, 2159–2165. [Google Scholar] [CrossRef] [PubMed]
- Bosi, C.; Bartha, Á.; Galbardi, B.; Notini, G.; Naldini, M.M.; Licata, L.; Viale, G.; Mariani, M.; Pistilli, B.; Ali, H.R.; et al. Pan-cancer analysis of antibody-drug conjugate targets and putative predictors of treatment response. Eur. J. Cancer 2023, 195, 113379. [Google Scholar] [CrossRef] [PubMed]
- Torka, P.; Barth, M.; Ferdman, R.; Hernandez-Ilizaliturri, F.J. Mechanisms of Resistance to Monoclonal Antibodies (mAbs) in Lymphoid Malignancies. Curr. Hematol. Malig. Rep. 2019, 14, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Khoury, R.; Saleh, K.; Khalife, N.; Saleh, M.; Chahine, C.; Ibrahim, R.; Lecesne, A. Mechanisms of Resistance to Antibody-Drug Conjugates. Int. J. Mol. Sci. 2023, 24, 9674. [Google Scholar] [CrossRef]
- Loganzo, F.; Tan, X.; Sung, M.; Jin, G.; Myers, J.S.; Melamud, E.; Wang, F.; Diesl, V.; Follettie, M.T.; Musto, S.; et al. Tumor cells chronically treated with a trastuzumab-maytansinoid antibody-drug conjugate develop varied resistance mechanisms but respond to alternate treatments. Mol. Cancer Ther. 2015, 14, 952–963. [Google Scholar] [CrossRef]
- Escrivá-de-Romaní, S.; Saura, C. The change of paradigm in the treatment of HER2-positive breast cancer with the development of new generation antibody-drug conjugates. Cancer Drug Resist. 2023, 6, 45–58. [Google Scholar] [CrossRef]
- Andre, F.; Fernanda, M.; Deluche, E.; Lusque, A.; Le-Bescond, L.; Filleron, T.; Pradat, Y.; Ducoulombier, A.; Pistilli, B.; Bachelot, T.; et al. Mechanism of Action and Resistance to Trastuzumab Deruxtecan in Patients with Metastatic Breast Cancer: The DAISY trial. In Review. September 2022. Available online: https://www.researchsquare.com/article/rs-2083650/v1 (accessed on 25 September 2024).
- Fekairi, S.; Scaglione, S.; Chahwan, C.; Taylor, E.R.; Tissier, A.; Coulon, S.; Dong, M.-Q.; Ruse, C.; Yates, J.R.; Russell, P.; et al. Human SLX4 Is a Holliday Junction Resolvase Subunit that Binds Multiple DNA Repair/Recombination Endonucleases. Cell 2009, 138, 78–89. [Google Scholar] [CrossRef]
- Winkler, C.; Armenia, J.; Jones, G.N.; Tobalina, L.; Sale, M.J.; Petreus, T.; Baird, T.; Serra, V.; Wang, A.T.; Lau, A.; et al. SLFN11 informs on standard of care and novel treatments in a wide range of cancer models. Br. J. Cancer 2021, 124, 951–962. [Google Scholar] [CrossRef]
- Sands, J.; Tolaney, S.M.; Ueno, N.T.; Spira, A.I.; Yamamoto, N.; Janjigian, Y.Y.; Naito, Y.; Damodaran, S.; Meric-Bernstam, F.; Modi, S.; et al. A phase 1b, multicenter, open-label study of valemetostat in combination with DXd antibody drug conjugates (ADCs), trastuzumab deruxtecan (T-DXd) or datopotamab deruxtecan (Dato-DXd), in patients with solid tumors. J. Clin. Oncol. 2024, 42, TPS4180. [Google Scholar] [CrossRef]
- Matsui, H.; Takeshita, A.; Naito, K.; Shinjo, K.; Shigeno, K.; Maekawa, M.; Yamakawa, Y.; Tanimoto, M.; Kobayashi, M.; Ohnishi, K.; et al. Reduced effect of gemtuzumab ozogamicin (CMA-676) on P-glycoprotein and/or CD34-positive leukemia cells and its restoration by multidrug resistance modifiers. Leukemia 2002, 16, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Xi, M.; Zhu, J.; Zhang, F.; Shen, H.; Chen, J.; Xiao, Z.; Huangfu, Y.; Wu, C.; Sun, H.; Xia, G. Antibody-drug conjugates for targeted cancer therapy: Recent advances in potential payloads. Eur. J. Med. Chem. 2024, 276, 116709. [Google Scholar] [CrossRef] [PubMed]
- Caimi, P.F.; Ai, W.; Alderuccio, J.P.; Ardeshna, K.M.; Hamadani, M.; Hess, B.; Kahl, B.S.; Radford, J.; Solh, M.; Stathis, A.; et al. Loncastuximab tesirine in relapsed or refractory diffuse large B-cell lymphoma (LOTIS-2): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2021, 22, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Zein, N.; Sinha, A.M.; McGahren, W.J.; Ellestad, G.A. Calicheamicin gamma 1I: An antitumor antibiotic that cleaves double-stranded DNA site specifically. Science 1988, 240, 1198–1201. [Google Scholar] [CrossRef] [PubMed]
- Gbadamosi, M.; Meshinchi, S.; Lamba, J.K. Gemtuzumab ozogamicin for treatment of newly diagnosed CD33-positive acute myeloid leukemia. Future Oncol. 2018, 14, 3199–3213. [Google Scholar] [CrossRef]
- McDonald, G.B.; Freston, J.W.; Boyer, J.L.; DeLeve, L.D. Liver Complications Following Treatment of Hematologic Malignancy With Anti-CD22-Calicheamicin (Inotuzumab Ozogamicin). Hepatology 2019, 69, 831–844. [Google Scholar] [CrossRef]
- Comereski, C.R.; Peden, W.M.; Davidson, T.J.; Warner, G.L.; Hirth, R.S.; Frantz, J.D. BR96-doxorubicin conjugate (BMS-182248) versus doxorubicin: A comparative toxicity assessment in rats. Toxicol. Pathol. 1994, 22, 473–488. [Google Scholar] [CrossRef]
- Wiedemeyer, W.R.; Gavrilyuk, J.; Schammel, A.; Zhao, X.; Sarvaiya, H.; Pysz, M.; Gu, C.; You, M.; Isse, K.; Sullivan, T.; et al. ABBV-011, A Novel, Calicheamicin-Based Antibody-Drug Conjugate, Targets SEZ6 to Eradicate Small Cell Lung Cancer Tumors. Mol. Cancer Ther. 2022, 21, 986–998. [Google Scholar] [CrossRef]
- Morgensztern, D.; Ready, N.E.; Johnson, M.L.; Dowlati, A.; Choudhury, N.J.; Carbone, D.P.; Schaefer, E.S.; Arnold, S.M.; Puri, S.; Piotrowska, Z.; et al. First-in-human study of ABBV-011, a seizure-related homolog protein 6 (SEZ6)–targeting antibody-drug conjugate, in patients with small cell lung cancer. J. Clin. Oncol. 2023, 41, 3002. [Google Scholar] [CrossRef]
- Mantaj, J.; Jackson, P.J.M.; Rahman, K.M.; Thurston, D.E. From Anthramycin to Pyrrolobenzodiazepine (PBD)-Containing Antibody-Drug Conjugates (ADCs). Angew. Chem. Int. Ed. Engl. 2017, 56, 462–488. [Google Scholar] [CrossRef]
- Staben, L.R.; Chen, J.; Cruz-Chuh, J.D.; del Rosario, G.; Go, M.A.; Guo, J.; Khojasteh, S.C.; Kozak, K.R.; Li, G.; Ng, C.; et al. Systematic Variation of Pyrrolobenzodiazepine (PBD)-Dimer Payload Physicochemical Properties Impacts Efficacy and Tolerability of the Corresponding Antibody–Drug Conjugates. J. Med. Chem. Am. Chem. Soc. 2020, 63, 9603–9622. [Google Scholar] [CrossRef] [PubMed]
- Hartley, J.A. Antibody-drug conjugates (ADCs) delivering pyrrolobenzodiazepine (PBD) dimers for cancer therapy. Expert Opin. Biol. Ther. 2021, 21, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Hartley, J.A.; Flynn, M.J.; Bingham, J.P.; Corbett, S.; Reinert, H.; Tiberghien, A.; Masterson, L.A.; Antonow, D.; Adams, L.; Chowdhury, S.; et al. Pre-clinical pharmacology and mechanism of action of SG3199, the pyrrolobenzodiazepine (PBD) dimer warhead component of antibody-drug conjugate (ADC) payload tesirine. Sci. Rep. 2018, 8, 10479. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; Walter, R.B.; Erba, H.P.; Fathi, A.T.; Advani, A.S.; Lancet, J.E.; Ravandi, F.; Kovacsovics, T.; DeAngelo, D.J.; Bixby, D.; et al. A phase 1 trial of vadastuximab talirine as monotherapy in patients with CD33-positive acute myeloid leukemia. Blood 2018, 131, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Tiberghien, A.C.; Levy, J.-N.; Masterson, L.A.; Patel, N.V.; Adams, L.R.; Corbett, S.; Williams, D.G.; Hartley, J.A.; Howard, P.W. Design and Synthesis of Tesirine, a Clinical Antibody-Drug Conjugate Pyrrolobenzodiazepine Dimer Payload. ACS Med. Chem. Lett. 2016, 7, 983–987. [Google Scholar] [CrossRef]
- Lashari, B.H.; Vallatharasu, Y.; Kolandra, L.; Hamid, M.; Uprety, D. Rovalpituzumab Tesirine: A Novel DLL3-Targeting Antibody-Drug Conjugate. Drugs R D 2018, 18, 255–258. [Google Scholar] [CrossRef]
- Blackhall, F.; Jao, K.; Greillier, L.; Cho, B.C.; Penkov, K.; Reguart, N.; Majem, M.; Nackaerts, K.; Syrigos, K.; Hansen, K.; et al. Efficacy and Safety of Rovalpituzumab Tesirine Compared With Topotecan as Second-Line Therapy in DLL3-High SCLC: Results From the Phase 3 TAHOE Study. J. Thorac. Oncol. 2021, 16, 1547–1558. [Google Scholar] [CrossRef]
- Johnson, M.L.; Zvirbule, Z.; Laktionov, K.; Helland, A.; Cho, B.C.; Gutierrez, V.; Colinet, B.; Lena, H.; Wolf, M.; Gottfried, M.; et al. Rovalpituzumab Tesirine as a Maintenance Therapy After First-Line Platinum-Based Chemotherapy in Patients With Extensive-Stage-SCLC: Results From the Phase 3 MERU Study. J. Thorac. Oncol. 2021, 16, 1570–1581. [Google Scholar] [CrossRef]
- Hartley, J.A. What’s new in the use of antibody-drug conjugates (ADCs) delivering pyrrolobenzodiazepine (PBD) DNA cross-linkers for cancer therapy? Expert Opin. Biol. Ther. 2023, 23, 1049–1052. [Google Scholar] [CrossRef]
- Caimi, P.F.; Ai, W.Z.; Alderuccio, J.P.; Ardeshna, K.M.; Hamadani, M.; Hess, B.; Kahl, B.S.; Radford, J.; Solh, M.; Stathis, A.; et al. Loncastuximab tesirine in relapsed/refractory diffuse large B-cell lymphoma: Long-term efficacy and safety from the phase II LOTIS-2 study. Haematologica 2024, 109, 1184–1193. [Google Scholar] [CrossRef]
- Archer, K.E.; Reid, E.E.; Shizuka, M.; Woods, J.; Harris, L.; Maloney, E.K.; Bartle, L.M.; Ab, O.; Wilhelm, A.; Setiady, Y.; et al. Synthesis of Highly Potent N-10 Amino-Linked DNA-Alkylating Indolinobenzodiazepine Antibody-Drug Conjugates (ADCs). ACS Med. Chem. Lett. 2019, 10, 1211–1215. [Google Scholar] [CrossRef] [PubMed]
- Angelova, E.; Audette, C.; Kovtun, Y.; Daver, N.; Wang, S.A.; Pierce, S.; Konoplev, S.N.; Khogeer, H.; Jorgensen, J.L.; Konopleva, M.; et al. CD123 expression patterns and selective targeting with a CD123-targeted antibody-drug conjugate (IMGN632) in acute lymphoblastic leukemia. Haematologica 2019, 104, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Kovtun, Y.; Jones, G.E.; Adams, S.; Harvey, L.; Audette, C.A.; Wilhelm, A.; Bai, C.; Rui, L.; Laleau, R.; Liu, F.; et al. A CD123-targeting antibody-drug conjugate, IMGN632, designed to eradicate AML while sparing normal bone marrow cells. Blood Adv. 2018, 2, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Cai, T.; Han, L.; Kuruvilla, V.M.; Adams, S.; Callum, S.M.; Harutyunyan, K.; Lane, A.A.; Kovtun, Y.; Daver, N.G.; et al. Pre-Clinical Efficacy of CD123-Targeting Antibody-Drug Conjugate IMGN632 in Blastic Plasmacytoid Dentritic Cell Neoplasm (BPDCN) Models. Blood 2018, 132, 3956. [Google Scholar] [CrossRef]
- Kim, R.; Leal, A.D.; Parikh, A.; Ryan, D.P.; Wang, S.; Bahamon, B.; Gupta, N.; Moss, A.; Pye, J.; Miao, H.; et al. A phase I, first-in-human study of TAK-164, an antibody-drug conjugate, in patients with advanced gastrointestinal cancers expressing guanylyl cyclase C. Cancer Chemother. Pharmacol. 2023, 91, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Sarkar, S.; Scott, L.; Danelisen, I.; Trush, M.A.; Jia, Z.; Li, Y.R. Doxorubicin Redox Biology: Redox Cycling, Topoisomerase Inhibition, and Oxidative Stress. React. Oxyg. Species 2016, 1, 189–198. [Google Scholar] [CrossRef]
- Hulst, M.B.; Grocholski, T.; Neefjes, J.J.C.; van Wezel, G.P.; Metsä-Ketelä, M. Anthracyclines: Biosynthesis, engineering and clinical applications. Nat. Prod. Rep. 2022, 39, 814–841. [Google Scholar] [CrossRef]
- Cardinale, D.; Iacopo, F.; Cipolla, C.M. Cardiotoxicity of Anthracyclines. Front. Cardiovasc. Med. 2020, 7, 26. [Google Scholar] [CrossRef]
- Sapra, P.; Stein, R.; Pickett, J.; Qu, Z.; Govindan, S.V.; Cardillo, T.M.; Hansen, H.J.; Horak, I.D.; Griffiths, G.L.; Goldenberg, D.M. Anti-CD74 antibody-doxorubicin conjugate, IMMU-110, in a human multiple myeloma xenograft and in monkeys. Clin. Cancer Res. 2005, 11, 5257–5264. [Google Scholar] [CrossRef]
- Saleh, M.N.; Sugarman, S.; Murray, J.; Ostroff, J.B.; Healey, D.; Jones, D.; Daniel, C.R.; LeBherz, D.; Brewer, H.; Onetto, N.; et al. Phase I Trial of the Anti–Lewis Y Drug Immunoconjugate BR96-Doxorubicin in Patients With Lewis Y–Expressing Epithelial Tumors. J. Clin. Oncol. 2000, 18, 2282–2292. [Google Scholar] [CrossRef]
- Quintieri, L.; Geroni, C.; Fantin, M.; Battaglia, R.; Rosato, A.; Speed, W.; Zanovello, P.; Floreani, M. Formation and antitumor activity of PNU-159682, a major metabolite of nemorubicin in human liver microsomes. Clin. Cancer Res. 2005, 11, 1608–1617. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Meric-Bernstam, F.; McKean, M.; Beerli, R.R.; Waldmeier, L.; Gebleux, R.; Hellmann, I.; Chrom, P.; Grawunder, U. NBE-002: A novel anthracycline-based antibody-drug conjugate (ADC) targeting ROR1 for the treatment of advanced solid tumors—A phase 1/2 clinical trial. J. Clin. Oncol. 2021, 39, TPS1108. [Google Scholar] [CrossRef]
- Gébleux, R.; Briendl, M.; Grawunder, U.; Beerli, R.R. Sortase A Enzyme-Mediated Generation of Site-Specifically Conjugated Antibody-Drug Conjugates. Methods Mol. Biol. 2019, 2012, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Boger, D.L.; Johnson, D.S. CC-1065 and the duocarmycins: Unraveling the keys to a new class of naturally derived DNA alkylating agents. Proc. Natl. Acad. Sci. USA 1995, 92, 3642–3649. [Google Scholar] [CrossRef] [PubMed]
- Pavlidis, N.; Aamdal, S.; Awada, A.; Calvert, H.; Fumoleau, P.; Sorio, R.; Punt, C.; Verweij, J.; van Oosterom, A.; Morant, R.; et al. Carzelesin phase II study in advanced breast, ovarian, colorectal, gastric, head and neck cancer, non-Hodgkin’s lymphoma and malignant melanoma: A study of the EORTC early clinical studies group (ECSG). Cancer Chemother. Pharmacol. 2000, 46, 167–171. [Google Scholar] [CrossRef]
- Yao, H.-P.; Zhao, H.; Hudson, R.; Tong, X.-M.; Wang, M.-H. Duocarmycin-based antibody-drug conjugates as an emerging biotherapeutic entity for targeted cancer therapy: Pharmaceutical strategy and clinical progress. Drug Discov. Today 2021, 26, 1857–1874. [Google Scholar] [CrossRef]
- Dokter, W.; Ubink, R.; van der Lee, M.; van der Vleuten, M.; van Achterberg, T.; Jacobs, D.; Loosveld, E.; van den Dobbelsteen, D.; Egging, D.; Mattaar, E.; et al. Preclinical Profile of the HER2-Targeting ADC SYD983/SYD985: Introduction of a New Duocarmycin-Based Linker-Drug Platform. Mol. Cancer Ther. 2014, 13, 2618–2629. [Google Scholar] [CrossRef]
- Scribner, J.A.; Brown, J.G.; Son, T.; Chiechi, M.; Li, P.; Sharma, S.; Li, H.; De Costa, A.; Li, Y.; Chen, Y.; et al. Preclinical Development of MGC018, a Duocarmycin-based Antibody–drug Conjugate Targeting B7-H3 for Solid Cancer. Mol. Cancer Ther. 2020, 19, 2235–2244. [Google Scholar] [CrossRef]
- Groothuis, P.G.; Jacobs, D.C.H.; Hermens, I.A.T.; Damming, D.; Berentsen, K.; Mattaar-Hepp, E.; Stokman, M.E.M.; Boekel, T.V.; Rouwette, M.; van der Vleuten, M.A.J.; et al. Preclinical Profile of BYON3521 Predicts an Effective and Safe MET Antibody–Drug Conjugate. Mol. Cancer Ther. 2023, 22, 765–777. [Google Scholar] [CrossRef]
- Phase III Clinical Trial SYD985 vs. Physician’s Choice in Participants with HER2-Positive Locally Advanced or Metastatic Breast Cancer (TULIP). Available online: https://clinicaltrials.gov/ct2/show/NCT03262935 (accessed on 21 September 2024).
- Saura Manich, C.; O’Shaughnessy, J.; Aftimos, P.G.; van den Tweel, E.; Oesterholt, M.; Escrivá-de-Romaní, S.I.; Quenel Tueux, N.; Tan, T.J.; Lim, J.S.; Ladoire, S.; et al. LBA15 Primary outcome of the phase III SYD985.002/TULIP trial comparing [vic-]trastuzumab duocarmazine to physician’s choice treatment in patients with pre-treated HER2-positive locally advanced or metastatic breast cancer. Ann. Oncol. 2021, 32, S1288. [Google Scholar] [CrossRef]
- Elgersma, R.C.; Coumans, R.G.E.; Huijbregts, T.; Menge, W.M.P.B.; Joosten, J.A.F.; Spijker, H.J.; de Groot, F.M.H.; van der Lee, M.M.C.; Ubink, R.; van den Dobbelsteen, D.J.; et al. Design, Synthesis, and Evaluation of Linker-Duocarmycin Payloads: Toward Selection of HER2-Targeting Antibody–Drug Conjugate SYD985. Mol. Pharm. 2015, 12, 1813–1835. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Llamas, S.; Caro-Magdaleno, M.; Mataix-Albert, B.; Avilés-Prieto, J.; Romero-Barranca, I.; Rodríguez-de-la-Rúa, E. Adverse events of antibody–drug conjugates on the ocular surface in cancer therapy. Clin. Transl. Oncol. 2023, 25, 3086–3100. [Google Scholar] [CrossRef] [PubMed]
- López de Sá, A.; Díaz-Tejeiro, C.; Poyatos-Racionero, E.; Nieto-Jiménez, C.; Paniagua-Herranz, L.; Sanvicente, A.; Calvo, E.; Pérez-Segura, P.; Moreno, V.; Moris, F.; et al. Considerations for the design of antibody drug conjugates (ADCs) for clinical development: Lessons learned. J. Hematol. Oncol. 2023, 16, 118. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Gunaydin, H. Probabilistic Approach to Generating MPOs and Its Application as a Scoring Function for CNS Drugs. ACS Med. Chem. Lett. 2016, 7, 89–93. [Google Scholar] [CrossRef]
- Zhao, H.; Atkinson, J.; Gulesserian, S.; Zeng, Z.; Nater, J.; Ou, J.; Yang, P.; Morrison, K.; Coleman, J.; Malik, F.; et al. Modulation of Macropinocytosis-Mediated Internalization Decreases Ocular Toxicity of Antibody-Drug Conjugates. Cancer Res. 2018, 78, 2115–2126. [Google Scholar] [CrossRef]
- Valsasina, B.; Orsini, P.; Caruso, M.; Albanese, C.; Ciavolella, A.; Cucchi, U.; Fraietta, I.; Melillo, N.; Fiorentini, F.; Rizzi, S.; et al. Novel Thienoduocarmycin-Trastuzumab ADC Demonstrates Strong Antitumor Efficacy with Favorable Safety Profile in Preclinical Studies. Mol. Cancer Ther. 2023, 22, 1465–1478. [Google Scholar] [CrossRef]
- Caruso, M.; Gasparri, F.; Valsasina, B.; Albanese, C.; Beria, I.; Candiani, I.; Ciomei, M.; Colombo, N.; Cribioli, S.; Cucchi, U.; et al. Abstract 734: Thienoindoles: New Highly Promising Agents for Antibody-Drug Conjugates Generation. Experimental and Molecular Therapeutics; American Association for Cancer Research: Philadelphia, PA, USA, 2018; p. 734. Available online: http://cancerres.aacrjournals.org/lookup/doi/10.1158/1538-7445.AM2018-734 (accessed on 31 October 2020).
- Tichenor, M.S.; MacMillan, K.S.; Stover, J.S.; Wolkenberg, S.E.; Pavani, M.G.; Zanella, L.; Zaid, A.N.; Spalluto, G.; Rayl, T.J.; Hwang, I.; et al. Rational Design, Synthesis, and Evaluation of Key Analogues of CC-1065 and the Duocarmycins. J. Am. Chem. Soc. 2007, 129, 14092–14099. [Google Scholar] [CrossRef]
- Colombo, R.; Rich, J.R. The therapeutic window of antibody drug conjugates: A dogma in need of revision. Cancer Cell 2022, 40, 1255–1263. [Google Scholar] [CrossRef]
- Orsini, P.; Salsa, M.; Rizzi, S.; Cucchi, U.; Faiardi, D.; Burocchi, A.; Ciavolella, A.; Lupi, R.; Gasparri, F.; Valsasina, B. Abstract 5805: A novel platform of diversified payloads to drive ADC innovation. Cancer Res. 2024, 84 (Suppl. S6), 5805. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Drug Names | Year of Approval | Target | Linker | Linker Catabolism | Payload | DAR | Conjugation Amino Acid | Mechanism of Action | Approved Disease Indications |
|---|---|---|---|---|---|---|---|---|---|
| Mylotarg™; Gemtuzumab ozogamicin | 2000 | CD33 | AcBut acyl hydrazone-disulfide | Cleavable | Calicheamicin | 2–3 | Lysine | DNA binder | Acute Myeloid Leukemia |
| Adcetris™; Brentuximab vedotin | 2011 | CD30 | Valine-Citrulline | Cleavable | MMAE | 4 | Cysteine | Tubulin binder | Classical Hodgkin Lymphoma; Cutaneous T-Cell Lymphoma; PTCL; sALCL |
| Kadcyla™; T-DM1; Trastuzumab emtansine | 2013 | HER-2 | SMCC | Non-Cleavable | DM1 | 3.5 | Lysine | Tubulin binder | HER2 Positive Early Breast Cancer; HER2+ Metastatic Breast Cancer |
| Besponsa™; Inotuzumab ozogamicin | 2017 | CD22 | AcBut acyl hydrazone-disulfide | Cleavable | Calicheamicin | 2–3 | Lysine | DNA binder | B-cell Precursor (Ph+ Negative) Acute Lymphoblastic Leukemia |
| PADCEV™; Enfortumab vedotin | 2019 | Nectin-4 | Valine-Citrulline | Cleavable | MMAE | 4 | Cysteine | Tubulin binder | Advanced Urothelial Cancer; Metastatic Urothelial Cancer |
| POLIVY™; Polatuzumab vedotin | 2019 | CD79b | Valine-Citrulline | Cleavable | MMAE | 3.5 | Cysteine | Tubulin binder | Diffuse Large B-cell lymphoma |
| ENHERTU™; Trastuzumab deruxtecan | 2019 | HER-2 | GGFG | Cleavable | DXd | 8 | Cysteine | TOPO I inhibitor | HER2 Mutant metastatic NSCLC HER-2 Positive Advanced/Metastatic Gastric Cancer; HER2-Positive Advanced/metastatic Gastroesophageal Cancer; Metastatic HER2 Positive Breast Cancer; |
| Trodelvy™; Sacituzumab govitecan | 2020 | TROP-2 | CL2A | Cleavable | SN-38 | 7.6 | Cysteine | TOPO I inhibitor | Advanced/metastatic TNBC Advanced/Metastatic Urothelial Cancer; |
| BLENREP™; Belantamab mafodotin | 2020 | BCMA | maleimido-caproyl | Non-Cleavable | MMAF | 4 | Cysteine | Tubulin binder | Myeloma/Multiple Myeloma/Kahler’s disease/Myelomatosis |
| ZYNLONTA™ Loncastuximab Tesirine | 2021 | CD19 | Valine-Alanine | Cleavable | SG3199 (PBD) | 2.3 ± 0.3 | Cysteine | DNA binder | Diffuse Large B-Cell Lymphoma |
| TIVDAK™ Tisotumab vedotin | 2021 | TF | Valine-Citrulline | Cleavable | MMAE | 4 | Cysteine | Tubulin binder | Metastatic Cervical Cancer; Recurrent Cervical Cancer |
| ELAHERE™ Mirvetuximab soravtansine | 2022 | FOLRA | Sulfo-SPDB | Cleavable | DM4 | 3.4 | Lysine | Tubulin binder | Platinum-Resistant Ovarian Cancer; Platinum-Resistant Fallopian Tube Carcinoma; Primary Peritoneal Cancer |
| Most Advanced ADC | Most Advanced Stage | Payload Chemical Class | Payload Mechanism of Action | Linker Catabolism | DAR | Conjugation Amino Acid | Target | Most Common Adverse Events |
|---|---|---|---|---|---|---|---|---|
| brentuximab vedotin | FDA approved | Auristatin | Tubulin binder | cleavable | 4 | Cysteine | CD30 | Peripheral neuropathy |
| enfortumab vedotin | FDA approved | Auristatin | Tubulin binder | cleavable | 4 | Cysteine | Nectin 4 | Peripheral neuropathy, Skin reactions |
| disitamab vedotin | China approved | Auristatin | Tubulin binder | cleavable | 4 | Cysteine | HER2 | ALT, AST elevation |
| tisitamab vedotin | FDA approved | Auristatin | Tubulin binder | cleavable | 4 | Cysteine | TF | Peripheral neuropathy, bleeding |
| polatuzumab vedotin | FDA approved | Auristatin | Tubulin binder | cleavable | 3.8 | Cysteine | CD79B | Peripheral neuropathy Anemia, neutropenia |
| Trastuzumab emtansine | FDA approved | Maytansine | Tubulin binder | uncleavable | 3.5 | Lysine | HER2 | Gastrointestinal disorders, thrombocytopenia |
| Mirvetuximab sorvantansine | FDA approved | Maytansine | Tubulin binder | uncleavable | 3.5 | Lysine | FOLRA | Ocular Tox, pneumonia Peripheral neuropathy |
| Trastuzumab deruxtecan | FDA approved | Camptothecin | Topoisomerase I inhibitor | cleavable | 8 | Cysteine | HER2 | Gastrointestinal disorders, ILD |
| Sacituzumab govitecan | FDA approved | Camptothecin | Topoisomerase I inhibitor | cleavable | 8 | Cysteine | TROP2 | Neutropenia, gastrointestinal disorders |
| Gemtuzumab ozogamicin | FDA approved | Calicheamicin | Minor groove binder and DNA double-strand break inducer | cleavable | 2–3 | Lysine | CD33 | Neutropenia VOD, hepatic failure |
| Inotuzumab ozogamicin | FDA approved | Calicheamicin | Minor groove binder and DNA double-strand break inducer | cleavable | 2–3 | Lysine | CD22 | Neutropenia VOD hepatic failure |
| Loncastuximab Tesirine | FDA approved | PBD dimer | Minor groove DNA binder and Guanine crosslinker | cleavable | 2.3 | Cysteine | CD19 | Respiratory disorders |
| Rovalpituzumab tesirine | Phase III | PBD dimer | DNA crosslinker | cleavable | 2 | Cysteine | DLL3 | Respiratory disorders, neutropenia, thrombocytopenia |
| Pivekimab Sunirine | Phase II | IGN | DNA alkylating | cleavable | Cysteine | CD123 | Respiratory disorders, neutropenia, thrombocytopenia | |
| TAK-164 | Phase II | IGN | DNA alkylating | cleavable | Cysteine | GCC | Thrombocytopenia hepatic tox | |
| NBE-002 | Phase I | Anthracycline | Topo II inhibition and DNA intercalation | uncleavable | 2 | C terminus | ROR1 | No data |
| SOT102 | Phase I | Anthracycline | Topo II inhibition and DNA intercalation | uncleavable | 2 | C terminus | CLDN18.2 | No data |
| SGN-15 | Phase II | Anthracycline | Topo II inhibition and DNA intercalation | cleavable | Cysteine | Lewis Y antigen | Gastrointestinal respiratory disorders | |
| IMMU 110 | Phase I/II | Anthracycline | Topo II inhibition and DNA intercalation | cleavable | 8 | Cysteine | CD74 | |
| Trastuzumab duocarmazine | Phase III | Duocarmycin | DNA minor groove binder and alkylating agent | cleavable | 2.8 | Cysteine | HER2 | Ocular tox, respiratory disorders |
| MGC018 | Phase II | Duocarmycin | DNA minor groove binder and alkylating agent | cleavable | 2.8 | Cysteine | B7-H3 | Skin disorders, neutropenia, respiratory disorders |
| Drug Name | MOA | MW | Complexity | ACD_ LogP | H-Bond Donors | H-Bond Acceptors | Rotatable Bonds | PSA | PSA_ SASA | MPO | sp3 Fractions | Sol (mol/L) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Desired Features | <500 | <5 | <5 | <10 | <10 | <140 | >3 | >0.42 | 0.001 | |||
| MMAE | tubulin binder | 718.99 | 1100 | 3.82 | 4 | 8 | 20 | 154.11 | 0.8 | 1.81 | 0.74 | 1.00 × 10−2 |
| DM1 | tubulin binder | 738.29 | 1340 | 3.87 | 2 | 11 | 8 | 195.27 | 0.74 | 2.14 | 0.6 | 4.40 × 10−7 |
| DM4 | tubulin binder | 780.37 | 1430 | 4.81 | 2 | 11 | 9 | 195.27 | 0.74 | 1.6 | 0.63 | 2.80 × 10−7 |
| SN38 | topo I inhibitor | 392.4 | 882 | 2.11 | 2 | 7 | 2 | 99.96 | 0.64 | 4.89 | 0.32 | 3.50 × 10−3 |
| DXd | topo I inhibitor | 493.48 | 1080 | 1.76 | 3 | 8 | 3 | 129.06 | 0.65 | 3.22 | 0.38 | 3.90 × 10−4 |
| N acetyl calicheamicin | DNA damage | 1478.44 | 2500 | 6.13 | 10 | 27 | 26 | 447.74 | 0.76 | 1 | 0.66 | 4.70 × 10−6 |
| SG3199 (PBD) | DNA crosslinker | 602.68 | 1080 | 1.8 | 2 | 24 | 10 | 122.16 | 0.85 | 3.5 | 0.42 | 7.70 × 10−5 |
| Sunirine | DNA alkylator | 788.84 | na | 1.05 | 5 | 9 | 9 | 193.2 | 0.72 | 3 | 0.24 | 3.50 × 10−2 |
| PNU | topo II inhibitor | 642.63 | 1200 | 3.64 | 4 | 14 | 6 | 191.95 | 0.7 | 2.11 | 0.53 | 3.40 × 10−6 |
| Seco DUBA | MGBAA | 526.97 | 885 | 3.76 | 3 | 5 | 4 | 107.16 | 0.63 | 2.37 | 0.14 | 4.50 × 10−6 |
| Payload Name | MOA | Efflux Pumps Sensitivity | Potency M | MTD Free Toxin (mg/kg) | Most Advanced ADC | Bystander Effect | MTD ADC Toxin Normalized (mg/kg) |
|---|---|---|---|---|---|---|---|
| MMAE | tubulin binder | ++ | 10−11–10−10 | Dolastatin 0.008 | Brentuximab Vedotin | Yes | 0.032 Q3W |
| Enfortumab Vedotin | Yes | 0.022 D1, 8, 15, 28 days cycle | |||||
| Disitamab Vedotin | Yes | 0.038 Q2W | |||||
| Tisitamab Vedotin | Yes | 0.037 Q3W | |||||
| Polatuzumab Vedotin | Yes | 0.034 Q3W | |||||
| DM1 | tubulin binder | ++ | 10−11–10−10 | Maytansine 0.054 Q3W | TDM1 (Kadcyla) | No | 0.062Q3W |
| DM4 | tubulin binder | ++ | 10−11–10−10 | Maytansine 0.054 Q3W | Mirvetuximab soravtansine (Elahere) | No | 0.11 Q3W |
| SN38 | topo I inhibitor | ++ | 10−9–10−8 | Irinotecan 0.12 QW | Sacituzumab govitecan (Trodelvy) | Yes | 0.19 D1, 8, 21 Days cycle |
| DXd | topo I inhibitor | + | 10−10–10−9 | Topotecan 0.20 QDX5 every three weeks | Trastuzumab deruxtecan (Enhertu) | Yes | 0.14 Q3W |
| N acetyl calicheamicin | DNA damage | ++ | 10−11–10−10 | not available | Gemtuzumab ozogamicine (Mylotarg) | Yes | 0.00087 fractionated dose on day 1, 8 15 |
| Inotuzumab ozogamicine (Besponsa) | Yes | ||||||
| SG3199 (PBD) | DNA crosslinker | + | 10−13–10−12 | SJG-136 0.0012 Q3W | Zynlonta | Yes | 0.0013 Q3W |
| Rovalpizumab tesirine | Yes | ||||||
| Sunirine | DNA alkylator | + | 10−13–10−12 | SJG-136 0.0012 Q3W | Pivekimab sunirine | Yes | 0.00042 Q3W |
| TAK164 | Yes | ||||||
| PNU | topo II inhibitor, DNA intercalation | - | 10−13–10−12 | Nemorubicin 0.022 Q4W | NBE002 | Yes | Not reported |
| SOT102 | Yes | Not reported | |||||
| Seco-DUBA | MGBAA | - | 10−11–10−10 | Carzelesine 0.0081Q4W | SYD985 | Yes | 0.011 Q3W |
| MGC018 | Yes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valsasina, B.; Orsini, P.; Terenghi, C.; Ocana, A. Present Scenario and Future Landscape of Payloads for ADCs: Focus on DNA-Interacting Agents. Pharmaceuticals 2024, 17, 1338. https://doi.org/10.3390/ph17101338
Valsasina B, Orsini P, Terenghi C, Ocana A. Present Scenario and Future Landscape of Payloads for ADCs: Focus on DNA-Interacting Agents. Pharmaceuticals. 2024; 17(10):1338. https://doi.org/10.3390/ph17101338
Chicago/Turabian StyleValsasina, Barbara, Paolo Orsini, Chiara Terenghi, and Alberto Ocana. 2024. "Present Scenario and Future Landscape of Payloads for ADCs: Focus on DNA-Interacting Agents" Pharmaceuticals 17, no. 10: 1338. https://doi.org/10.3390/ph17101338
APA StyleValsasina, B., Orsini, P., Terenghi, C., & Ocana, A. (2024). Present Scenario and Future Landscape of Payloads for ADCs: Focus on DNA-Interacting Agents. Pharmaceuticals, 17(10), 1338. https://doi.org/10.3390/ph17101338