

Chemical Investigation and Regulation of Adipogenic Differentiation of Cultivated Moringa oleifera

 , , , and
, , , and
Abstract
1. Introduction
2. Results and Discussion
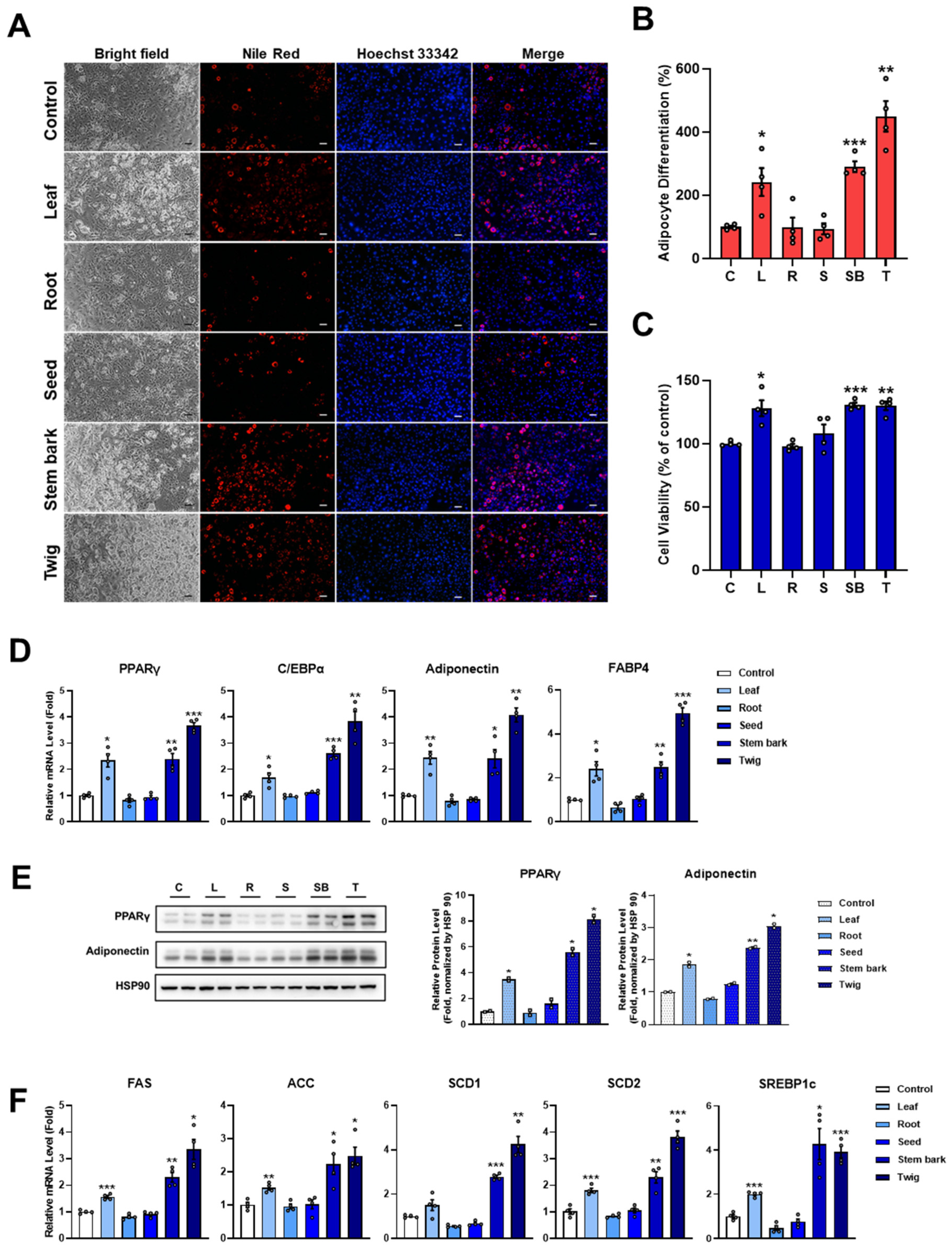
2.1. Moringa Leaf, Stem Bark, and Twig Extracts Stimulate Adipocyte Differentiation
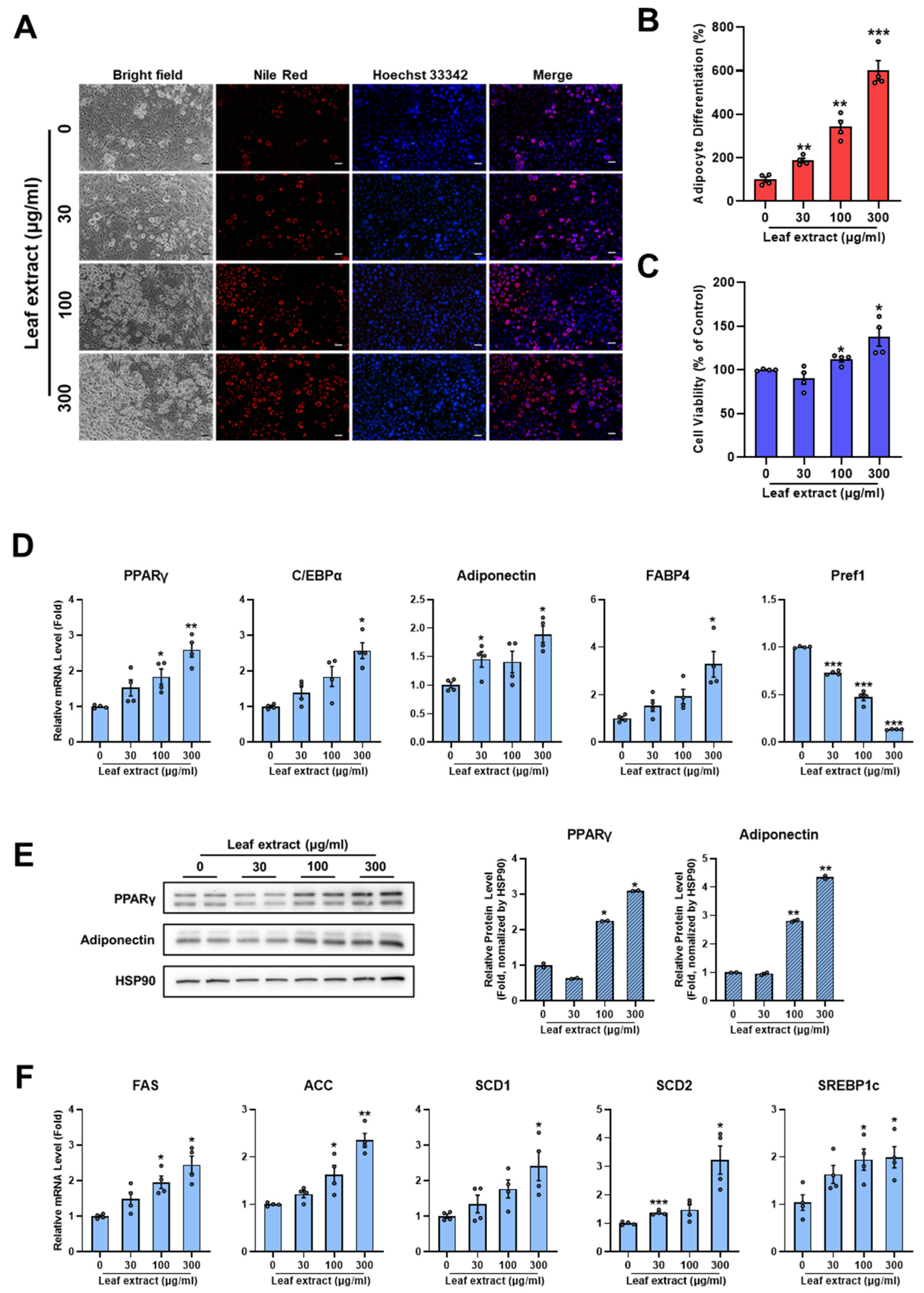
2.2. Leaf Extract of M. oleifera Stimulates Adipocyte Differentiation
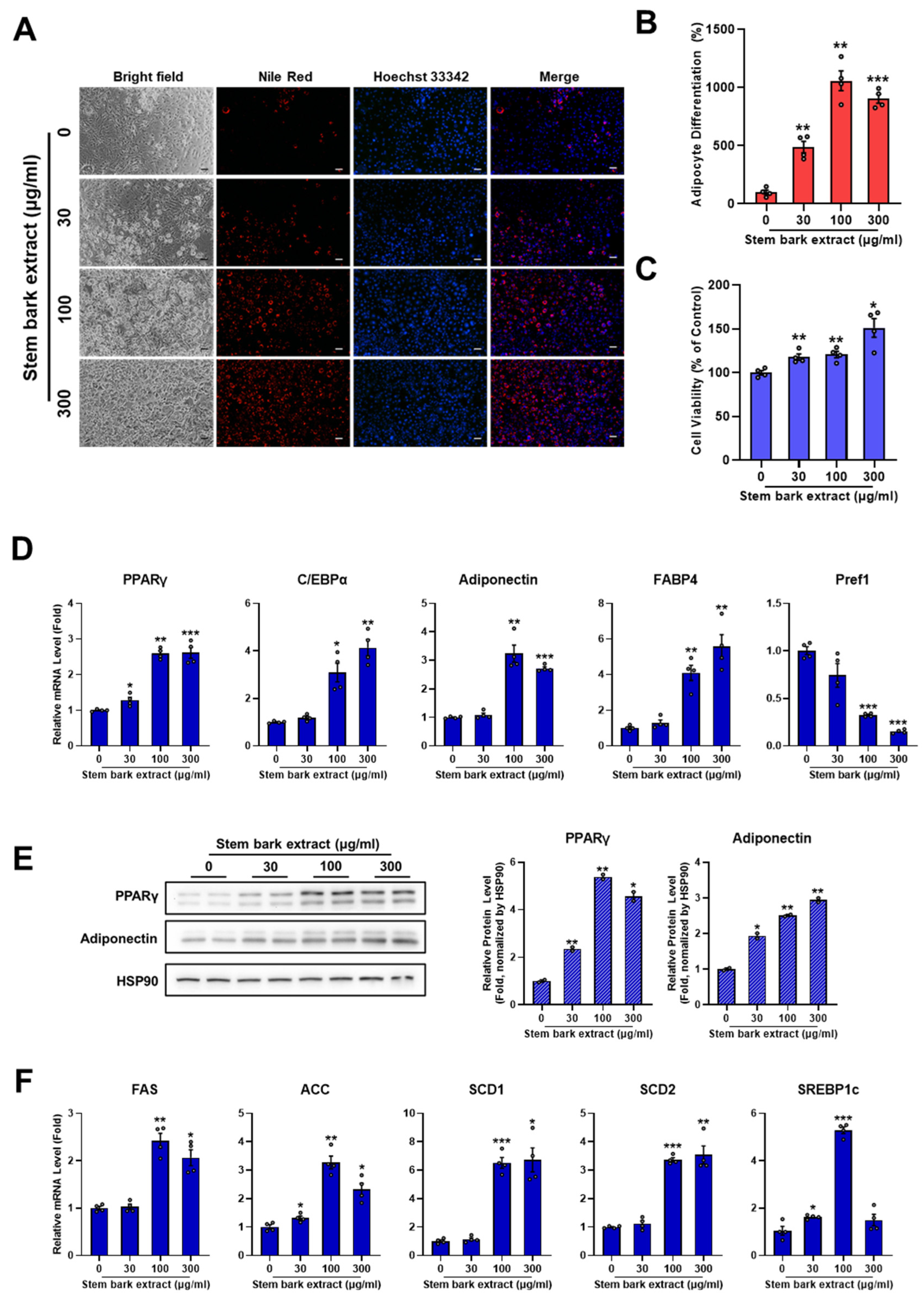
2.3. Stem Bark Extract of M. oleifera Induces Adipocyte Differentiation
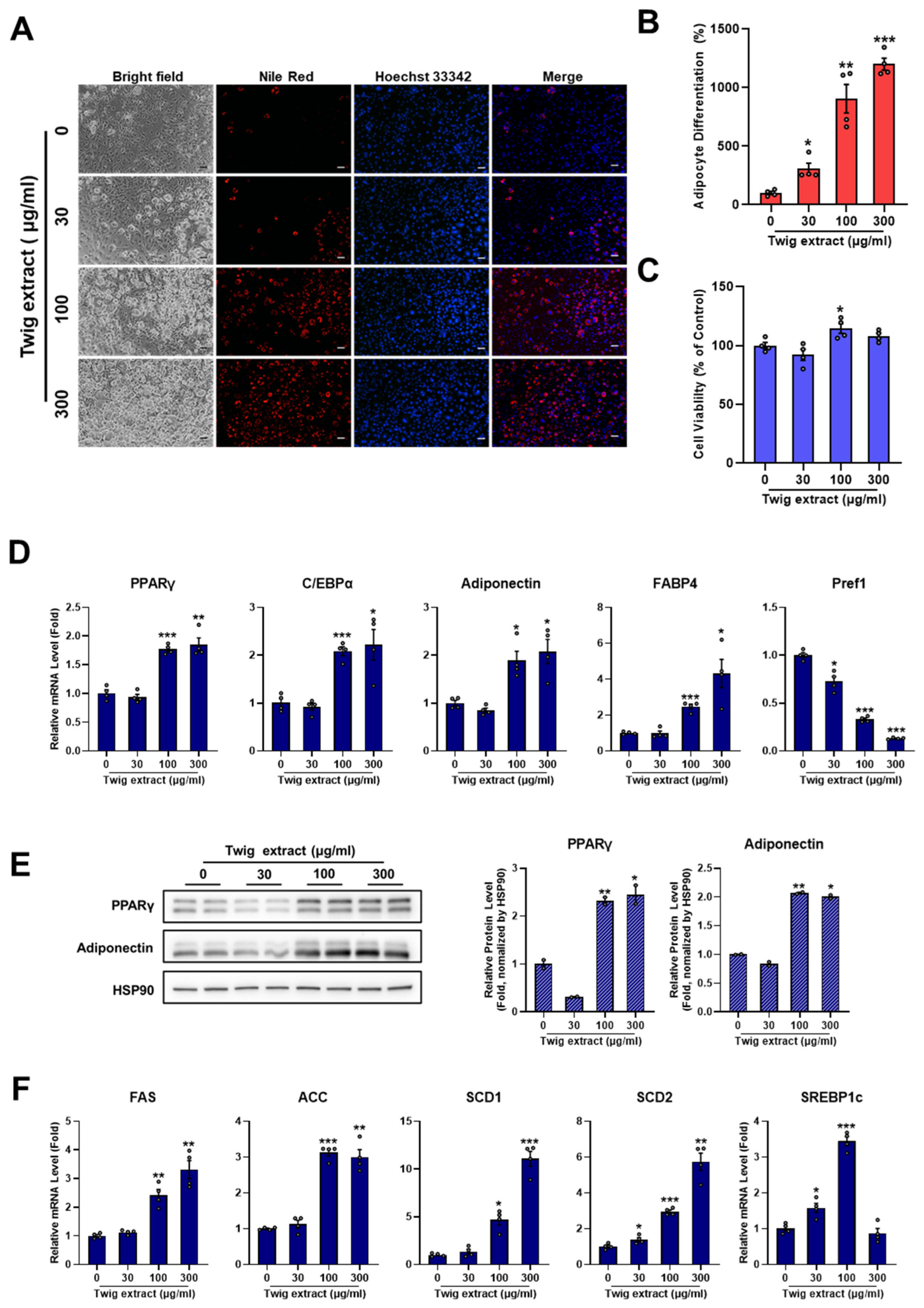
2.4. Twig Extract of M. oleifera Increases Adipocyte Differentiation
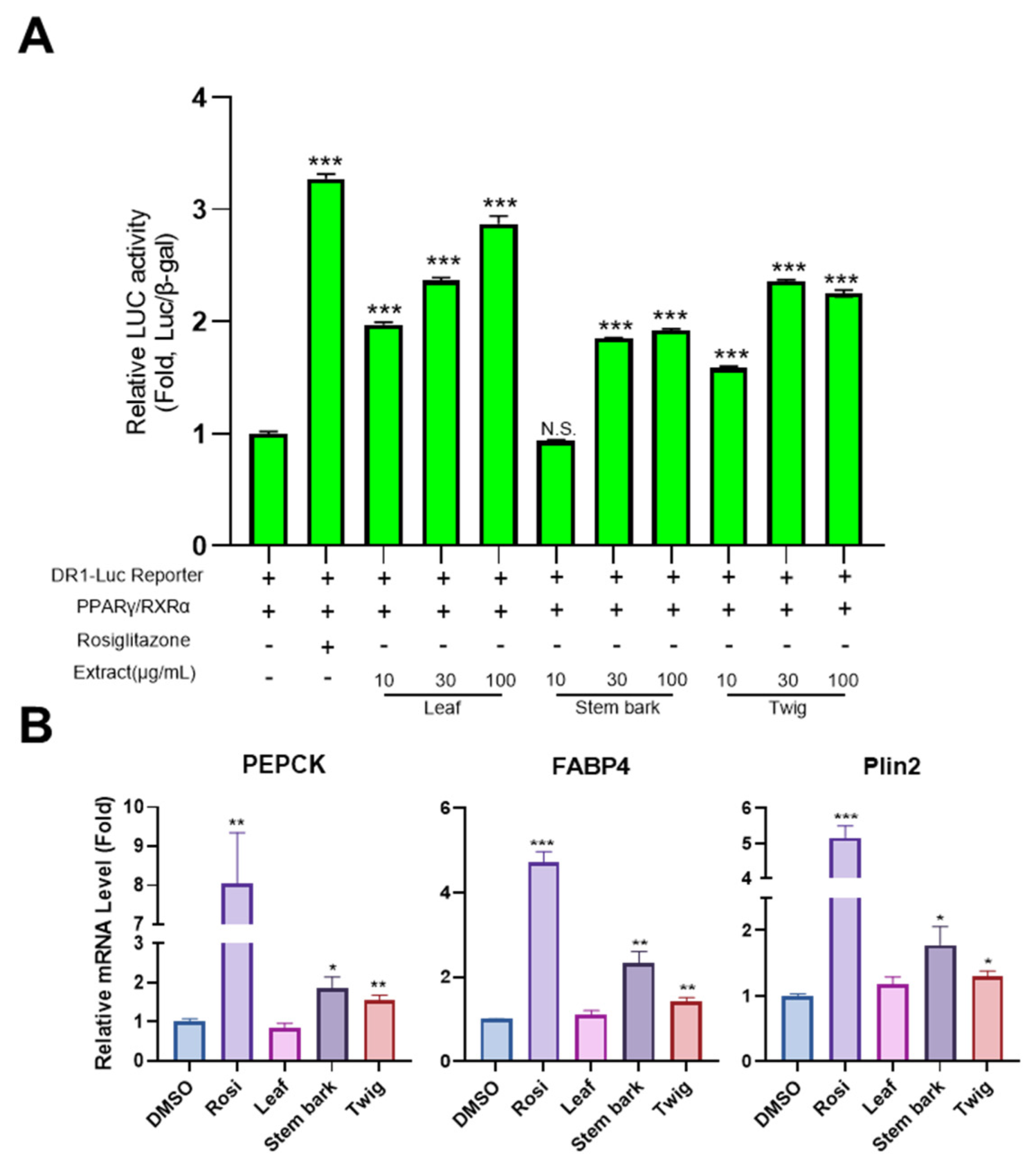
2.5. Leaf, Stem Bark, and Twig Extracts of M. oleifera Enhance PPARγ Activity and the Expression of PPARγ Target Genes
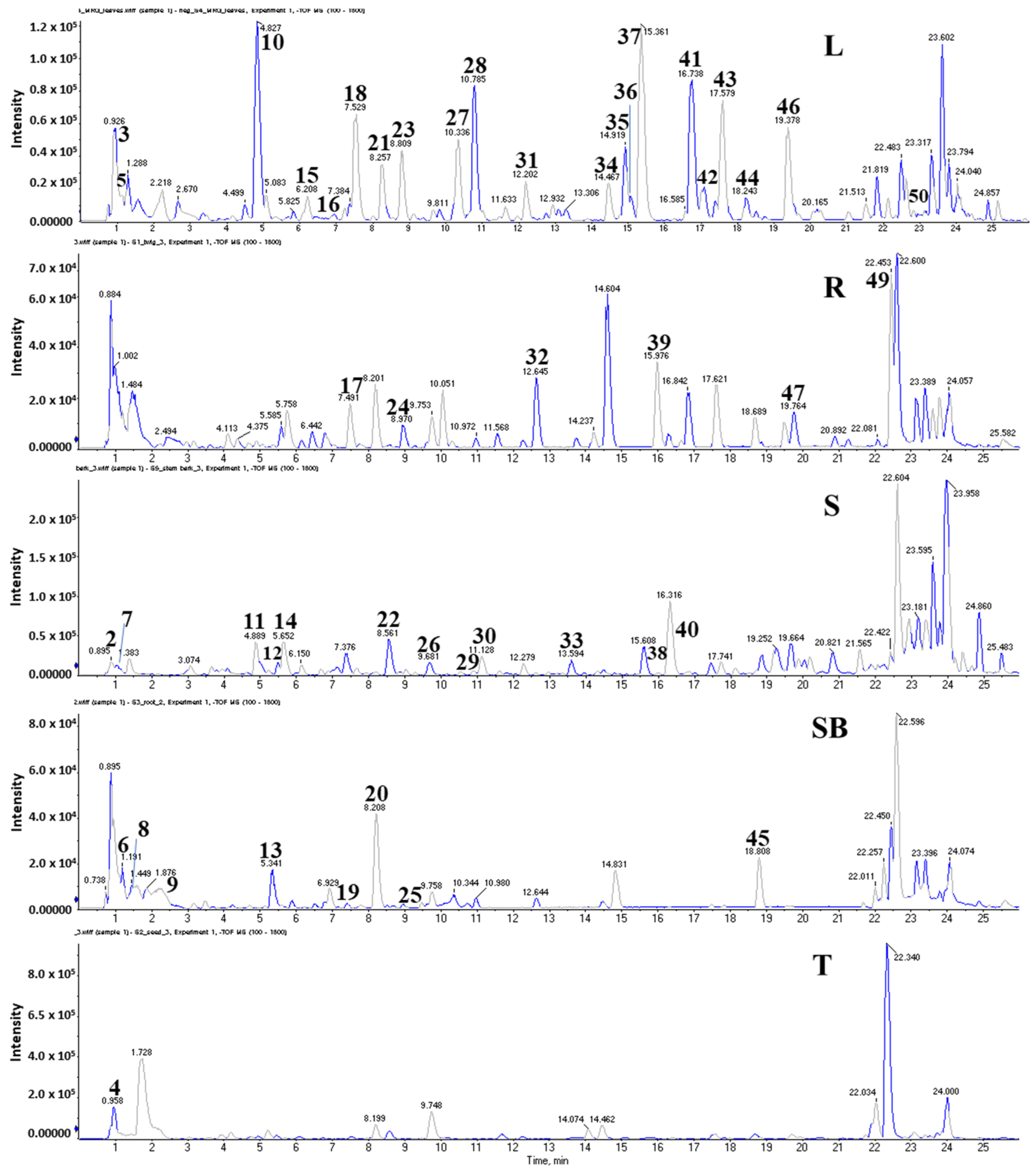
2.6. Identification of Metabolites from Five Organs of M. oleifera
2.6.1. Glycosides
2.6.2. Flavones
2.6.3. Fatty Acid
2.6.4. Phenolics
2.6.5. Other Compounds
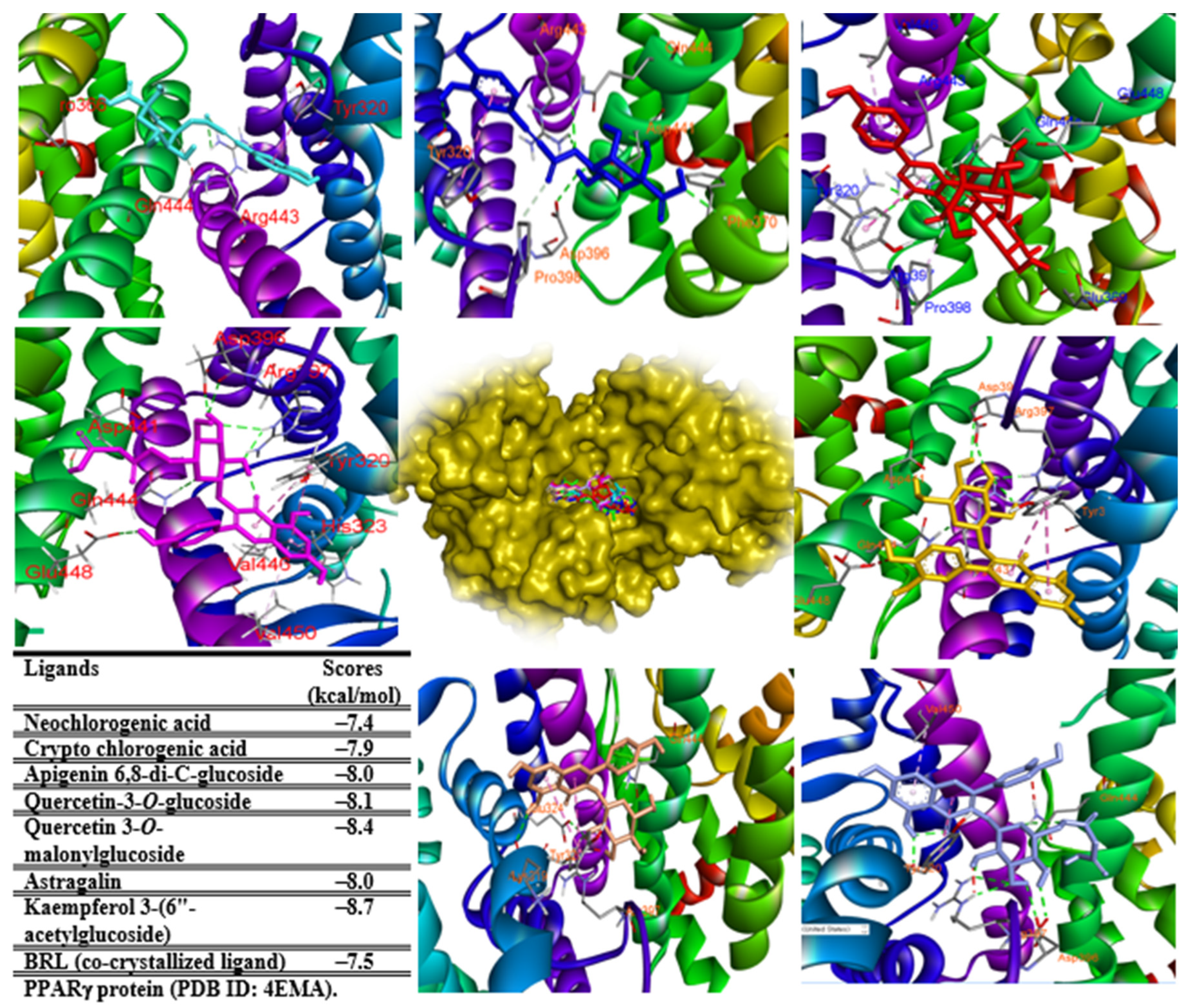
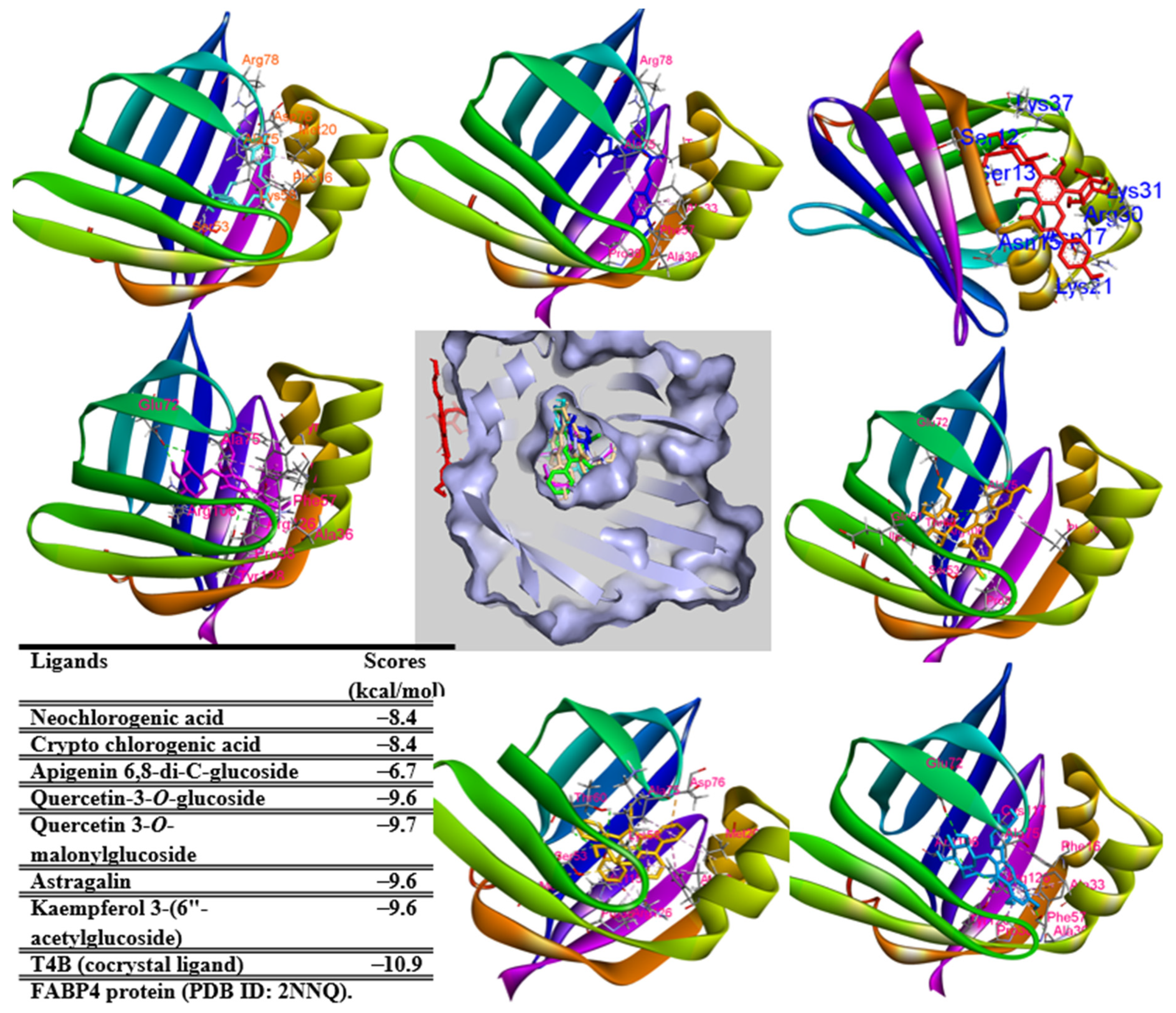
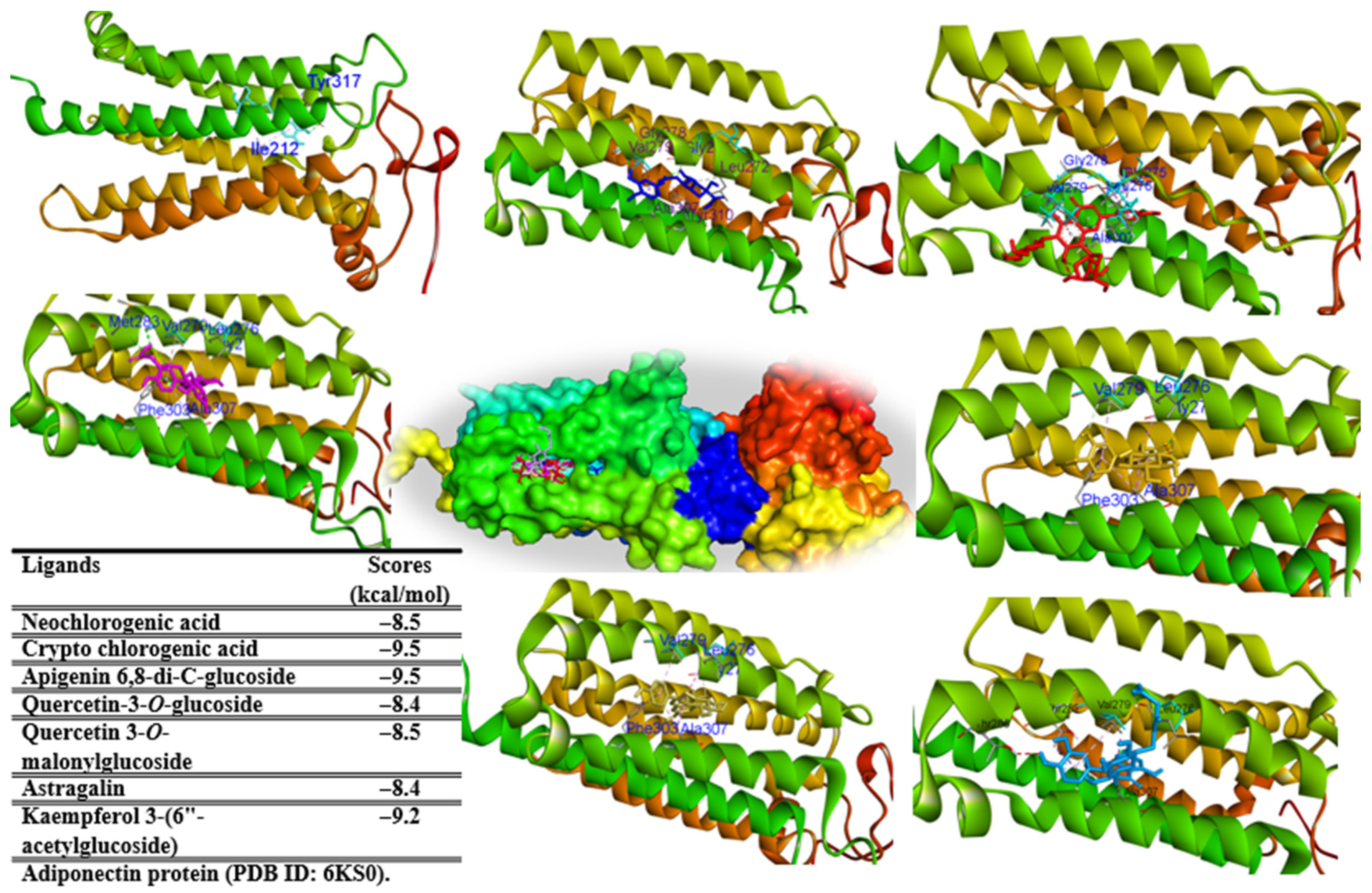
2.7. In Silico Study
3. Materials and Methods
3.1. Plant Materials
Extraction of Samples
3.2. Cell Culture
3.3. Adipocyte Differentiation
3.4. Nile Red and Hoechst 33342 Staining
3.5. Quantitative Real-Time PCR Analysis
3.6. Western Blotting Analysis
3.7. Luciferase and β-Galactosidase Assay
3.8. Analysis of Chemical Composition from Total Extracts of Leaves, Twigs, Stem Bark, Seeds, and Roots
3.9. Molecular Docking
3.10. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anwar, F.; Latif, S.; Ashraf, M.; Gilani, A.H. Moringa oleifera: A Food Plant with Multiple Medicinal Uses. Phytother. Res. 2007, 21, 17–25. [Google Scholar] [CrossRef]
- Gautier, A.; Duarte, C.M.; Sousa, I. Moringa oleifera Seeds Characterization and Potential Uses as Food. Foods 2022, 11, 1629. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.-L.; Xu, Y.-B.; Wu, J.-L.; Li, N.; Gou, M.-Q. Hypoglycemic and Hypolipidemic Effects of Moringa oleifera Leaves and Their Functional Chemical Constituents. Food Chem. 2020, 333, 127478. [Google Scholar] [CrossRef] [PubMed]
- Pareek, A.; Pant, M.; Gupta, M.M.; Kashania, P.; Ratan, Y.; Jain, V.; Pareek, A.; Chuturgoon, A.A. Moringa oleifera: An updated comprehensive review of its pharmacological activities, ethnomedicinal, phytopharmaceutical formulation, clinical, phytochemical, and toxicological aspects. Int. J. Mol. Sci. 2023, 24, 2098. [Google Scholar] [CrossRef]
- Klimek-Szczykutowicz, M.; Gaweł-Bęben, K.; Rutka, A.; Blicharska, E.; Tatarczak-Michalewska, M.; Kulik-Siarek, K.; Kukula-Koch, W.; Malinowska, M.A.; Szopa, A. Moringa oleifera (Drumstick tree)—Nutraceutical, Cosmetological and Medicinal Importance: A Review. Front. Pharmacol. 2024, 15, 1288382. [Google Scholar] [CrossRef]
- Mahato, D.K.; Kargwal, R.; Kamle, M.; Sharma, B.; Pandhi, S.; Mishra, S.; Gupta, A.; Mahmud, M.M.C.; Gupta, M.K.; Singha, L.B.; et al. Ethnopharmacological Properties and Nutraceutical Potential of Moringa oleifera. Phytomedicine 2022, 2, 100168. [Google Scholar] [CrossRef]
- Zhang, F.-X.; Li, M.; Yao, Z.-H.; Li, C.; Qiao, L.-R.; Shen, X.-Y.; Yu, K.; Dai, Y.; Yao, X.-S. A Target and Nontarget Strategy for Identification or Characterization of The Chemical Ingredients in Chinese Herb Preparation Shuang-Huang-Lian Oral Liquid by Ultra-performance Liquid Chromatography–quadrupole Time-of-flight Mass Spectrometry. Biomed. Chromatogr. 2018, 32, e4110. [Google Scholar] [CrossRef]
- Hruby, A.; Hu, F.B. The Epidemiology of Obesity: A Big Picture. Pharmaco. Econ. 2015, 33, 673–689. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.T.; Hochfeld, W.E.; Myburgh, R.; Pepper, M.S. Adipocyte and Adipogenesis. Eur. J. Cell Biol. 2013, 92, 229–236. [Google Scholar] [CrossRef]
- Lefterova, M.I.; Lazar, M.A. New Developments in Adipogenesis. Trends Endocrinol. Metab. 2009, 20, 107–114. [Google Scholar] [CrossRef]
- Giralt, M.; Cereijo, R.; Villarroya, F. Adipokines and the Endocrine Role of Adipose Tissues. Handb. Exp. Pharmacol. 2016, 233, 265–282. [Google Scholar] [PubMed]
- Luo, L.; Liu, M. Adipose Tissue in Control of Metabolism. J. Endocrinol. 2016, 231, R77–R99. [Google Scholar] [CrossRef] [PubMed]
- Koerner, A.; Kratzsch, J.; Kiess, W. Adipocytokines: Leptin the Classical, Resistin the Controversical, Adiponectin the Promising, and More to Come. Best. Pract. Res. Clin. Endocrinol. Metab. 2005, 19, 525–546. [Google Scholar] [CrossRef]
- Rosen, E.D.; Walkey, C.J.; Puigserver, P.; Spiegelman, B.M. Transcriptional Regulation of Adipogenesis. Genes Dev. 2000, 14, 1293–1307. [Google Scholar] [CrossRef]
- Tontonoz, P.; Hu, E.; Spiegelman, B.M. Stimulation of Adipogenesis in Fibroblasts by PPARγ2, a Lipid-activated Transcription Factor. Cell 1994, 79, 1147–1156. [Google Scholar] [CrossRef]
- Wu, Z.; Puigserver, P.; Spiegelman, B.M. Transcriptional Activation of Adipogenesis. Curr. Opin. Cell Biol. 1999, 11, 689–694. [Google Scholar] [CrossRef]
- Hamm, J.K.; Park, B.H.; Farmer, S.R. A Role for C/EBPβ in Regulating Peroxisome Proliferator-activated Receptor γ Activity during Adipogenesis in 3T3-L1 Preadipocytes. J. Biol. Chem. 2001, 276, 18464–18471. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, W. The journey of thiazolidinediones as modulators of PPARs for the management of diabetes: A current perspective. Curr. Pharm. Des. 2019, 25, 2540–2554. [Google Scholar] [CrossRef]
- Song, Z.; Xiaoli, A.M.; Yang, F. Regulation and metabolic significance of de novo lipogenesis in adipose Tissues. Nutrients 2018, 10, 1383. [Google Scholar] [CrossRef]
- Nallamuthu, I.; Jain, A.; Anand, T. Comparative evaluation of Brassica oleracea, Ocimum basilicum, and Moringa oleifera leaf extracts on lipase inhibition and adipogenesis in 3T3-L1 adipocytes. J. Food Biochem. 2022, 46, e14158. [Google Scholar] [CrossRef]
- Xie, J.; Wang, Y.; Jiang, W.-W.; Luo, X.-F.; Dai, T.-Y.; Peng, L.; Song, S.; Li, L.-F.; Tao, L.; Shi, C.-Y.; et al. Moringa oleifera leaf petroleum ether extract inhibits lipogenesis by activating the AMPK signaling pathway. Front. Pharmacol. 2018, 9, 1447. [Google Scholar] [CrossRef]
- Minh, T.N.; Minh, B.Q.; Duc, T.H.; Thinh, P.V.; Anh, L.V.; Dat, N.T.; Nhan, L.V.; Trung, N.Q. Potential use of Moringa oleifera Twigs Extracts as an Anti-hyperuricemic and Anti-microbial Source. Processes 2022, 10, 563. [Google Scholar] [CrossRef]
- Taylor, V.F.; March, R.E.; Longerich, H.P.; Christopher, J.S. A Mass Spectrometric Study of Glucose, Sucrose, and Fructose using an Inductively Coupled Plasma and Electrospray Ionization. Int. J. Mass. Spectrom. 2005, 243, 71–84. [Google Scholar] [CrossRef]
- Kets, E.; Galinski, E.; Wit, M.D.; Bont, J.A.M.D.; Heipieper, H.J. Mannitol, a Novel Bacterial Compatible Solute in Pseudomonas putida S12. J. Bacteriol. 2014, 178, 6665–6670. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.-B.; Chen, G.-L.; Guo, M.-Q. Antioxidant and Anti-Inflammatory Activities of the Crude Extracts of Moringa oleifera from Kenya and Their Correlations with Flavonoids. Antioxidants 2019, 8, 296. [Google Scholar] [CrossRef]
- Liu, G.; Ma, J.; Chen, Y.; Tian, Q.; Shen, Y.; Wang, X.; Chen, B.; Yao, S. Investigation of Flavonoid Profile of Scutellaria bacalensis Georgi by High Performance Liquid Chromatography with Diode Array Detection and Electrospray Ion Trap Mass Spectrometry. J. Chromatograp. A 2009, 1216, 4809–4814. [Google Scholar] [CrossRef]
- Lin, W.; Liu, S.; Wu, B. Structural Identification of Chemical Constituents from Scutellaria baicalensis by HPLC-ESI-MS/MS and NMR Spectroscopy. Asian J. Chem. 2013, 25, 3799–3805. [Google Scholar] [CrossRef]
- Corradini, E.; Foglia, P.; Giansanti, P.; Gubbiotti, R.; Samperi, R.; Lagana, A. Flavonoids: Chemical Properties and Analytical Methodologies of Identification and Quantitation in Foods and Plants. Nat. Prod. Res. 2011, 25, 469–495. [Google Scholar] [CrossRef] [PubMed]
- Sagandykova, G.; Walczak-Skierska, J.; Monedeiro, F.; Pomastowski, P.; Buszewski, B. New Methodology for the Identification of Metabolites of Saccharides and Cyclitols by Off-Line EC-MALDI-TOF-MS. Int. J. Mol. Sci. 2020, 21, 5265. [Google Scholar] [CrossRef]
- He, Z.-H.; Liu, M.; Ren, J.-X.; Ouyang, D.-W. Structural Characterization of Chemical Compounds Based on Their Fragmentation Rules in Sophorae Fructus by UPLC-QTOF-MS/MS. Pharm. Front. 2022, 4, e162–e178. [Google Scholar] [CrossRef]
- Śliwka-Kaszyńska, M.; Anusiewicz, I.; Skurski, P. The mechanism of a Retro-Diels–Alder Fragmentation of Luteolin: Theoretical Studies Supported by Electrospray Ionization Tandem Mass Spectrometry Results. Molecules 2022, 27, 1032. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, S.; Xu, C.; Yu, L.; Chu, S.; Bao, J.; Wang, J.; Wang, J. Identification of Diagnostic Biomarkers for Compensatory Liver Cirrhosis Based on Gut Microbiota and Urine Metabolomics Analyses. Mol. Biotechnol. 2023; Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Cui, S.; Li, Z.; Yan, Y.; Li, C.; Li, R. A Combination of Metabolite Profiling and Network Pharmacology to Explore the Potential Pharmacological Changes of Secoisolariciresinol-diglycoside. RSC Adv. 2020, 10, 34847–34858. [Google Scholar] [CrossRef] [PubMed]
- Shahzad, M.N.; Ahmad, S.; Tousif, M.I.; Ahmad, H.R.; Ahmad, B.; Basit, A. Profiling of Phytochemicals from aerial Parts of Terminalia neotaliala using LC-ESI-MS2 and Determination of Antioxidant and Enzyme Inhibition Activities. PLoS ONE 2022, 17, e0266094. [Google Scholar] [CrossRef] [PubMed]
- Ricciutelli, M.; Moretti, S.; Galarini, R.; Sagratini, G.; Mari, M.; Lucarini, S.; Vittori, S.; Caprioli, G. Identification and Quantification of New Isomers of Isopropyl-malic acid in Wine by LC-IT and LC-Q-Orbitrap. Food Chem. 2019, 294, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Kikuzaki, H.; Jin, Y.; Nakatani, N.; Zhu, N.; Csiszar, K.; Boyd, C.; Rosen, R.T.; Ghai, G.; Ho, D.T. Novel Glycosides from Noni (Morinda citrifolia). J. Nat. Prod. 2000, 63, 1182–1183. [Google Scholar] [CrossRef]
- Hung, T.-W.; Yang, M.-Y.; Yu, M.-H.; Tsai, I.N.; Tsai, Y.-C.; Chan, K.-C.; Wang, C.-J. Mulberry leaf extract and neochlorogenic acid ameliorate glucolipotoxicity-induced diabetic nephropathy in high-fat diet-fed db/db mice. Food Funct. 2023, 14, 8975–8986. [Google Scholar] [CrossRef]
- Zhou, Y. The protective effects of cryptochlorogenic acid on β-cells function in diabetes in vivo and vitro via inhibition of ferroptosis. Diabetes Metab. Syndr. Obes. 2020, 13, 1921–1931. [Google Scholar] [CrossRef]
- Takeda, Y.; Dai, P. Functional roles of pantothenic acid, riboflavin, thiamine, and choline in adipocyte browning in chemically induced human brown adipocytes. Sci. Rep. 2024, 14, 18252. [Google Scholar] [CrossRef]
- Sun, M.-Y.; Ye, H.-J.; Zheng, C.; Jin, Z.-J.; Yuan, Y.; Weng, H.-B. Astragalin ameliorates renal injury in diabetic mice by modulating mitochondrial quality control via AMPK-dependent PGC1α pathway. Acta Pharmacol. Sin. 2023, 44, 1676–1686. [Google Scholar] [CrossRef]
- Jeon, H.-J.; Choi, D.K.; Choi, J.; Lee, S.; Lee, H.; Yu, J.H.; Min, S.-H. D-Mannitol induces a brown fat-like phenotype via a β3-adrenergic receptor-dependent mechanism. Cells 2021, 10, 768. [Google Scholar] [CrossRef] [PubMed]
- Agudelo, L.Z.; Ferreira, D.M.S.; Cervenka, I.; Bryzgalova, G.; Dadvar, S.; Jannig, P.R.; Pettersson-Klein, A.T.; Lakshmikanth, T.; Sustarsic, E.G.; Porsmyr-Palmertz, M.; et al. Kynurenic Acid and Gpr35 Regulate Adipose Tissue Energy Homeostasis and Inflammation. Cell Metab. 2018, 27, 378–392.e5. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.-G.; Pang, Y.-L.; Zhu, Q.; Kang, J.-H.; Liu, M.-X.; Wang, Z. Chlorogenic acid functions as a novel agonist of PPARγ2 during the differentiation of mouse 3T3-L1 preadipocytes. Biomed. Res. Int. 2018, 2018, 8594767. [Google Scholar] [CrossRef]
- Yeon, J.; Suh, S.-S.; Youn, U.-J.; Bazarragcha, B.; Enebish, G.; Seo, J.B. Methanol extract of Mongolian Iris bungei Maxim. Stimulates 3T3-L1 adipocyte differentiation. J. Nanosci. Nanotechnol. 2021, 21, 3943–3949. [Google Scholar] [CrossRef] [PubMed]
- Eom, J.; Choi, J.; Suh, S.-S.; Seo, J.B. SLC3A2 and SLC7A2 Mediate the Exogenous Putrescine-induced Adipocyte Differentiation. Mol. Cells 2022, 45, 963–975. [Google Scholar] [CrossRef]
- Yeon, J.; Kim, E.; Bazarragchaa, B.; Kim, S.-Y.; Hub, J.Y.; Park, H.; Suh, S.-S.; Seo, J.B. Stellera chamaejasme L. Extract Inhibits Adipocyte Differentiation Through Activation of The Extracellular Signal-regulated Kinase Pathway. PLoS ONE 2024, 19, e0300520. [Google Scholar] [CrossRef] [PubMed]
- Le, D.D.; Kim, Y.-R.; Yu, S.; Dang, T.; Lee, K.-T.; Lee, M. Phytochemical and bioactivities of promising flavonoid glycosides and their content from unmatured fruits of Vicia bungei with their bioactivity. Food Chem. 2024, 460, 140541. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Compound | Leaves | Twigs | Stem Bark | Roots | Seeds | Formula | Adduct | m/z (Da) | RT (min) | Error (ppm) | Class |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Glucose # | − | + | − | + | + | C6H12O6 | [M-H]- | 179.05636 | 0.84 | 1.4 | Glycoside |
| 2 | Mannitol # | − | − | + | − | − | C7H12O6 | [M-H]- | 181.07231 | 0.88 | 0.2 | Glycoside |
| 3 | Quinic acid # | + | + | + | + | − | C7H12O6 | [M-H]- | 191.05626 | 0.93 | 0.8 | Quinic |
| 4 | Unknown * | − | − | − | − | + | C19H25NO19 | [M-H]- | 341.10893 | 0.95 | 1.0 | Unknown |
| 5 | Sucrose # | + | + | − | + | + | C12H22O11 | [M-H]- | 341.10893 | 0.98 | 0.0 | Glycoside |
| 6 | Citric acid # | − | − | − | + | − | C6H8O7 | [M-H]- | 191.02004 | 1.21 | 1.6 | Carboxylic acid |
| 7 | N-Acetyl-DL-glutamic acid # | − | − | + | − | − | C7H11NO5 | [M-H]- | 188.05645 | 1.30 | 0.0 | Amino acid |
| 8 | Adenosin # | − | − | − | + | − | C10H13N5O4 | [M+H]+ | 268.10422 | 1.32 | 0.7 | Nucleoside |
| 9 | N-(1-Deoxy-1-fructosyl)phenylalanine # | − | − | − | + | − | C10H19NO7 | [M+H]+ | 328.13957 | 2.50 | 0.4 | Phenolic glycoside |
| 10 | Neochlorogenic acid † | + | + | − | − | − | C16H18O9 | [M-H]- | 353.08751 | 4.83 | 0.8 | Quinic |
| 11 | Unknown * | − | − | + | − | − | C14H20O8 | [M-H]- | 315.10825 | 4.89 | 0.9 | Unknown |
| 12 | Kynurenic acid # | − | − | + | − | − | C10H7NO3 | [M-H]- | 188.03538 | 5.29 | 0.4 | Alkaloid |
| 13 | 2-Isopropylmalic acid # | − | − | − | + | − | C7H12O5 | [M-H]- | 175.0615 | 5.34 | 1.7 | Carboxylic acid |
| 14 | Unknown * | − | − | + | − | − | C13H16O7 | [M-H]- | 283.0822 | 5.65 | 0.4 | Unknown |
| 15 | p-Coumaroylquinic acid # | + | + | − | − | − | C16H18O8 | [M-H]- | 337.09319 | 6.21 | 0.9 | Quinic |
| 16 | Chlorogenic acid † | + | − | − | − | − | C16H18O9 | [M-H]- | 353.08745 | 6.90 | 1.0 | Quinic |
| 17 | 3-Methoxytyrosine # | − | + | − | + | − | C10H13NO4 | [M-H]- | 210.07746 | 7.49 | 1.3 | Phenolic |
| 18 | Cryptochlorogenic acid † | + | + | − | − | − | C16H18O9 | [M-H]- | 353.08751 | 7.53 | 0.8 | Quinic |
| 19 | Unknown * | − | − | − | + | − | C19H27NO11 | [M-H]- | 444.15071 | 7.55 | 1.0 | Unknown |
| 20 | Unknown * | − | − | − | + | − | C18H29NO10 | [M+H]+ | 420.18629 | 8.21 | 0.3 | Unknown |
| 21 | Acetyl-4-(α-L-rhamnopyranosyloxy) benzyl glucosinolate ϕ | + | − | − | − | − | C22H31NO15S2 | [M-H]- | 612.10619 | 8.26 | 0.1 | Phenolic glycoside |
| 22 | Unknown * | − | − | + | − | − | C14H18O8 | [M-H]- | 313.09267 | 8.56 | 0.7 | Unknown |
| 23 | Orcinol gentiobioside * | + | + | + | + | + | C19H28O12 | [M-H]- | 447.15036 | 8.81 | 1.0 | Phenolic glycoside |
| 24 | 4-Methylumbelliferyl-α-D-galactopyranoside * | − | + | − | − | − | C16H18O8 | [M-H]- | 337.09254 | 8.97 | 1.0 | Phenolic glycoside |
| 25 | Pantothenic acid # | − | + | + | + | − | C9H17NO5 | [M-H]- | 218.10356 | 9.10 | 0.8 | Alkaloid |
| 26 | 4-Hydroxyhydrocinnamic acid * | − | − | + | − | − | C9H10O3 | [M+H]+ | 165.05603 | 9.68 | 1.9 | Phenolic |
| 27 | 2-Formylphenyl 2-acetamido-2-deoxy-β-D-glucopyranoside * | + | + | + | − | + | C15H19NO7 | [M-H]- | 324.10871 | 10.34 | 0.5 | Phenolic glucoside |
| 28 | Apigenin-6,8-di-C-glucoside (Vitexin-2) # | + | + | + | − | − | C27H30O15 | [M-H]- | 593.15097 | 10.79 | 0.4 | Flavone-di-C-glycosides |
| 29 | Acetyl-L-phenylalanine # | − | − | + | − | − | C11H13NO3 | [M-H]- | 206.08244 | 10.91 | 0.8 | Amino acid |
| 30 | Suberic acid # | − | − | + | − | − | C8H14O4 | [M-H]- | 173.08207 | 11.13 | 0.8 | Fatty acid |
| 31 | Nonioside D * | + | + | − | − | − | C18H32O12 | [M-H]- | 439.18181 | 12.2 | 0.7 | Butanolic acid glycosides |
| 32 | Unknown * | − | + | − | − | − | C20H25NO9 | [M-H]- | 422.14524 | 12.65 | 1.0 | Unknown |
| 33 | 9-Oxo-nonanoic acid | − | − | + | − | − | C9H16O3 | [M-H]- | 171.1024 | 13.59 | 0.8 | Fatty acid |
| 34 | Vitexin # | + | + | − | − | − | C21H20O10 | [M-H]- | 431.09828 | 14.46 | 0.2 | Flavone-C-glucose |
| 35 | Rutin # | + | + | − | − | − | C27H30O16 | [M-H]- | 609.14672 | 14.92 | 1.0 | Flavone-Di-O-glycosides |
| 36 | Isovitexin # | + | + | − | − | − | C21H20O10 | [M-H]- | 431.09859 | 14.95 | 0.5 | Flavone-Di-O-glycosides |
| 37 | Quercetin-3-glucoside † | + | − | − | + | − | C21H20O12 | [M-H]- | 463.0886 | 15.36 | 0.9 | Flavone glucoside |
| 38 | 9-[2,3-Dihydroxypropoxy]-9-oxononanoic acid # | − | − | + | + | − | C12H22O6 | [M-H]- | 261.13411 | 15.72 | 1.0 | Fatty acid |
| 39 | Quercetin 3-(6″-acetylglucoside) # | − | + | − | − | − | C23H22O13 | [M-H]- | 505.09856 | 15.98 | 0.4 | Flavone glucoside |
| 40 | Azelaic acid # | − | + | + | + | + | C9H16O4 | [M-H]- | 187.09775 | 16.34 | 0.9 | Fatty acid |
| 41 | Quercetin 3-O-malonylglucoside # | + | + | − | − | − | C24H22O15 | [M-H]- | 549.08917 | 16.74 | 1.0 | Flavone-O-glucoside |
| 42 | Kaempferol-3-rutinoside # | + | + | − | − | − | C27H30O15 | [M-H]- | 593.1516 | 17.09 | 0.7 | Flavone-O-glycoside |
| 43 | Astragalin † | + | + | − | + | − | C27H30O15 | [M-H]- | 447.09308 | 17.58 | 0.5 | Flavone-O-glycoside |
| 44 | Isorhamnetin-3-glucoside # | + | + | − | + | − | C22H22O12 | [M-H]- | 477.10425 | 18.25 | 0.8 | Flavone-O-glycoside |
| 45 | Secoisolariciresinol # | − | − | − | + | − | C20H26O6 | [M-H]- | 361.16596 | 18.63 | 0.8 | Phenolic |
| 46 | Kaempferol 3-(6″-acetylglucoside) ϕ | + | + | − | − | − | C22H22O12 | [M-H]- | 489.10414 | 19.38 | 0.6 | Flavone-O-glycoside |
| 47 | Unknown * | − | + | − | − | − | C12H17NO4 | [M-H]- | 238.10863 | 19.77 | 0.6 | Unknown |
| 48 | Decanedioic acid # | − | − | + | − | − | C10H18O4 | [M-H]- | 201.11326 | 21.56 | 0.1 | Fatty acid |
| 49 | 9,12,13-Trihydroxy-10,15-octadecadienoic acid # | − | − | − | + | − | C18H32O5 | [M-H]- | 327.21799 | 22.47 | 0.9 | Fatty acid |
| 50 | Tridecanedioic acid | + | − | − | − | − | C13H24O4 | [M-H]- | 243.1588 | 22.937 | 0.3 | Fatty acid |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, D.D.; Kim, E.; Dang, T.; Lee, J.; Shin, C.H.; Park, J.W.; Lee, S.-g.; Seo, J.B.; Lee, M. Chemical Investigation and Regulation of Adipogenic Differentiation of Cultivated Moringa oleifera. Pharmaceuticals 2024, 17, 1310. https://doi.org/10.3390/ph17101310
Le DD, Kim E, Dang T, Lee J, Shin CH, Park JW, Lee S-g, Seo JB, Lee M. Chemical Investigation and Regulation of Adipogenic Differentiation of Cultivated Moringa oleifera. Pharmaceuticals. 2024; 17(10):1310. https://doi.org/10.3390/ph17101310
Chicago/Turabian StyleLe, Duc Dat, Eunbin Kim, Thinhulinh Dang, Jiseok Lee, Choon Ho Shin, Jin Woo Park, Seul-gi Lee, Jong Bae Seo, and Mina Lee. 2024. "Chemical Investigation and Regulation of Adipogenic Differentiation of Cultivated Moringa oleifera" Pharmaceuticals 17, no. 10: 1310. https://doi.org/10.3390/ph17101310
APA StyleLe, D. D., Kim, E., Dang, T., Lee, J., Shin, C. H., Park, J. W., Lee, S.-g., Seo, J. B., & Lee, M. (2024). Chemical Investigation and Regulation of Adipogenic Differentiation of Cultivated Moringa oleifera. Pharmaceuticals, 17(10), 1310. https://doi.org/10.3390/ph17101310