
Therapeutic, Clinicopathological, and Molecular Correlates of PRKACA Expression in Gastrointestinal Cancers


Abstract
1. Introduction
2. Results
2.1. Enrichment of Gene Targets of Multiple Drug Agents in PRKACA-Low and -High Cancers
2.1.1. Gastric Cancer Cohort
2.1.2. Colorectal Cancer Cohort
2.2. Clinicopathological and Molecular Features of PRKACA Expression
2.2.1. Gastric Cancer Cohort
2.2.2. Colorectal Cancer Cohort
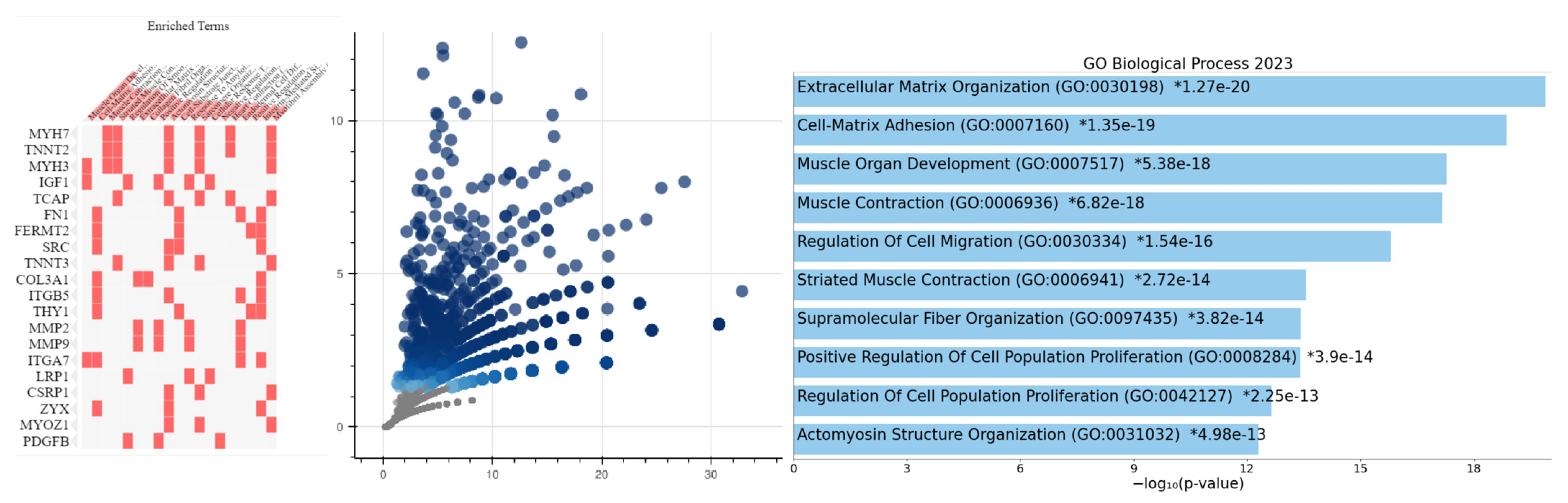
2.3. Biological Significance of PRKACA Expression
2.3.1. Gastric Cancer Cohort
2.3.2. Colorectal Cancer Cohort
2.4. Differential Core Enrichment Gene Sets between GC and CRC PRKACA-High Subsets
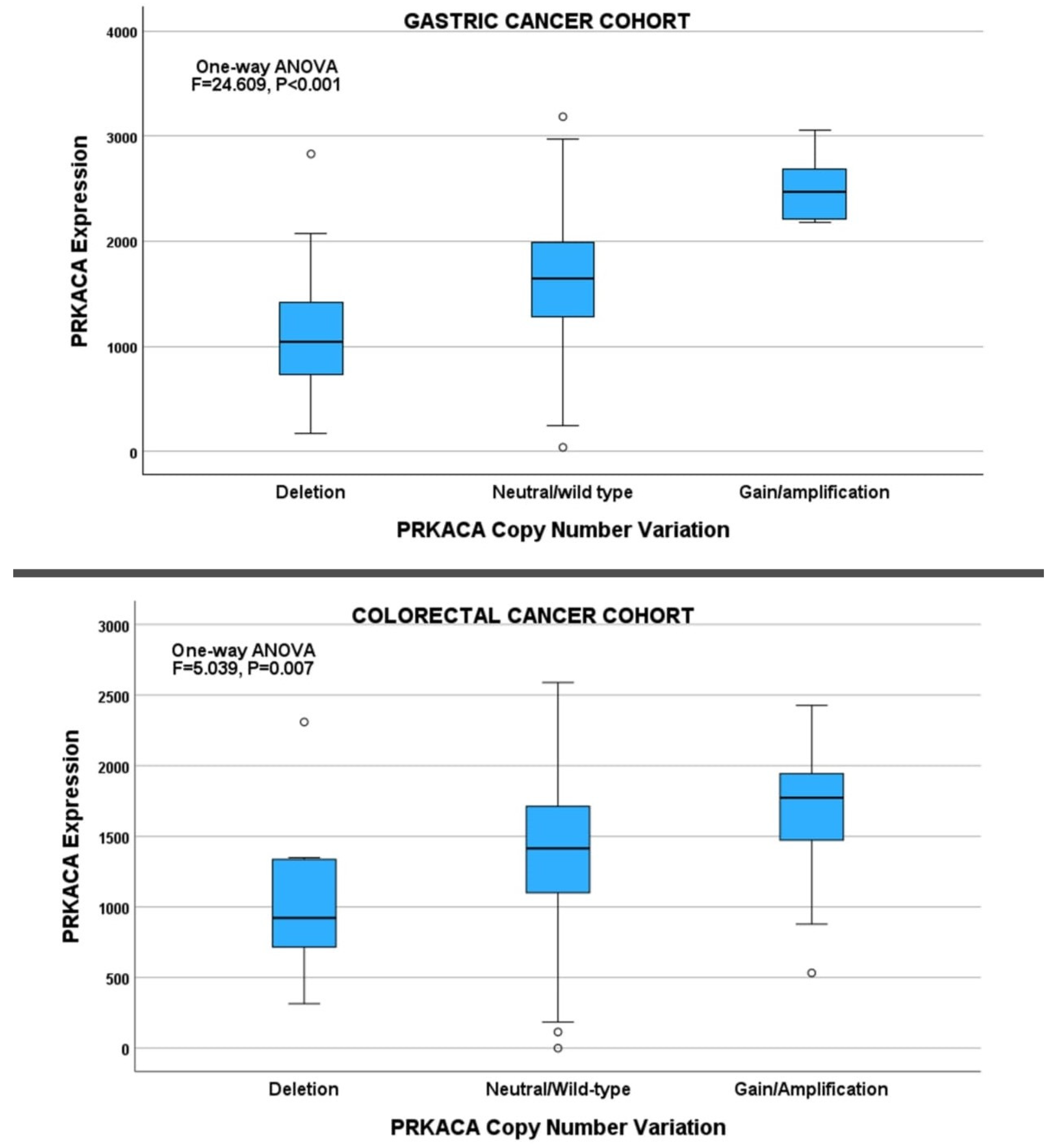
2.5. Deregulation of PRKACA Expression in Gastrointestinal Cancers
2.5.1. Gastric Cancer Cohort
2.5.2. Colorectal Cancer Cohort
3. Discussion
4. Materials and Methods
4.1. Study Cohorts
4.2. Data Processing
4.3. Study Approach
4.4. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jardim, S.R.; de Souza, L.M.P.; de Souza, H.S.P. The Rise of Gastrointestinal Cancers as a Global Phenomenon: Unhealthy Behavior or Progress? Int. J. Environ. Res. Public Health 2023, 20, 3640. [Google Scholar] [CrossRef] [PubMed]
- Li, M.M.; Yuan, J.; Guan, X.Y.; Ma, N.F.; Liu, M. Molecular subclassification of gastrointestinal cancers based on cancer stem cell traits. Exp. Hematol. Oncol. 2021, 10, 53. [Google Scholar] [CrossRef]
- Minciuna, C.-E.; Tanase, M.; Manuc, T.E.; Tudor, S.; Herlea, V.; Dragomir, M.P.; Calin, G.A.; Vasilescu, C. The seen and the unseen: Molecular classification and image based-analysis of gastrointestinal cancers. CSBJ 2022, 20, 5065–5075. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Piña-Sánchez, P.; Chávez-González, A.; Ruiz-Tachiquín, M.; Vadillo, E.; Monroy-García, A.; Montesinos, J.J.; Grajales, R.; Gutiérrez de la Barrera, M.; Mayani, H. Cancer Biology, Epidemiology, and Treatment in the 21st Century: Current Status and Future Challenges From a Biomedical Perspective. Cancer Control 2021, 28, 10732748211038735. [Google Scholar] [CrossRef]
- Chehelgerdi, M.; Chehelgerdi, M.; Allela, O.Q.B.; Pecho, R.D.C.; Jayasankar, N.; Rao, D.P.; Thamaraikani, T.; Vasanthan, M.; Viktor, P.; Lakshmaiya, N.; et al. Progressing nanotechnology to improve targeted cancer treatment: Overcoming hurdles in its clinical implementation. Mol. Cancer 2023, 22, 169. [Google Scholar] [CrossRef]
- Shimozaki, K.; Nakayama, I.; Hirota, T.; Yamaguchi, K. Current Strategy to Treat Immunogenic Gastrointestinal Cancers: Perspectives for a New Era. Cells 2023, 12, 1049. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.W.; Sood, A.K. Molecular Pathways: Beta-Adrenergic Signaling in Cancer. Clin. Cancer Res. 2012, 18, 1201–1206. [Google Scholar] [CrossRef]
- Stratakis, C.A. Cyclic AMP-dependent protein kinase catalytic subunit A (PRKACA): The expected, the unexpected, and what might be next. J. Pathol. 2018, 244, 257–259. [Google Scholar] [CrossRef]
- Hou, J.Y.; Gao, L.J.; Shen, J.; Zhou, L.; Shi, J.Y.; Sun, T.; Hao, S.L.; Wang, D.P.; Cao, J.M. Crotonylation of PRKACA enhances PKA activity and promotes colorectal cancer development via the PKA-FAK-AKT pathway. Genes Dis. 2022, 10, 332–335. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kong, Q.; Wang, J.; Jiang, Y.; Hua, H. Complex roles of cAMP–PKA–CREB signaling in cancer. Exp. Hematol. Oncol. 2020, 9, 32. [Google Scholar] [CrossRef] [PubMed]
- Neumayer, C.; Ng, D.; Jiang, C.S.; Qureshi, A.; Lalazar, G.; Vaughan, R.; Simon, S.M. Oncogenic Addiction of Fibrolamellar Hepatocellular Carcinoma to the Fusion Kinase DNAJB1-PRKACA. Clin. Cancer Res. 2023, 29, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.; Köhler, N.; Maringer, Y.; Bucher, P.; Bilich, T.; Zwick, M.; Dicks, S.; Nelde, A.; Dubbelaar, M.; Scheid, J.; et al. The oncogenic fusion protein DNAJB1-PRKACA can be specifically targeted by peptide-based immunotherapy in fibrolamellar hepatocellular carcinoma. Nat. Commun. 2022, 13, 6401. [Google Scholar] [CrossRef] [PubMed]
- Kastenhuber, E.R.; Lalazar, G.; Houlihan, S.L.; Tschaharganeh, D.F.; Baslan, T.; Chen, C.C.; Requena, D.; Tian, S.; Bosbach, B.; Wilkinson, J.E.; et al. DNAJB1-PRKACA fusion kinase interacts with β-catenin and the liver regenerative response to drive fibrolamellar hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2017, 114, 13076–13084. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, Y.; Liu, J.; Chen, M.; Li, W.; Chen, Y.; Xu, M. The diagnostic value of extracellular protein kinase A (ECPKA) in serum for gastric and colorectal cancer. Transl. Cancer Res. 2020, 9, 3870–3878. [Google Scholar] [CrossRef]
- Moody, S.E.; Schinzel, A.C.; Singh, S.; Chen, M.; Li, W.; Chen, Y.; Xu, M. PRKACA mediates resistance to HER2-targeted therapy in breast cancer cells and restores anti-apoptotic signaling. Oncogene 2015, 34, 2061–2071. [Google Scholar] [CrossRef]
- Schalm, S.S.; O’Hearn, E.; Wilson, K.; LaBranche, T.P.; Silva, G.; Zhang, Z.; DiPietro, L.; Bifulco, N.; Woessner, R.; Stransky, N.; et al. Evaluation of Protein Kinase cAMP-Activated Catalytic Subunit Alpha as a Therapeutic Target for Fibrolamellar Carcinoma. Gastro Hep Adv. 2023, 2, 307–321. [Google Scholar] [CrossRef]
- Tang, J.; Aittokallio, T. Network pharmacology strategies toward multi-target anticancer therapies: From computational models to experimental design principles. Curr. Pharm. Des. 2014, 20, 23–36. [Google Scholar] [CrossRef]
- Ferlier, T.; Coulouarn, C. Regulation of Gene Expression in Cancer—An Overview. Cells 2022, 11, 4058. [Google Scholar] [CrossRef]
- Bayat-Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Kulesskiy, E.; Saarela, J.; Turunen, L.; Wennerberg, K.; Aittokallio, T.; Tang, J. Methods for High-throughput Drug Combination Screening and Synergy Scoring. Methods Mol. Biol. 2018, 1711, 351–398. [Google Scholar] [CrossRef]
- McKenzie, A.J.; Svec, K.V.; Williams, T.F.; Howe, A.K. Protein kinase A activity is regulated by actomyosin contractility during cell migration and is required for durotaxis. Mol. Biol. Cell 2020, 31, 45–58. [Google Scholar] [CrossRef]
- Tonucci, F.M.; Almada, E.; Borini-Etichetti, C.; Pariani, A.; Hidalgo, F.; Rico, M.J.; Girardini, J.; Favre, C.; Goldenring, J.R. Identification of a CIP4 PKA phosphorylation site involved in the regulation of cancer cell invasiveness and metastasis. Cancer Lett. 2019, 461, 65–77. [Google Scholar] [CrossRef]
- Cheng, Y.; Gao, X.H.; Li, X.J.; Cao, Q.H.; Zhao, D.D.; Zhou, J.R.; Wu, H.X.; Wang, Y.; You, L.J. Depression promotes prostate cancer invasion and metastasis via a sympathetic-cAMP–FAK signaling pathway. Oncogene 2018, 37, 2953–2966. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Li, J.; Wang, F.; Wei, J.; Yang, Z.; Sun, M.; Liu, J.; Wen, M.; Huang, W.; Chen, Z.; et al. Protein modifications throughout the lung cancer proteome unravel the cancer-specific regulation of glycolysis. Cell Rep. 2021, 37, 110137. [Google Scholar] [CrossRef] [PubMed]
- Berthon, A.S.; Szarek, E.; Stratakis, C.A. PRKACA: The catalytic subunit of protein kinase A and adrenocortical tumors. Front. Cell Dev. Biol. 2015, 3, 26. [Google Scholar] [CrossRef] [PubMed]
- Di Dalmazi, G.; Kisker, C.; Calebiro, D.; Mannelli, M.; Canu, L.; Arnaldi, G.; Quinkler, M.; Rayes, N.; Tabarin, A.; Jullié, M.L.; et al. Novel somatic mutations in the catalytic subunit of the protein kinase A as a cause of adrenal Cushing’s syndrome: A European multicentric study. J. Clin. Endocrinol. Metab. 2014, 99, E2093–E2100. [Google Scholar] [CrossRef]
- Goh, G.; Scholl, U.I.; Healy, J.M.; Choi, M.; Prasad, M.L.; Nelson-Williams, C.; Kuntsman, J.W.; Korah, R.; Suttorp, A.C.; Dietrich, D.; et al. Recurrent activating mutation in PRKACA in cortisol-producing adrenal tumors. Nat. Genet. 2014, 46, 613–617. [Google Scholar] [CrossRef]
- Sato, Y.; Maekawa, S.; Ishii, R.; Sanada, M.; Morikawa, T.; Shiraishi, Y.; Yoshida, K.; Nagata, Y.; Sato-Otsubo, A.; Yoshizato, T.; et al. Recurrent somatic mutations underlie corticotropin-independent Cushing’s syndrome. Science 2014, 344, 917–920. [Google Scholar] [CrossRef]
- Hatzikirou, H.; Basanta, D.; Simon, M.; Schaller, K.; Deutsch, A. ‘Go or Grow’: The key to the emergence of invasion in tumour progression? Math. Med. Biol. A J. IMA 2012, 29, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Hoek, K.S.; Eichhoff, O.M.; Schlegel, N.C.; Döbbeling, U.; Kobert, N.; Schaerer, L.; Hemmi, S.; Dummer, R. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 2008, 68, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Gil-Henn, H.; Patsialou, A.; Wang, Y.; Warren, M.S.; Condeelis, J.S.; Koleske, A.J. Arg/Abl2 promotes invasion and attenuates proliferation of breast cancer in vivo. Oncogene 2013, 32, 2622–2630. [Google Scholar] [CrossRef] [PubMed]
- Patsialou, A.; Wang, Y.; Pignatelli, J.; Chen, X.; Entenberg, D.; Oktay, M.; Condeelis, J.S. Autocrine CSF1R signaling mediates switching between invasion and proliferation downstream of TGFβ in claudin-low breast tumor cells. Oncogene 2015, 34, 2721–2731. [Google Scholar] [CrossRef]
- Yang, Y.; Ye, W.L.; Zhang, R.N.; He, X.S.; Wang, J.R.; Liu, Y.X.; Wang, Y.; Yang, X.M.; Zhang, Y.J.; Gan, W.J. The Role of TGF-β Signaling Pathways in Cancer and Its Potential as a Therapeutic Target. Evid. Based Complement. Altern. Med. 2021, 2021, 6675208. [Google Scholar] [CrossRef]
- Li, J.; Poi, M.J.; Tsai, M.D. Regulatory mechanisms of tumor suppressor P16(INK4A) and their relevance to cancer. Biochemistry 2011, 50, 5566–5582. [Google Scholar] [CrossRef]
- Li, S.; Zhu, Y.; Liang, Z.; Wang, X.; Meng, S.; Xu, X.; Xu, X.; Wu, J.; Ji, A.; Hu, Z.; et al. Up-regulation of p16 by miR-877-3p inhibits proliferation of bladder cancer. Oncotarget 2016, 7, 51773–51783. [Google Scholar] [CrossRef]
- Jung, A.; Schrauder, M.; Oswald, U.; Knoll, C.; Sellberg, P.; Palmqvist, R.; Niedobitek, G.; Brabletz, T.; Kirchner, T. The invasion front of human colorectal adenocarcinomas shows co-localization of nuclear beta-catenin, cyclin D1, and p16INK4A and is a region of low proliferation. Am. J. Pathol. 2001, 159, 1613–1617. [Google Scholar] [CrossRef]
- Itoh, T.; Omori, Y.; Seino, M.; Hirose, K.; Date, F.; Ono, Y.; Mizukami, Y.; Aoki, S.; Ishida, M.; Mizuma, M.; et al. Gene Rearrangement and Expression of PRKACA and PRKACB Govern Morphobiology of Pancreatobiliary Oncocytic Neoplasms. Mod. Pathol. 2024, 3, 100358. [Google Scholar] [CrossRef]
- Saviana, M.; Le, P.; Micalo, L.; Del Valle-Morales, D.; Romano, G.; Acunzo, M.; Li, H.; Nana-Sinkam, P. Crosstalk between miRNAs and DNA Methylation in Cancer. Genes 2023, 14, 1075. [Google Scholar] [CrossRef]
- Liu, Y.; Sethi, N.S.; Hinoue, T.; Schneider, B.G.; Cherniack, A.D.; Sanchez-Vega, F.; Seoane, J.A.; Farshidfar, F.; Bowlby, R.; Islam, M.; et al. Comparative Molecular Analysis of Gastrointestinal Adenocarcinomas. Cancer Cell 2018, 33, 721–735.e8. [Google Scholar] [CrossRef] [PubMed]
- Ebili, H.O.; Agboola, A.O.; Rakha, E. MSI-WES: A simple approach for microsatellite instability testing using whole exome sequencing. Future Oncol. 2021, 17, 3595–3606. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Reich, M.; Liefeld, T.; Gould, J.; Lerner, J.; Tamayo, P.; Mesirov, J. GenePattern 2.0. Nat. Genet. 2006, 38, 500–501. [Google Scholar] [CrossRef]
- Templeton, G.F. A Two-Step Approach for Transforming Continuous Variables to Normal: Implications and Recommendations for IS Research. Commun. Assoc. Inf. Syst. 2011, 28, 41–58. [Google Scholar] [CrossRef]
- Yoo, M.; Shin, J.; Kim, J.; Ryall, K.A.; Lee, K.; Lee, S.; Jeon, M.; Kang, J.; Tan, A.C. DSigDB: Drug Signatures Database for Gene Set Analysis. Bioinformatics 2015, 31, 3069–3071. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene set knowledge discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef] [PubMed]
- Pujato, M.; Kieken, F.; Skiles, A.A.; Tapinos, N.; Fiser, A. Prediction of DNA binding motifs from 3D models of transcription factors; identifying TLX3 regulated genes. Nucleic Acids Res. 2014, 42, 13500–13512. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PRKACA Expression | |||||||
|---|---|---|---|---|---|---|---|
| Clinicopathological Features | Low PRKACA | High PRKACA | Total | X2 * | p Value | Adjusted p Value | |
| Gastric cancer cohort | |||||||
| Age Group | 30–60 yrs | 52 | 79 | 131 | 7.630 | 0.006 | 0.016 |
| 61–90 yrs | 150 | 126 | 276 | ||||
| Total | 202 | 205 | 407 | ||||
| Gender | Male | 137 | 128 | 265 | 0.857 | 0.355 | 0.441 |
| Female | 69 | 78 | 147 | ||||
| Total | 206 | 206 | 412 | ||||
| Race/Ethnicity | White | 126 | 133 | 259 | 2.609 | 0.112 | 0.179 |
| Native Americans | 1 | 0 | 1 | ||||
| Black | 10 | 2 | 12 | ||||
| Asians | 31 | 56 | 87 | ||||
| Total | 168 | 191 | 359 | ||||
| Pathological tumor stage | pT1 | 13 | 9 | 22 | 0.753 | 0.386 | 0.441 |
| pT2 | 38 | 49 | 87 | ||||
| pT3 | 85 | 96 | 181 | ||||
| pT4 | 63 | 51 | 114 | ||||
| Total | 199 | 205 | 404 | ||||
| Pathological nodal stage | N0 | 46 | 77 | 123 | 9.775 | 0.002 | 0.008 |
| N1 | 54 | 55 | 109 | ||||
| N2 | 48 | 31 | 79 | ||||
| N3 | 46 | 36 | 82 | ||||
| Total | 194 | 199 | 393 | ||||
| Pathological metastasis stage | M0 | 183 | 181 | 364 | 0.280 | 0.596 | 0.596 |
| M1 and above | 15 | 12 | 27 | ||||
| Total | 198 | 193 | 391 | ||||
| TNM stage | Early Stage | 45 | 81 | 126 | 13.226 | <0.001 | 0.002 |
| Late Stage | 152 | 123 | 275 | ||||
| Total | 197 | 204 | 401 | ||||
| Histological type of gastric cancer | Diffuse type Adenocarcinoma | 10 | 3 | 13 | 3.892 | 0.049 | 0.098 |
| Intestinal type Adenocarcinoma | 196 | 203 | 399 | ||||
| Total | 206 | 206 | 412 | ||||
| Colorectal cancer cohort | |||||||
| Age Group | 31–60 yrs | 70 | 103 | 173 | 9.295 | 0.002 | 0.007 |
| 61–90 yrs | 198 | 165 | 363 | ||||
| Total | 268 | 268 | 536 | ||||
| Gender | Male | 141 | 142 | 283 | 0.007 | 0.931 | 0.931 |
| Female | 127 | 126 | 253 | ||||
| Total | 268 | 268 | 536 | ||||
| Race/Ethnicity | White | 116 | 167 | 283 | 3.876 | 0.049 | 0.106 |
| Asian/Native American | 7 | 6 | 13 | ||||
| Black | 34 | 29 | 63 | ||||
| Total | 157 | 202 | 359 | ||||
| Pathological tumor stage | pTis and pT1 | 5 | 10 | 15 | 0.120 | 0.729 | 0.790 |
| pT2 | 55 | 33 | 88 | ||||
| pT3 | 172 | 196 | 368 | ||||
| pT4 | 36 | 29 | 65 | ||||
| Total | 268 | 268 | 536 | ||||
| Pathological nodal stage | N0 | 170 | 134 | 304 | 7.160 | 0.007 | 0.018 |
| N1 | 55 | 76 | 131 | ||||
| N2 | 43 | 56 | 99 | ||||
| Total | 268 | 266 | 534 | ||||
| Pathological metastasis stage | M0 | 202 | 192 | 394 | 0.272 | 0.602 | 0.711 |
| M1 | 35 | 38 | 73 | ||||
| Total | 237 | 230 | 467 | ||||
| TNM stage | Early Stage | 153 | 129 | 282 | 3.956 | 0.0467 | 0.106 |
| Late Stage | 115 | 137 | 252 | ||||
| Total | 268 | 266 | 534 | ||||
| Histological type of colorectal cancer | Adenocarcinoma NOS | 225 | 240 | 465 | 3.452 | 0.063 | 0.117 |
| Mucinous Adenocarcinoma | 40 | 26 | 66 | ||||
| Total | 265 | 266 | 531 | ||||
| Colonic tumor site 1 | Right colon | 164 | 92 | 256 | 23.553 | <0.001 | <0.001 |
| Left colon | 92 | 128 | 220 | ||||
| Total | 256 | 220 | 476 | ||||
| Primary tumor site | Colon | 268 | 177 | 445 | 108.404 | <0.001 | <0.001 |
| Rectum | 0 | 90 | 90 | ||||
| Total | 268 | 267 | 535 | ||||
| Vascular Invasion (VI) | VI absent | 180 | 174 | 354 | 1.746 | 0.186 | 0.269 |
| VI present | 48 | 62 | 110 | ||||
| Total | 228 | 236 | 464 | ||||
| Lymphovascular Invasion | LVI absent | 153 | 146 | 299 | 0.513 | 0.474 | 0.616 |
| LVI present | 87 | 95 | 182 | ||||
| Total | 240 | 241 | 481 | ||||
| Perineural Invasion (PNI) | PNI absent | 75 | 95 | 170 | 1.884 | 0.170 | 0.269 |
| PNI present | 20 | 39 | 59 | ||||
| Total | 95 | 134 | 229 | ||||
| PRKACA Expression | |||||||
|---|---|---|---|---|---|---|---|
| Molecular Correlates | Low PRKACA | High PRKACA | Total | X2 * | p Value | Adjusted p Value | |
| Gastric cancer cohort | |||||||
| Molecular subtypes | CIN | 120 | 102 | 222 | 7.231 | 0.007 | 0.014 |
| MSI | 42 | 31 | 73 | ||||
| GS | 18 | 32 | 50 | ||||
| EBV | 11 | 19 | 30 | ||||
| POLE | 2 | 5 | 7 | ||||
| Total | 193 | 189 | 382 | ||||
| MSS vs. MSI | MSS | 140 | 138 | 278 | 1.192 | 0.275 | 0.275 |
| MSI | 42 | 31 | 73 | ||||
| Total | 182 | 169 | 351 | ||||
| Aneuploidy score | Low Aneuploidy Score | 88 | 110 | 198 | 5.563 | 0.018 | 0.024 |
| High Aneuploidy Score | 112 | 87 | 199 | ||||
| Total | 200 | 197 | 397 | ||||
| Global methylation score | Low Global methylation score | 128 | 74 | 202 | 28.455 | <0.001 | <0.001 |
| High Global methylation score | 77 | 131 | 208 | ||||
| Total | 205 | 205 | 410 | ||||
| Colorectal cancer cohort | |||||||
| Molecular subtypes | CIN | 112 | 178 | 290 | 21.901 | <0.001 | <0.001 |
| MSI | 42 | 21 | 63 | ||||
| GS | 39 | 16 | 55 | ||||
| POLE | 3 | 4 | 7 | ||||
| Total | 196 | 219 | 415 | ||||
| MSS vs. MSI | MSS | 154 | 198 | 352 | 11.260 | 0.001 | 0.002 |
| MSI | 42 | 21 | 63 | ||||
| Total | 196 | 219 | 415 | ||||
| Mutation Count | Low Mutation count | 93 | 120 | 213 | 6.168 | 0.013 | 0.017 |
| High Mutation count | 136 | 110 | 246 | ||||
| Total | 229 | 230 | 459 | ||||
| Fraction of Genome Altered | Low Fraction Genome Altered | 151 | 112 | 263 | 10.592 | 0.001 | 0.002 |
| High Fraction Genome Altered | 102 | 136 | 238 | ||||
| Total | 253 | 248 | 501 | ||||
| MANTIS Score | Low MANTIS Score | 109 | 116 | 225 | 0.724 | 0.395 | 0.395 |
| High MANTIS Score | 135 | 123 | 258 | ||||
| Total | 244 | 239 | 483 | ||||
| MSI Sensor | Low MSI Sensor Score | 106 | 151 | 257 | 19.578 | <0.001 | <0.001 |
| High MSI Sensor Score | 150 | 96 | 246 | ||||
| Total | 256 | 247 | 503 | ||||
| Aneuploidy Score | Low Aneuploidy Score | 120 | 89 | 209 | 8.537 | 0.003 | 0.005 |
| High Aneuploidy Score | 92 | 121 | 213 | ||||
| Total | 212 | 210 | 422 | ||||
| Tumor Mutational Burden | Low TMB | 83 | 108 | 191 | 3.701 | 0.054 | 0.061 |
| High TMB | 115 | 102 | 217 | ||||
| Total | 198 | 210 | 408 | ||||
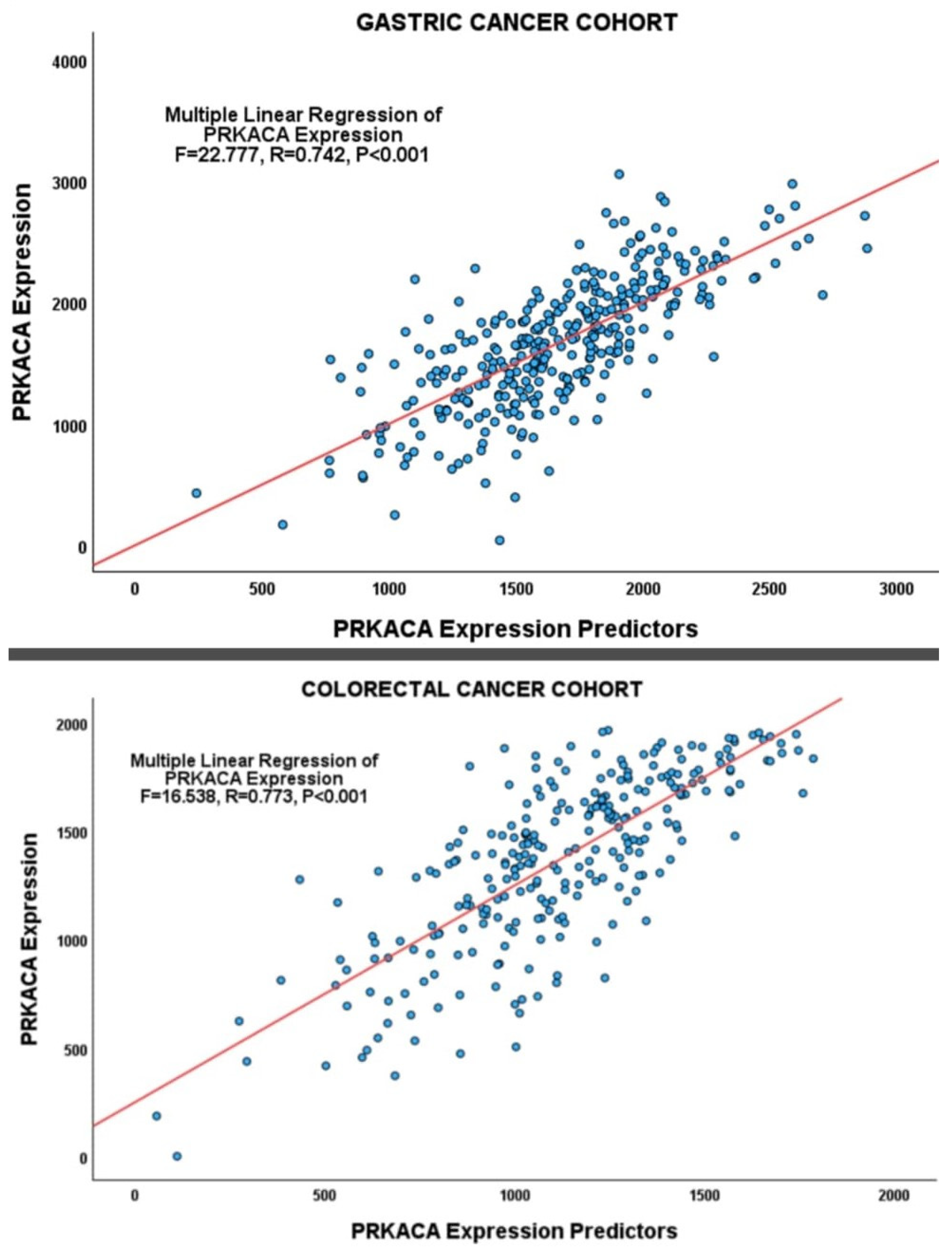
| Gastric Cancer | ||||
|---|---|---|---|---|
| R | R2 | Adjusted R2 | S.E. of Estimate | |
| 0.742 | 0.550 | 0.526 | 366.808 | |
| Coefficients | ||||
| Unstandardized Coefficients | t | p | ||
| B | S. E. | |||
| (Constant) | 2453.432 | 240.529 | 10.200 | <0.001 |
| hsa-mir-490 | 0.586 | 0.130 | 4.514 | <0.001 |
| PRKACA CNV | 450.825 | 69.936 | 6.446 | <0.001 |
| ZNF644 Expression | −60.541 | 15.418 | −3.927 | <0.001 |
| ZNF557 Expression | 79.896 | 24.840 | 3.216 | 0.001 |
| cg17818798 | 3007.832 | 745.072 | 4.037 | <0.001 |
| cg26613742 | −2233.574 | 373.632 | −5.978 | <0.001 |
| cg01596520 | 1031.285 | 338.217 | 3.049 | 0.002 |
| HEY2 Expression | 71.020 | 18.869 | 3.764 | <0.001 |
| CDC5L Expression | −8.872 | 4.218 | −2.103 | 0.036 |
| TFEB Expression | 21.175 | 7.597 | 2.787 | 0.006 |
| cg19621460 | 447.432 | 160.599 | 2.786 | 0.006 |
| hsa-mir-7641-1 | 13.596 | 4.805 | 2.830 | 0.005 |
| SP8 Expression | −61.200 | 21.970 | −2.786 | 0.006 |
| ZNF101 Expression | 45.030 | 16.502 | 2.729 | 0.007 |
| ZKSCAN3 Expression | 128.247 | 46.163 | 2.778 | 0.006 |
| ZNF451 Expression | −97.305 | 34.028 | −2.860 | 0.005 |
| cg01010868 | −29,345.397 | 12879.446 | −2.278 | 0.023 |
| ANOVA | ||||
| df | F | p | ||
| Regression | 17 | 22.777 | <0.001 | |
| Residual | 317 | |||
| Total | 334 | |||
| R | R2 | Adjusted R2 | S.E. of Estimate | ||
|---|---|---|---|---|---|
| 0.773 | 0.598 | 0.561 | 259.799 | ||
| Coefficients | |||||
| Unstandardized Coefficients | t | p | |||
| B | S. E. | ||||
| (Constant) | −520.078 | 360.444 | −1.443 | 0.150 | |
| ZBTB7A Expression | 31.065 | 4.177 | 7.437 | <0.001 | |
| hsa-mir-143 | 0.001 | 0.000 | 2.461 | 0.015 | |
| cg15814923 | −6980.818 | 2832.849 | −2.464 | 0.014 | |
| hsa-mir-577 | −0.505 | 0.196 | −2.578 | 0.011 | |
| TSHZ3 Expression | 92.833 | 19.104 | 4.859 | <0.001 | |
| DHX34 Expression | 18.602 | 6.649 | 2.798 | 0.006 | |
| cg17818798 | −996.038 | 497.031 | −2.004 | 0.046 | |
| cg19586199 | −610.229 | 186.442 | −3.273 | 0.001 | |
| ZNF451 Expression | −124.101 | 27.985 | −4.434 | <0.001 | |
| ZNF442 Expression | 324.096 | 163.862 | 1.978 | 0.049 | |
| ZKSCAN8 Expression | 68.663 | 15.481 | 4.435 | <0.001 | |
| MAFK Expression | −15.686 | 4.159 | −3.772 | <0.001 | |
| HOXA1 Expression | 80.336 | 23.028 | 3.489 | 0.001 | |
| ZNF516 Expression | −111.062 | 23.760 | −4.674 | <0.001 | |
| cg17119568 | 2626.810 | 942.395 | 2.787 | 0.006 | |
| hsa-mir-216a | 20.726 | 7.769 | 2.668 | 0.008 | |
| ZNF140 Expression | −37.664 | 20.633 | −1.825 | 0.069 | |
| ZNF557 Expression | 89.689 | 34.613 | 2.591 | 0.010 | |
| hsa-mir-1-2 | −1.278 | 0.643 | −1.986 | 0.048 | |
| cg20110535 | 846.904 | 368.866 | 2.296 | 0.023 | |
| hsa-mir-5092 | −78.546 | 31.141 | −2.522 | 0.012 | |
| ZNF235 Expression | −162.614 | 78.187 | −2.080 | 0.039 | |
| ANOVA | |||||
| df | F | p | |||
| Regression | 22 | 16.538 | <0.001 | ||
| Residual | 245 | ||||
| Total | 267 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Othaim, A.A.; Alasiri, G.; Alfahed, A. Therapeutic, Clinicopathological, and Molecular Correlates of PRKACA Expression in Gastrointestinal Cancers. Pharmaceuticals 2024, 17, 1263. https://doi.org/10.3390/ph17101263
Othaim AA, Alasiri G, Alfahed A. Therapeutic, Clinicopathological, and Molecular Correlates of PRKACA Expression in Gastrointestinal Cancers. Pharmaceuticals. 2024; 17(10):1263. https://doi.org/10.3390/ph17101263
Chicago/Turabian StyleOthaim, Ayoub Al, Glowi Alasiri, and Abdulaziz Alfahed. 2024. "Therapeutic, Clinicopathological, and Molecular Correlates of PRKACA Expression in Gastrointestinal Cancers" Pharmaceuticals 17, no. 10: 1263. https://doi.org/10.3390/ph17101263
APA StyleOthaim, A. A., Alasiri, G., & Alfahed, A. (2024). Therapeutic, Clinicopathological, and Molecular Correlates of PRKACA Expression in Gastrointestinal Cancers. Pharmaceuticals, 17(10), 1263. https://doi.org/10.3390/ph17101263