

Total Synthesis and Anti-Inflammatory Evaluation of Osajin, Scandenone and Analogues
Abstract
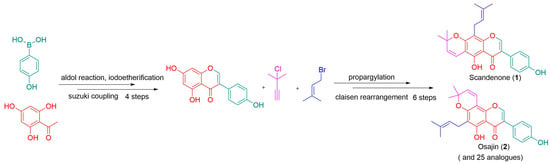
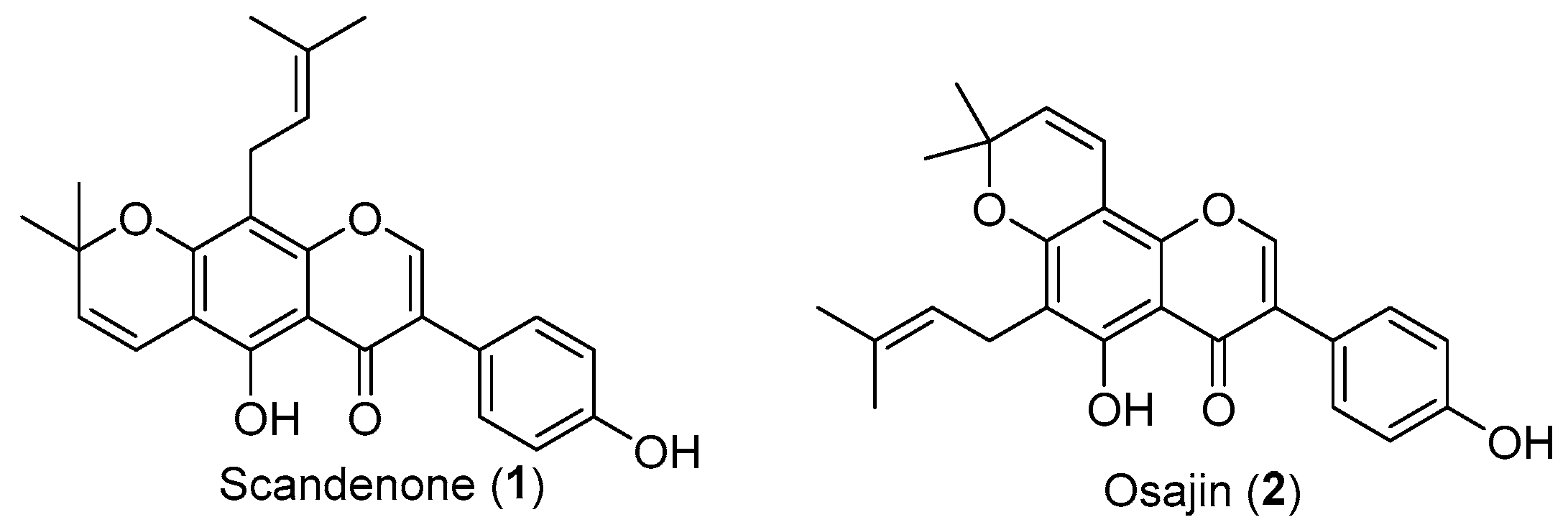
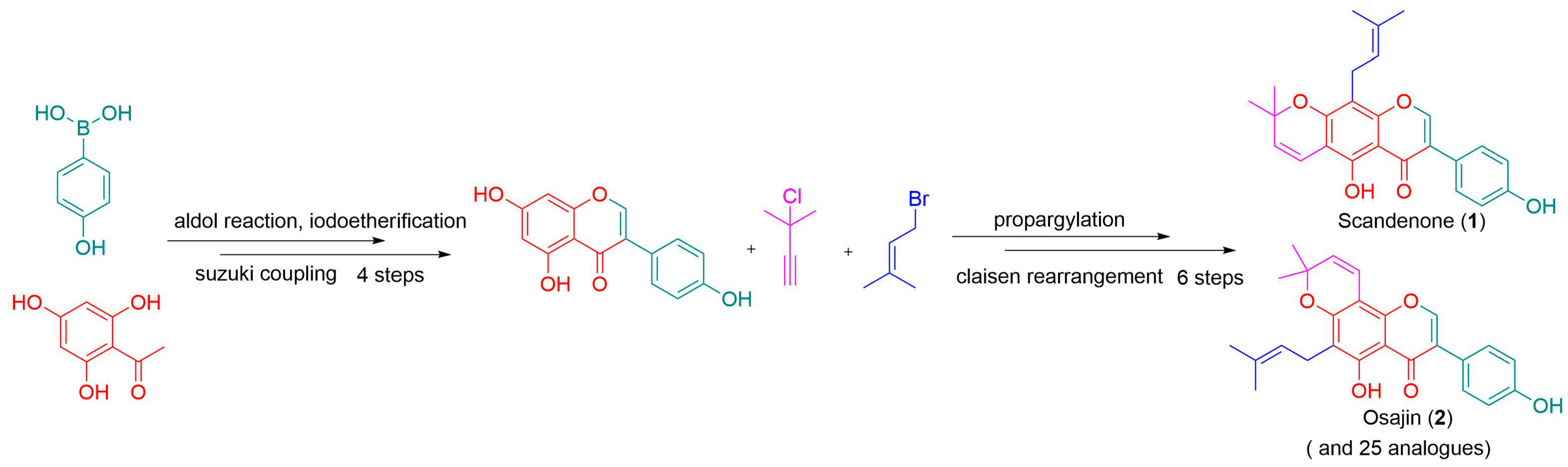
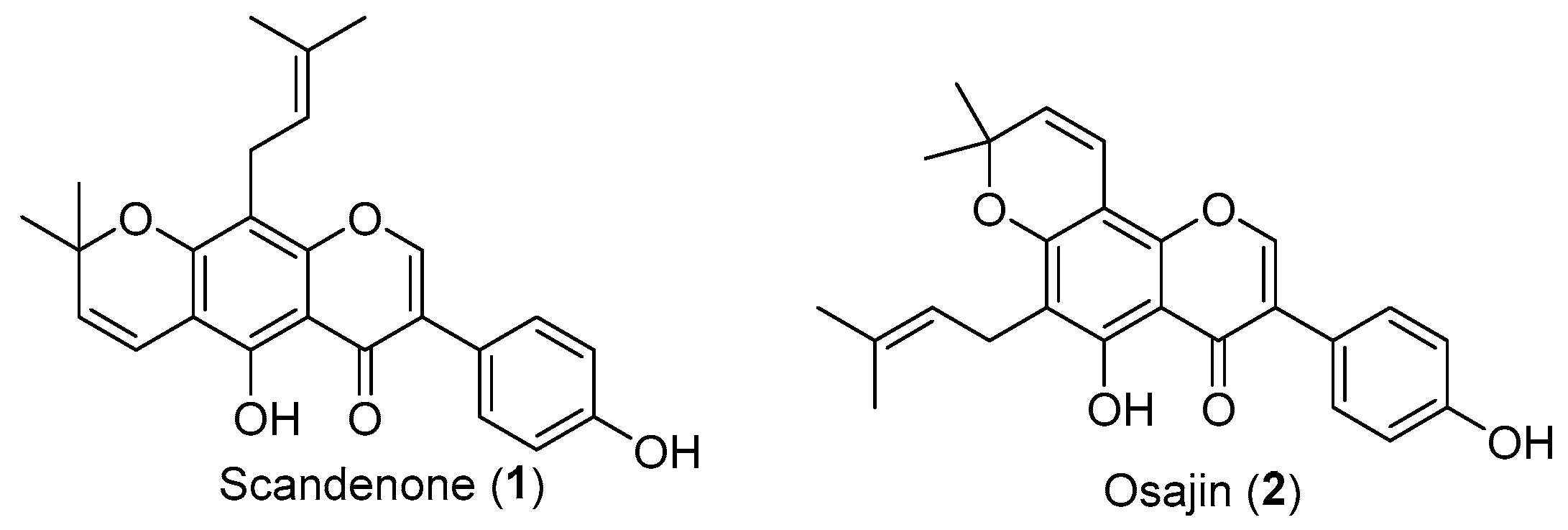
1. Introduction
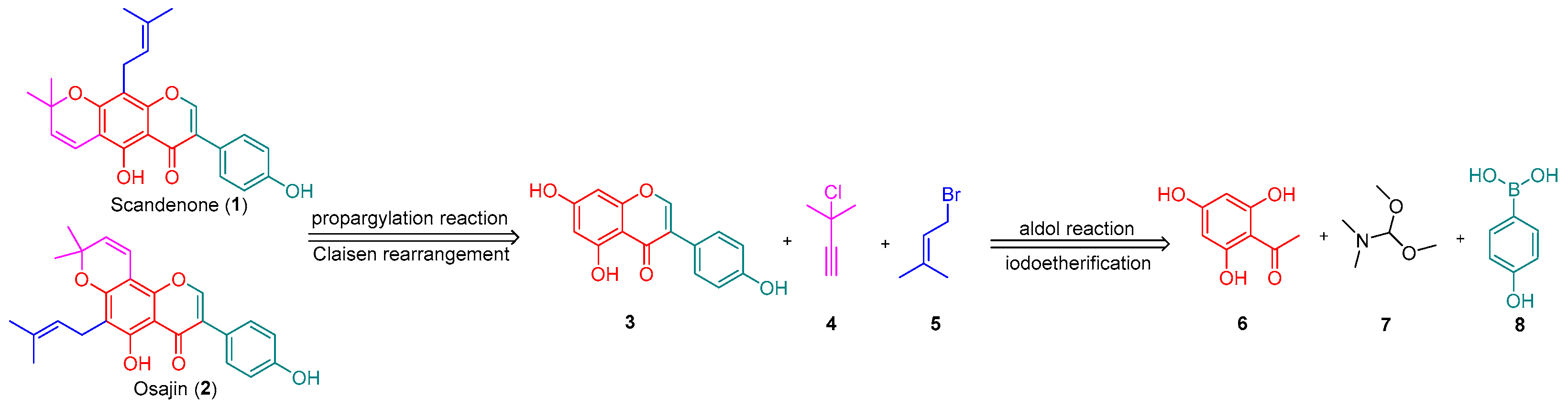
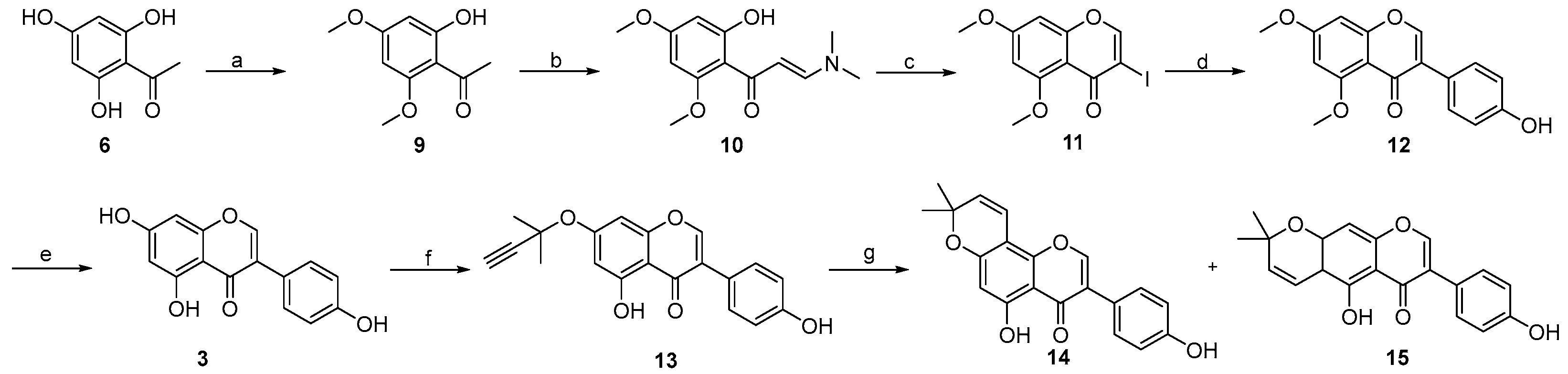
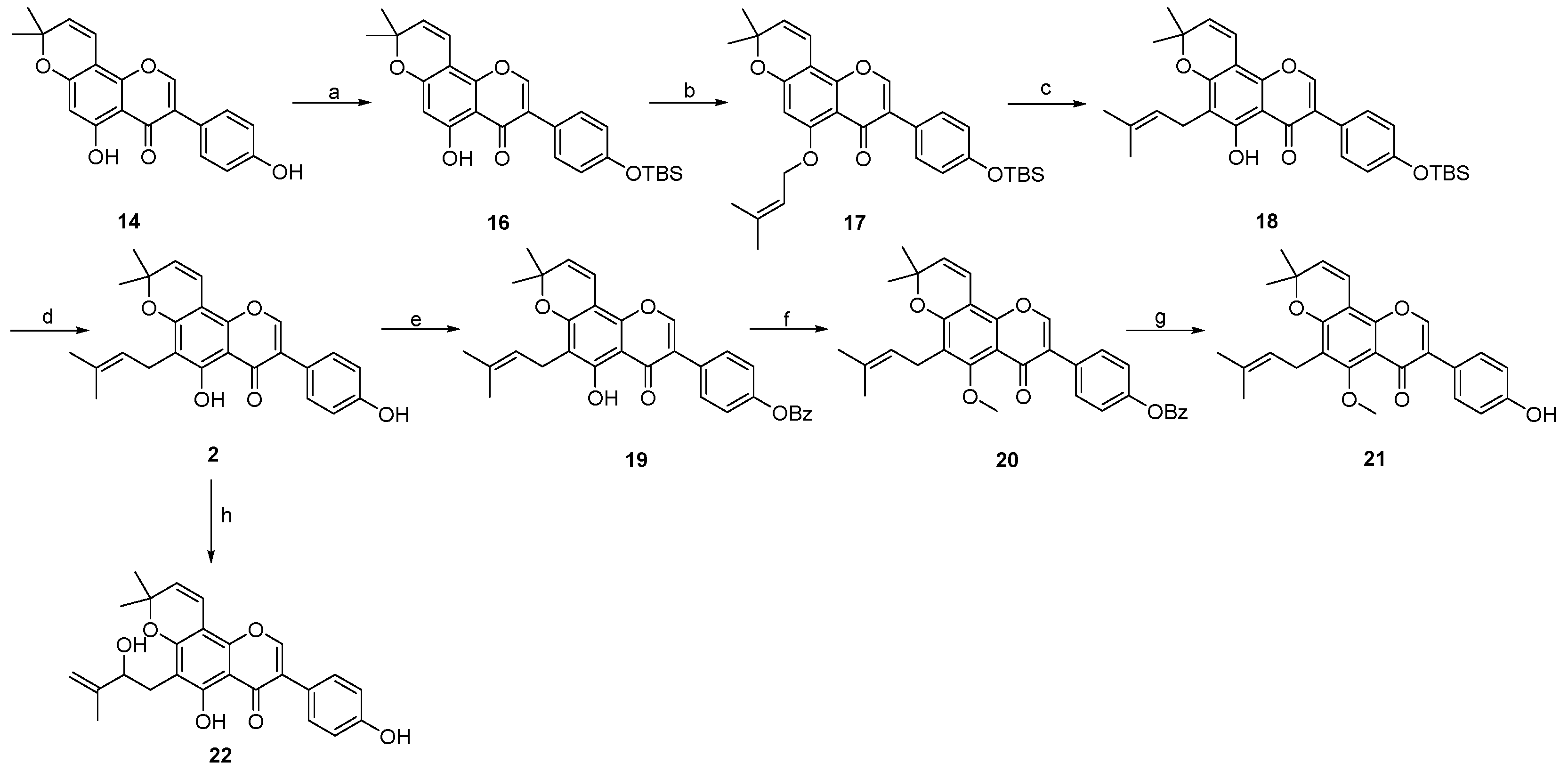
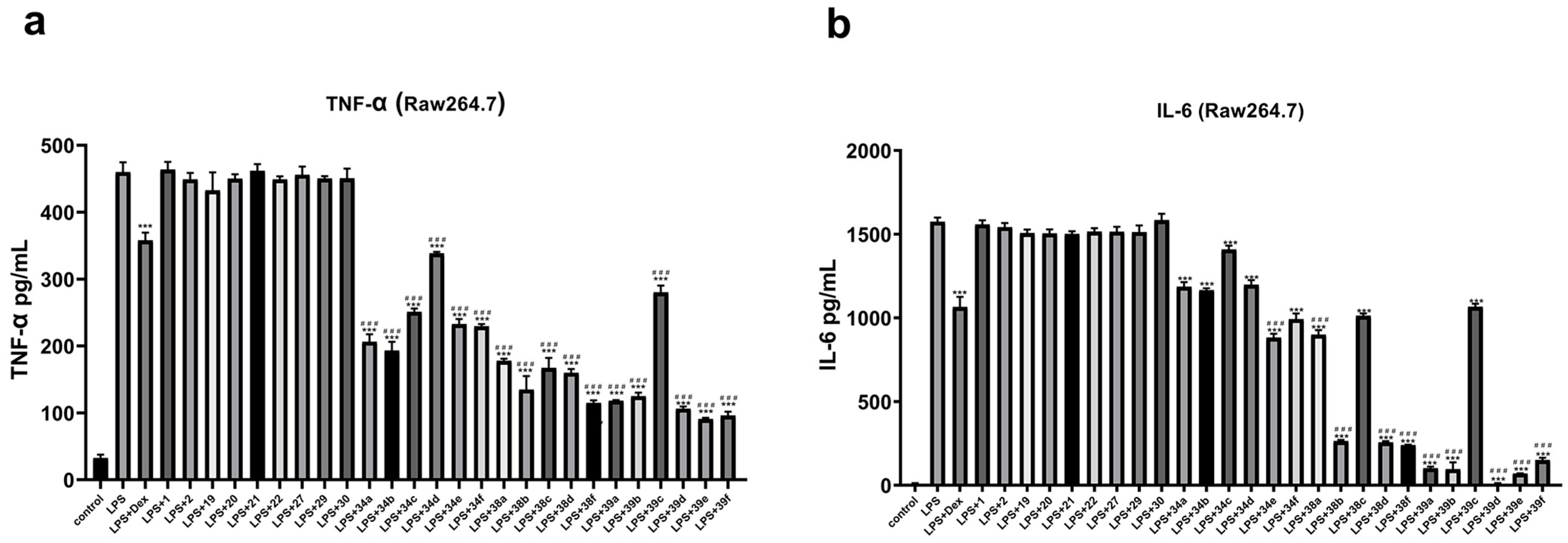
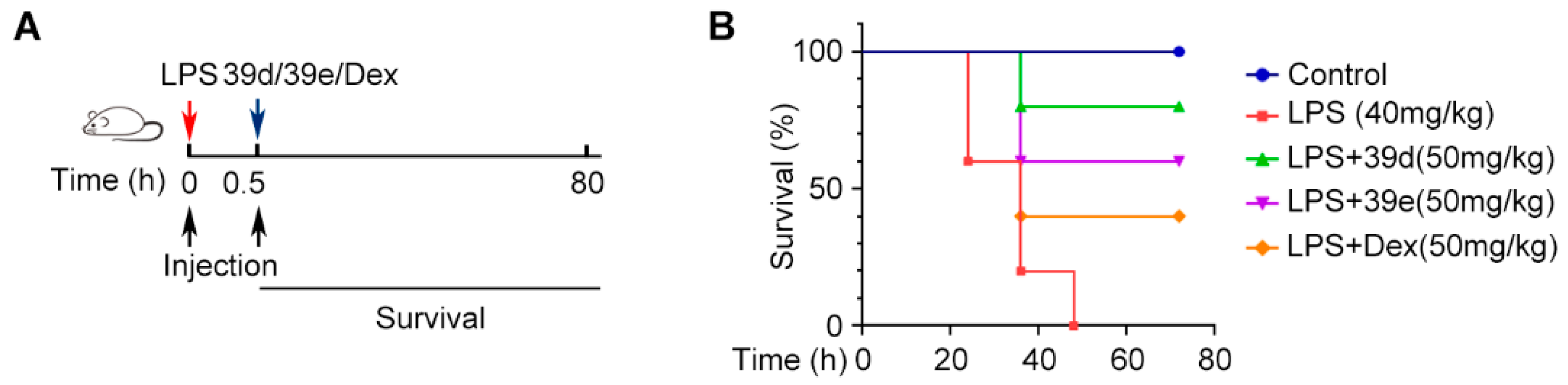
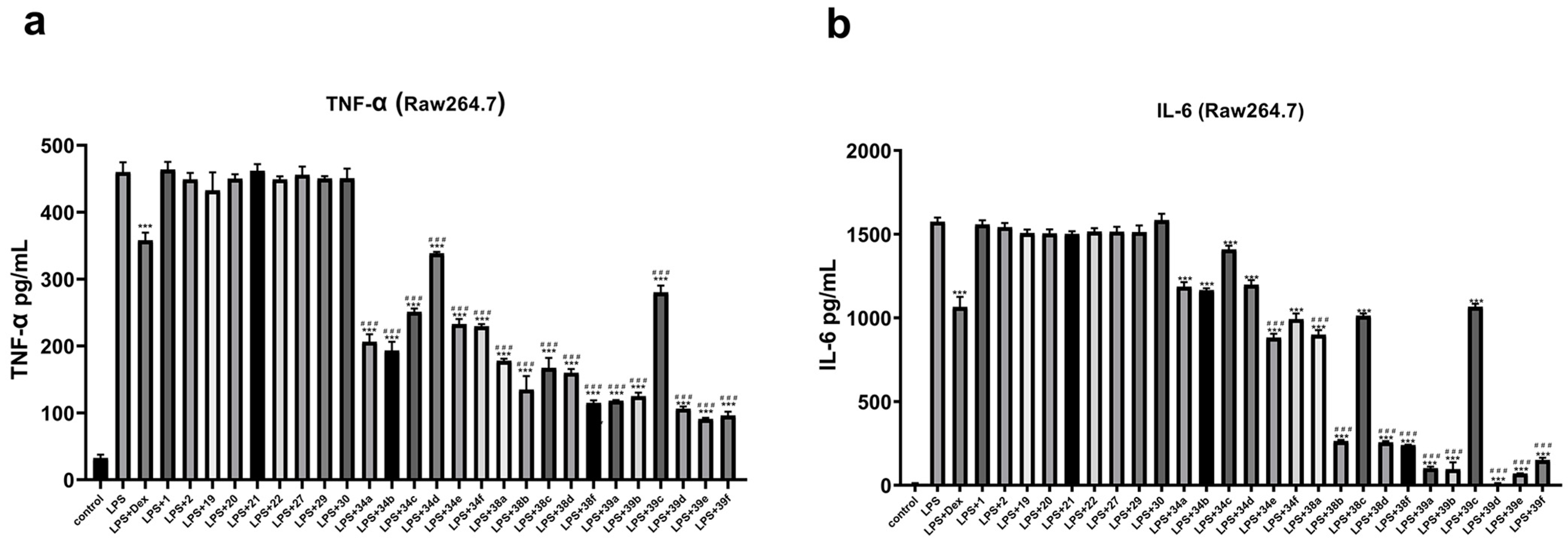
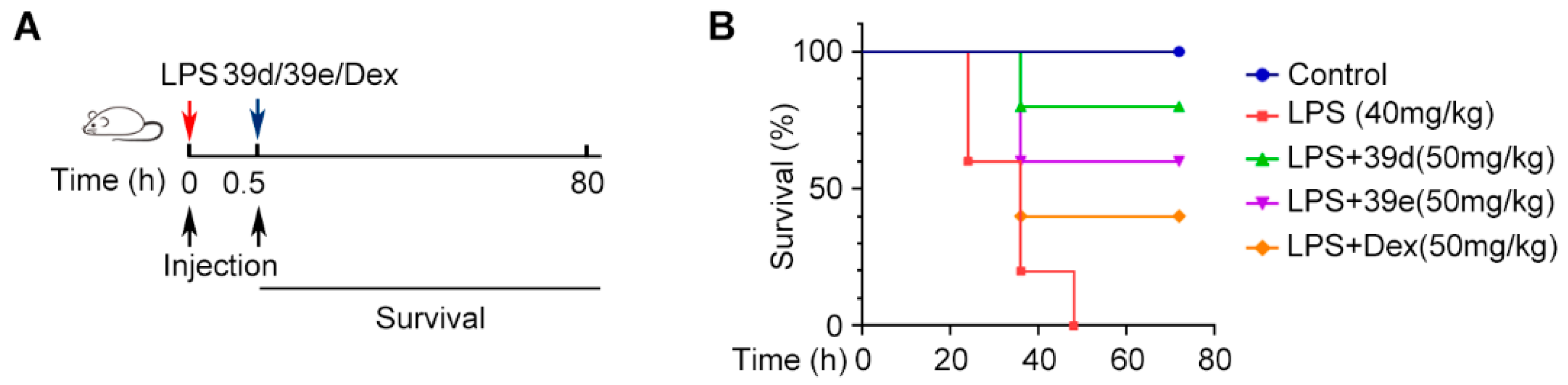
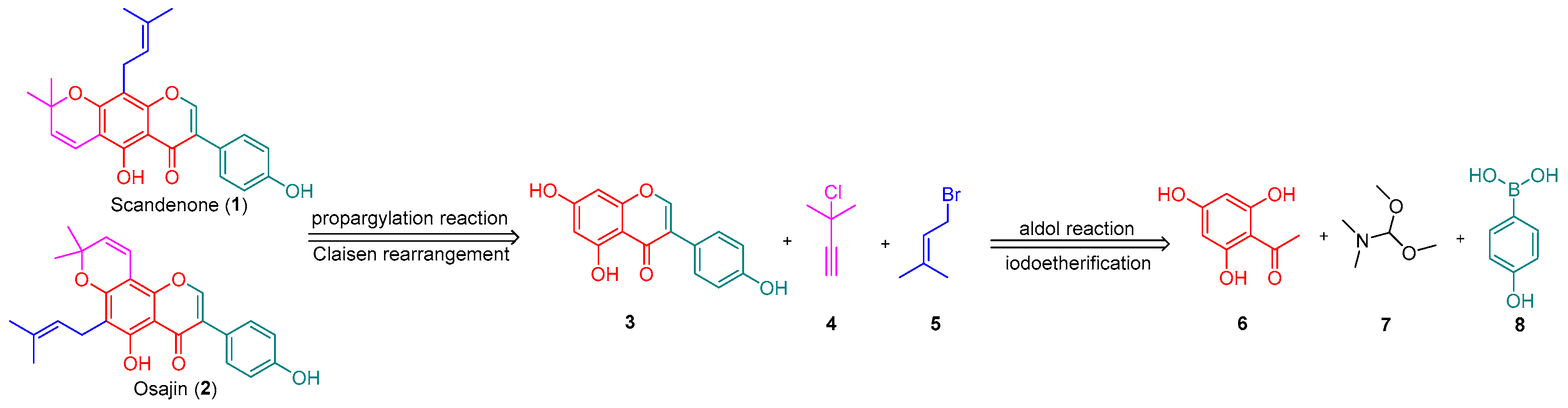
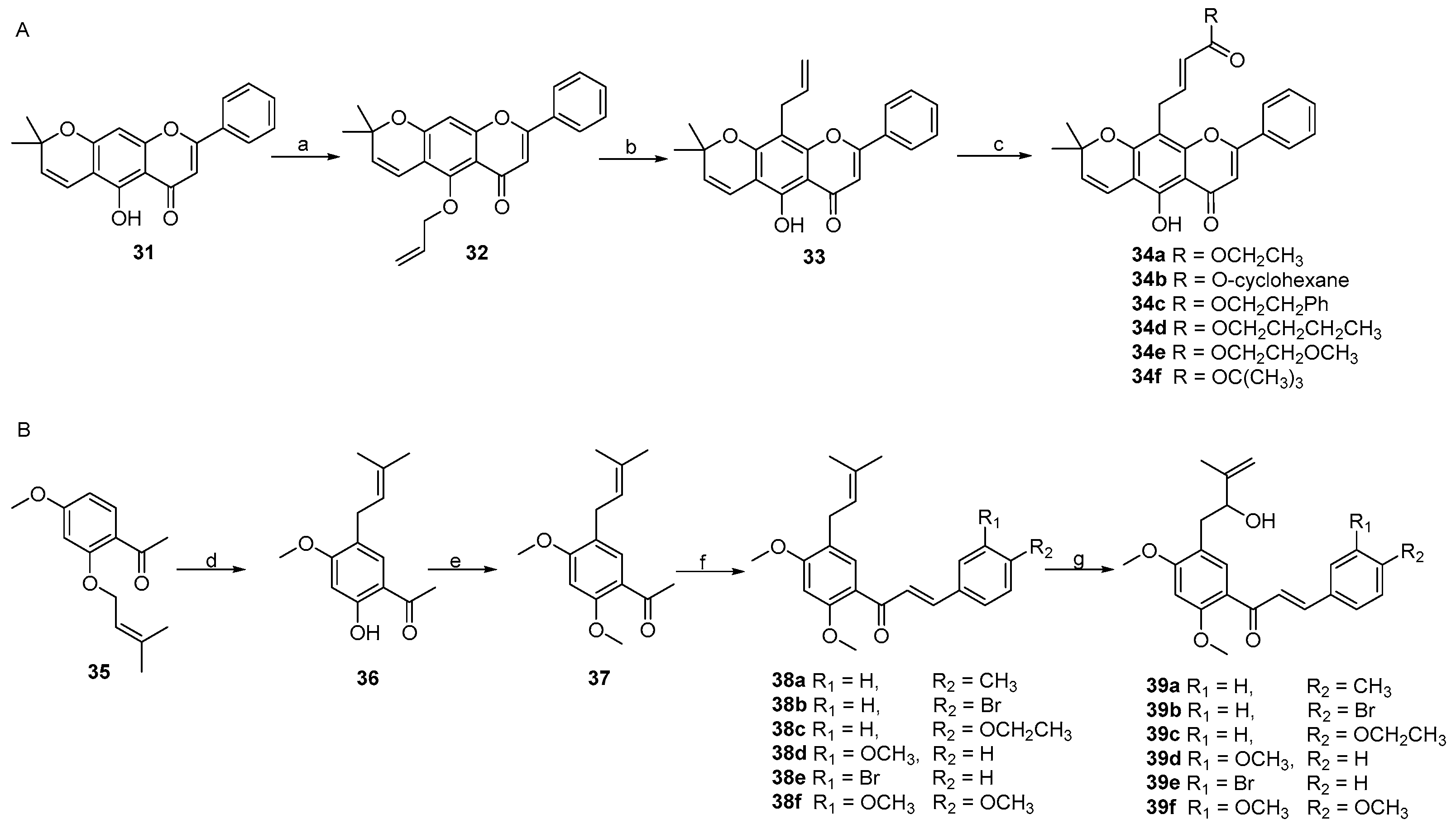
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Materials
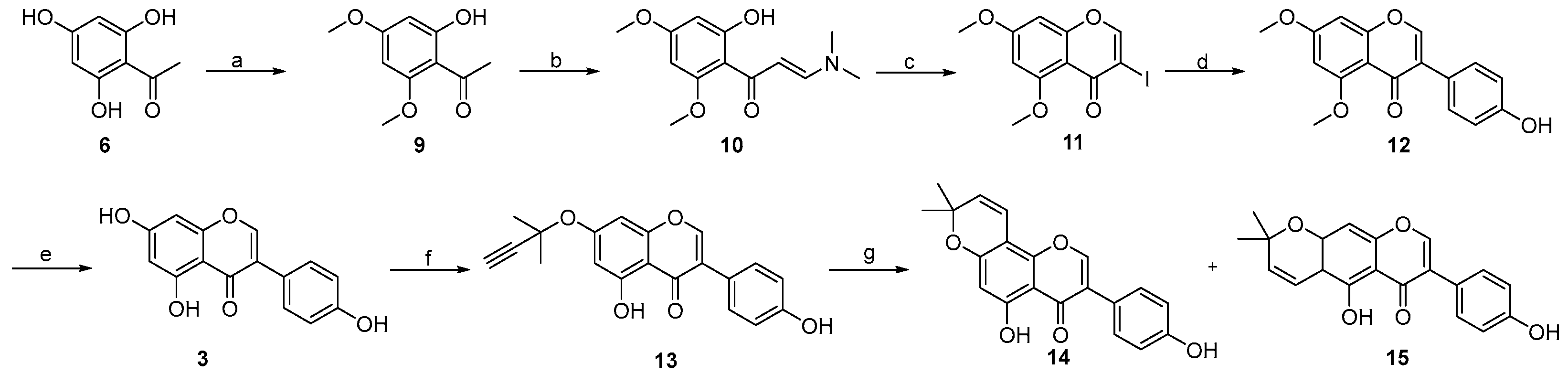
3.3. Procedure for the Synthesis of Osajin and Scandenone
- 1-(2-hydroxy-4,6-dimethoxyphenyl)ethan-1-one 9:
- (E)-3-(dimethylamino)-1-(2-hydroxy-4,6-dimethoxyphenyl)prop-2-en-1-one 10:
- 3-iodo-5,7-dimethoxy-4H-chromen-4-one 11:
- 3-(4-hydroxyphenyl)-5,7-dimethoxy-4H-chromen-4-one 12:
- 5,7-dihydroxy-3-(4-hydroxyphenyl)-4H-chromen-4-one 3:
- 5-hydroxy-3-(4-hydroxyphenyl)-7-((2-methylbut-3-yn-2-yl)oxy)-4H-chromen-4-one 13:
- 5-hydroxy-3-(4-hydroxyphenyl)-8,8-dimethyl-4H,8H-pyrano [2,3-f]chromen-4-one 14:
- 5-hydroxy-7-(4-hydroxyphenyl)-2,2-dimethyl-2H,6H-pyrano [3,2-g]chromen-6-one 15:
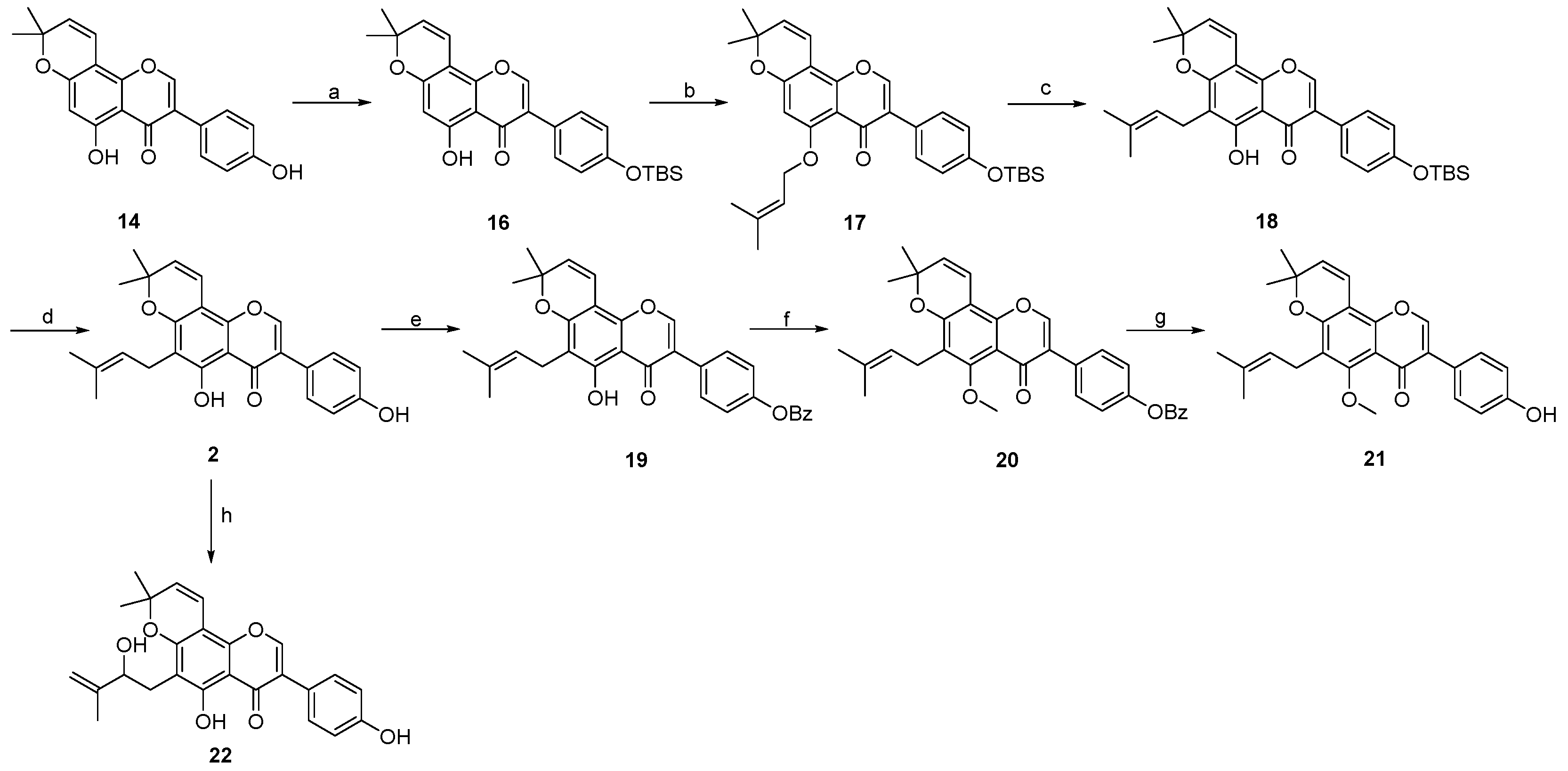
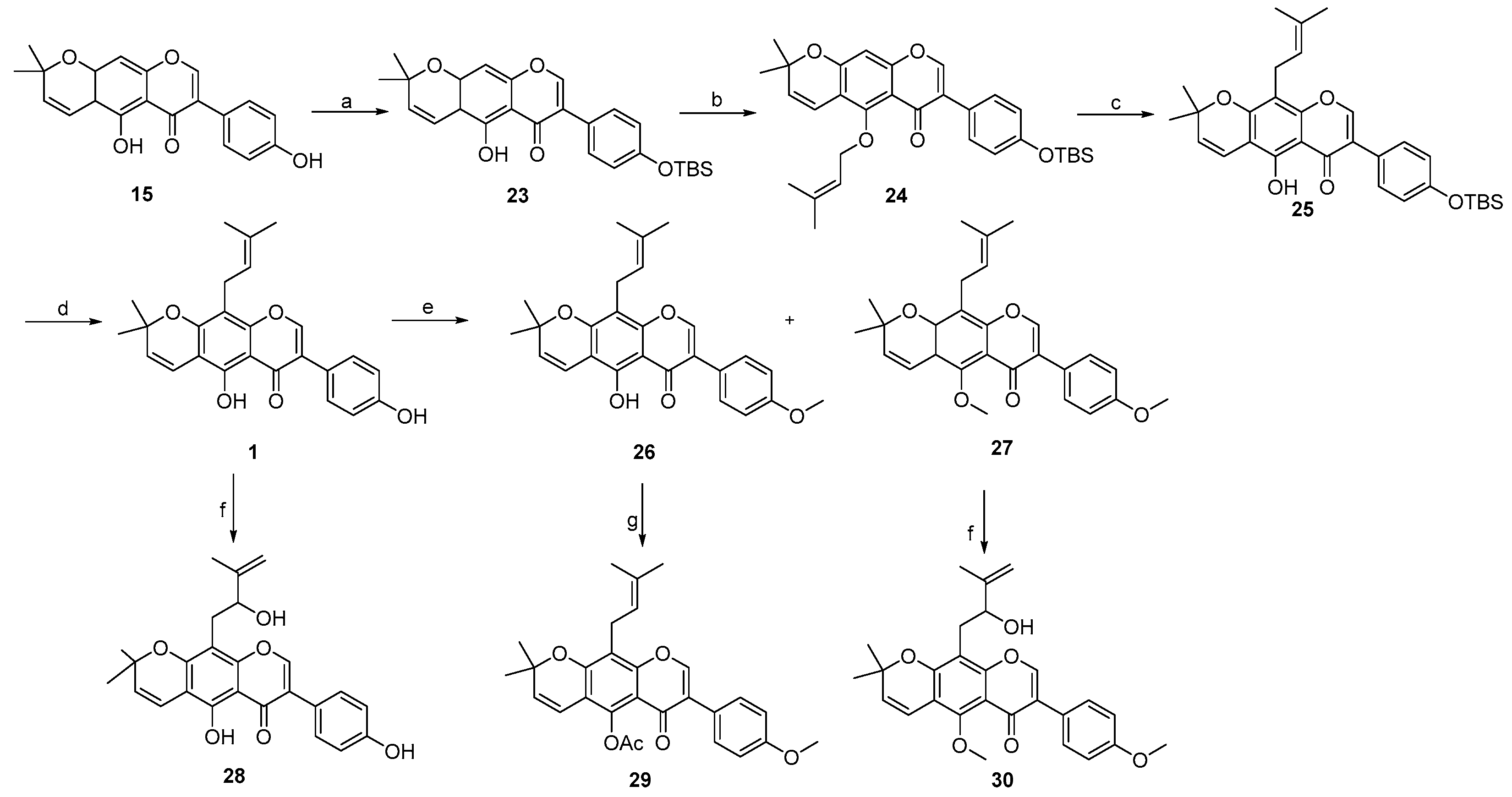
- 7-(4-((tert-butyldimethylsilyl)oxy)phenyl)-5-hydroxy-2,2-dimethyl-2H,6H-pyrano [3,2-g]chromen-6-one 23:
- 7-(4-((tert-butyldimethylsilyl)oxy)phenyl)-2,2-dimethyl-5-((3-methylbut-2-en-1-yl)oxy)-2H,6H-pyrano [3,2-g]chromen-6-one 24:
- 7-(4-((tert-butyldimethylsilyl)oxy)phenyl)-5-hydroxy-2,2-dimethyl-10-(3-methylbut-2-en-1-yl)-2H,6H-pyrano [3,2-g]chromen-6-one 25:
- 3-(4-((tert-butyldimethylsilyl)oxy)phenyl)-5-hydroxy-8,8-dimethyl-4H,8H-pyrano [2,3-f]chromen-4-one 16: Compound 16 was synthesized by following a similar procedure as that of 23.
- 3-(4-((tert-butyldimethylsilyl)oxy)phenyl)-8,8-dimethyl-5-((3-methylbut-2-en-1-yl)oxy)-4H,8H-pyrano [2,3-f]chromen-4-one 17: Compound 17 was synthesized by following a similar procedure as that of 24.
- 3-(4-((tert-butyldimethylsilyl)oxy)phenyl)-5-hydroxy-8,8-dimethyl-6-(3-methylbut-2-en-1-yl)-4H,8H-pyrano [2,3-f]chromen-4-one 18:
- 5-hydroxy-3-(4-hydroxyphenyl)-8,8-dimethyl-6-(3-methylbut-2-en-1-yl)-4H,8H-pyrano [2,3-f]chromen-4-one 2: Compound 2 was synthesized by following a similar procedure as that of 1.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maleki, S.J.; Crespo, J.F.; Cabanillas, B. Anti-inflammatory effects of flavonoids. Food Chem. 2019, 299, 125124. [Google Scholar] [CrossRef]
- Sánchez, M.; Romero, M.; Gómez-Guzmán, M.; Tamargo, J.; Pérez-Vizcaino, F.; Duarte, J. Cardiovascular Effects of Flavonoids. Curr. Med. Chem. 2019, 26, 6991–7034. [Google Scholar] [CrossRef]
- Li, X.; Yin, M.; Yang, X.; Yang, G.; Gao, X. Flavonoids from Mirabilis himalaica. Fitoterapia 2018, 127, 89–95. [Google Scholar] [CrossRef]
- Liskova, A.; Koklesova, L.; Samec, M.; Smejkal, K.; Samuel, S.M.; Varghese, E.; Abotaleb, M.; Biringer, K.; Kudela, E.; Danko, J.; et al. Flavonoids in Cancer Metastasis. Cancers 2020, 12, 1498. [Google Scholar] [CrossRef]
- Lv, Y.; Marsafari, M.; Koffas, M.; Zhou, J.; Xu, P. Optimizing Oleaginous Yeast Cell Factories for Flavonoids and Hydroxylated Flavonoids Biosynthesis. ACS Synth. Biol. 2019, 8, 2514–2523. [Google Scholar] [CrossRef]
- Raffa, D.; Maggio, B.; Raimondi, M.V.; Plescia, F.; Daidone, G. Recent discoveries of anticancer flavonoids. Eur. J. Med. Chem. 2017, 142, 213–228. [Google Scholar] [CrossRef]
- Oh, T.W.; Do, H.J.; Jeon, J.-H.; Kim, K. Quercitrin inhibits platelet activation in arterial thrombosis. Phytomedicine 2021, 80, 153363. [Google Scholar] [CrossRef]
- Zhang, D.; Jiang, X.; Xiao, L.; Lu, Y.; Sang, S.; Lv, L.; Dong, W. Mechanistic studies of inhibition on acrolein by myricetin. Food Chem. 2020, 323, 126788. [Google Scholar] [CrossRef]
- Sreelatha, T.; Hymavathi, A.; Rama Subba Rao, V.; Devanand, P.; Usha Rani, P.; Madhusudana Rao, J.; Suresh Babu, K. A new benzil derivative from Derris scandens: Structure-insecticidal activity study. Bioorg. Med. Chem. Lett. 2010, 20, 549–553. [Google Scholar] [CrossRef]
- Wang, Y.; Curtis-Long, M.J.; Yuk, H.J.; Kim, D.W.; Tan, X.F.; Park, K.H. Bacterial neuraminidase inhibitory effects of prenylated isoflavones from roots of Flemingia philippinensis. Bioorg. Med. Chem. 2013, 21, 6398–6404. [Google Scholar] [CrossRef]
- Cheenpracha, S.; Chokchaisiri, R.; Laphookhieo, S.; Limtharakul, T.; Thepmalee, C. Rare prenylated isoflavonoids from the young twigs of Millettia extensa and their cytotoxic activities. RSC Adv. 2022, 12, 30359–30364. [Google Scholar] [CrossRef]
- Ribaudo, G.; Coghi, P.; Zanforlin, E.; Law, B.Y.K.; Wu, Y.Y.J.; Han, Y.; Qiu, A.C.; Qu, Y.Q.; Zagotto, G.; Wong, V.K.W. Semi-synthetic isoflavones as BACE-1 inhibitors against Alzheimer’s disease. Bioorg. Chem. 2019, 87, 474–483. [Google Scholar] [CrossRef]
- Adem, F.A.; Mbaveng, A.T.; Kuete, V.; Heydenreich, M.; Ndakala, A.; Irungu, B.; Yenesew, A.; Efferth, T. Cytotoxicity of isoflavones and biflavonoids from Ormocarpum kirkii towards multi-factorial drug resistant cancer. Phytomedicine 2019, 58, 152853. [Google Scholar] [CrossRef]
- Xue, L.-L.; Wu, W.-S.; Ma, X.; Pei, H.-Y.; Tang, M.-H.; Kuang, S.; Cai, X.-Y.; Wang, L.; Li, Y.; Zhang, R.-J.; et al. Modulation of LPS-induced inflammation in RAW264.7 murine cells by novel isoflavonoids from Millettia pulchra. Bioorg. Chem. 2020, 97, 103693. [Google Scholar] [CrossRef]
- Ito, S.; Kitamura, T.; Arulmozhiraja, S.; Manabe, K.; Tokiwa, H.; Suzuki, Y. Total Synthesis of Termicalcicolanone A via Organocatalysis and Regioselective Claisen Rearrangement. Org. Lett. 2019, 21, 2777–2781. [Google Scholar] [CrossRef]
- Boruah, J.J.; Das, S.P. Palladium (Pd)-based Photocatalysts for Suzuki Coupling Reactions: An Overview. Mini-Rev. Org. Chem. 2023, 20, 687–699. [Google Scholar] [CrossRef]
- Yi, J.; Du, G.; Zhao, Y.; Zhang, L.; Li, B.; Zhu, W.; Huang, C.; Li, Y.; Guo, F. Bavachinin analogues as agonists of pan-peroxisome proliferator-activated receptors. Med. Chem. Res. 2018, 27, 1851–1862. [Google Scholar] [CrossRef]
- Orsi, D.L.; Easley, B.J.; Lick, A.M.; Altman, R.A. Base Catalysis Enables Access to α,α-Difluoroalkylthioethers. Org. Lett. 2017, 19, 1570–1573. [Google Scholar] [CrossRef]
- Bensinger, D.; Stubba, D.; Cremer, A.; Kohl, V.; Waßmer, T.; Stuckert, J.; Engemann, V.; Stegmaier, K.; Schmitz, K.; Schmidt, B. Virtual Screening Identifies Irreversible FMS-like Tyrosine Kinase 3 Inhibitors with Activity toward Resistance-Conferring Mutations. J. Med. Chem. 2019, 62, 2428–2446. [Google Scholar] [CrossRef]
- Bulman Page, P.C.; Chan, Y.; Noor Armylisas, A.H.; Alahmdi, M. Asymmetric epoxidation of chromenes mediated by iminium salts: Synthesis of mollugin and (3S,4R)-trans-3,4-dihydroxy-3,4-dihydromollugin. Tetrahedron 2016, 72, 8406–8416. [Google Scholar] [CrossRef]
- Amolak, C.J.; Bhola, N.S. Synthesis of Alpinum Isoflavone, Osajin, and Warangalone. J. Org. Chem. 1974, 39, 2215–2217. [Google Scholar]
- Helesbeux, J.-J.; Duval, O.; Guilet, D.; Séraphin, D.; Rondeau, D.; Richomme, P. Regioselectivity in the ene reaction of singlet oxygen with ortho-prenylphenol derivatives. Tetrahedron 2003, 59, 5091–5104. [Google Scholar] [CrossRef]
- Parmar, V.S.; Jain, S.C.; Bisht, K.S.; Sharma, N.K.; Gupta, S.; Prasad, A.K.; Jha, A.; Malhotra, S.; Sharma, S.K.; Bracke, M.E. Synthesis and anti-invasive activity of novel 1,3-diarylpropenones. Indian J. Chem. 1998, 37B, 628–643. [Google Scholar]
- Dong, T.; Li, C.; Wang, X.; Dian, L.; Zhang, X.; Li, L.; Chen, S.; Cao, R.; Li, L.; Huang, N.; et al. Ainsliadimer A selectively inhibits IKKα/β by covalently binding a conserved cysteine. Nat. Commun. 2015, 6, 6522. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | Solvent | Base | Temperature (°C) | Yield (%) c |
|---|---|---|---|---|---|
| 1 | Pd(OAc)2 | MeOH | Na2CO3 | rt b | 40 |
| 2 | Pd(OAc)2 | DMF | Na2CO3 | rt | 45 |
| 3 | Pd(OAc)2 | DMF | K2CO3 | rt | 43 |
| 4 | Pd(OAc)2 | DMF | K2CO3 | 50 | 60 |
| 5 | Pd(PPh3)4 | DMF | K2CO3 | rt | 46 |
| 6 | Pd(PPh3)4 | DMF | K2CO3 | 40 | 57 |
| 7 | Pd(PPh3)4 | DMF | K2CO3 | 60 | 67 |
| 8 | Pd(PPh3)4 | DMF | K2CO3 | 80 | 70 |
| 9 | PdCl2(dppf) | 1,4-Dioxane/H2O d | K2CO3 | 50 | 84 |
| 10 | PdCl2(dppf) | 1,4-Dioxane/H2O | K2CO3 | 80 | 86 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, R.; Ma, R.; Feng, K.; Lu, H.; Zhao, W.; Jin, H. Total Synthesis and Anti-Inflammatory Evaluation of Osajin, Scandenone and Analogues. Pharmaceuticals 2024, 17, 86. https://doi.org/10.3390/ph17010086
Wang R, Ma R, Feng K, Lu H, Zhao W, Jin H. Total Synthesis and Anti-Inflammatory Evaluation of Osajin, Scandenone and Analogues. Pharmaceuticals. 2024; 17(1):86. https://doi.org/10.3390/ph17010086
Chicago/Turabian StyleWang, Rui, Ran Ma, Ke Feng, Hongchen Lu, Wei Zhao, and Hongzhen Jin. 2024. "Total Synthesis and Anti-Inflammatory Evaluation of Osajin, Scandenone and Analogues" Pharmaceuticals 17, no. 1: 86. https://doi.org/10.3390/ph17010086
APA StyleWang, R., Ma, R., Feng, K., Lu, H., Zhao, W., & Jin, H. (2024). Total Synthesis and Anti-Inflammatory Evaluation of Osajin, Scandenone and Analogues. Pharmaceuticals, 17(1), 86. https://doi.org/10.3390/ph17010086