


Use of Exposure Data to Establish Causality in Drug–Adverse Event Relationships: An Example with Desvenlafaxine

 ,
,  ,
,  and
and
Abstract
1. Introduction
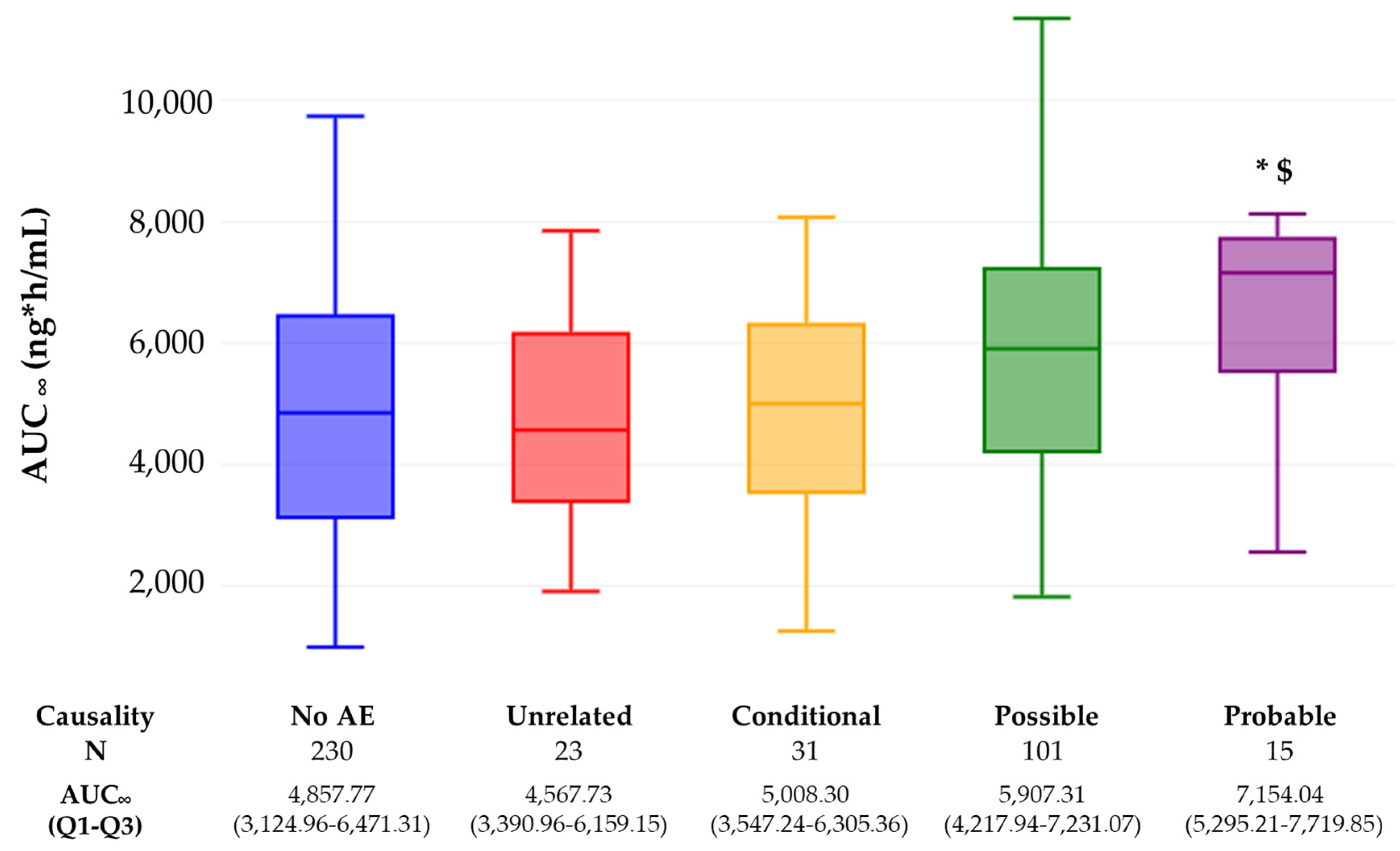
2. Results
2.1. Pharmacokinetics
2.2. Safety
3. Discussion
4. Materials and Methods
4.1. Study Population and Study Design
4.2. Pharmacokinetic Parameters
4.3. Safety
4.4. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boletín Oficial del Estado (BOE). Real Decreto 577/2013 de 26 de Julio, Por El Que Se Regula La Farmacovigilancia de Medicamentos de Uso Humano; Agentia Estatal: Madrid, Spain, 2013; Volume 179, pp. 55066–55092. [Google Scholar]
- Mejía, G.; Saiz-Rodríguez, M.; Gómez de Olea, B.; Ochoa, D.; Abad-Santos, F. Urgent Hospital Admissions Caused by Adverse Drug Reactions and Medication Errors-A Population-Based Study in Spain. Front. Pharmacol. 2020, 11, 734. [Google Scholar] [CrossRef] [PubMed]
- Montané, E.; Santesmases, J. Adverse Drug Reactions. Med. Clin. 2020, 154, 178–184. [Google Scholar] [CrossRef]
- Gawai, P.P. Introduction and Evaluation of Pharmacovigilance for Beginners. Int. J. Sci. Rep. 2020, 6, 425. [Google Scholar] [CrossRef]
- WHO. WHO Policy Perspectives on Medicines; WHO: Geneva, Switzerland, 2004.
- Jones, J.K. Adverse Drug Reactions in the Community Health Setting: Approaches to Recognizing, Counseling, and Reporting. Fam. Community Health 1982, 5, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Naranjo, C.A.; Busto, U.; Sellers, E.M.; Sandor, P.; Ruiz, I.; Roberts, E.A.; Janecek, E.; Domecq, C.; Greenblatt, D.J. A Method for Estimating the Probability of Adverse Drug Reactions. Clin. Pharmacol. Ther. 1981, 30, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.S.; Hutchinson, T.A. The Yale Algorithm. Special Workshop-Clinical. Drug Inf. J. 1984, 18, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Bégaud, B.; Evreux, J.C.; Jouglard, J.; Lagier, G. Imputation of the unexpected or toxic effects of drugs. Actualization of the method used in France. Therapie 1985, 40, 111–118. [Google Scholar] [PubMed]
- Aguirre, C.; García, M. Causality assessment in reports on adverse drug reactions. Algorithm of Spanish pharmacovigilance system. Med. Clin. 2016, 147, 461–464. [Google Scholar] [CrossRef]
- Karch, F.E.; Lasagna, L. Toward the Operational Identification of Adverse Drug Reactions. Clin. Pharmacol. Ther. 1977, 21, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Doherty, M.J. Algorithms for Assessing the Probability of an Adverse Drug Reaction. Respir. Med. CME 2009, 2, 63–67. [Google Scholar] [CrossRef]
- Calleja, S.; Zubiaur, P.; Ochoa, D.; Villapalos-García, G.; Mejia-Abril, G.; Soria-Chacartegui, P.; Navares-Gómez, M.; de Miguel, A.; Román, M.; Martín-Vílchez, S.; et al. Impact of Polymorphisms in CYP and UGT Enzymes and ABC and SLCO1B1 Transporters on the Pharmacokinetics and Safety of Desvenlafaxine. Front. Pharmacol. 2023, 14, 1110460. [Google Scholar] [CrossRef] [PubMed]
- Spanish Drug Agency PRISTIQ® (Desvenlafaxine) Extended-Release Tablets, Oral—Drug Label. 2012. Available online: https://cima.aemps.es/cima/pdfs/es/ft/75560/75560_ft.pdf (accessed on 20 October 2023).
- Liebowitz, M.R.; Manley, A.L.; Padmanabhan, S.K.; Ganguly, R.; Tummala, R.; Tourian, K.A. Efficacy, safety, and tolerability of desvenlafaxine 50 mg/day and 100 mg/day in outpatients with major depressive disorder. Curr. Med. Res. Opin. 2008, 24, 1877–1890. [Google Scholar] [CrossRef]
- Lieberman, D.Z.; Massey, S.H. Desvenlafaxine in major depressive disorder: An evidence-based review of its place in therapy. Core Evid. 2010, 4, 67–82. [Google Scholar] [CrossRef][Green Version]
- Lense, X.M.; Hiemke, C.; Funk, C.S.M.; Havemann-Reinecke, U.; Hefner, G.; Menke, A.; Mössner, R.; Riemer, T.G.; Scherf-Clavel, M.; Schoretsanitis, G.; et al. Venlafaxine’s Therapeutic Reference Range in the Treatment of Depression Revised: A Systematic Review and Meta-Analysis. Psychopharmacology 2023. [Google Scholar] [CrossRef] [PubMed]
- Suwała, J.; Machowska, M.; Wiela-Hojeńska, A. Venlafaxine pharmacogenetics: A comprehensive review. Pharmacogenomics 2019, 20, 829–845. [Google Scholar] [CrossRef] [PubMed]
- Soldin, O.P.; Mattison, D.R. Sex Differences in Pharmacokinetics and Pharmacodynamics. Clin. Pharmacokinet. 2009, 48, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Nichols, A.I.; Richards, L.S.; Behrle, J.A.; Posener, J.A.; Paul, J. Effect of Food on the Pharmacokinetics of Desvenlafaxine in Healthy Subjects. J. Bioequivalence Bioavailab. 2012, 4, 24–29. [Google Scholar] [CrossRef]
- Cipriani, A.; Furukawa, T.A.; Salanti, G.; Chaimani, A.; Atkinson, L.Z.; Ogawa, Y.; Leucht, S.; Ruhe, H.G.; Turner, E.H.; Higgins, J.P.T.; et al. Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: A systematic review and network meta-analysis. Lancet 2018, 391, 1357–1366. [Google Scholar] [CrossRef] [PubMed]
- Morais, J.A.G.; do Lobato, M.R. The New European Medicines Agency Guideline on the Investigation of Bioequivalence. Basic Clin. Pharmacol. Toxicol. 2010, 106, 221–225. [Google Scholar] [CrossRef]
- European Medicines Agency Guideline on Bioanalytical Method Validation 2011. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf (accessed on 25 October 2023).
- Vijayananthan, A.; Nawawi, O. The Importance of Good Clinical Practice Guidelines and Its Role in Clinical Trials. Biomed. Imaging Interv. J. 2008, 4, e5. [Google Scholar] [CrossRef] [PubMed]
- World Medical Association WMA Declaration of HelsinkiI—Ethical Principles for Medical Research Involving Human Subjects. Available online: https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/ (accessed on 15 September 2023).
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Medical Dictionary for Regulatory Activities (MedDRA). Available online: https://www.meddra.org/ (accessed on 15 September 2023).


{kind=link}
| Variable | N | Age (Years) | Height (m) | Weight (kg) | BMI (kg/m2) | |
|---|---|---|---|---|---|---|
| Median (Q1–Q3) | Median (Q1–Q3) | Median (Q1–Q3) | Mean (SD) | |||
| Sex | Male | 96 | 29 (25–34) | 1.75 (1.70–1.80) | 75.80 (66.75–83.98) | 24.68 (2.77) |
| Female | 82 | 29 (24–36) | 1.61 (1.60–1.67) * | 62.30 (57.45–67.53) * | 23.90 (2.53) | |
| Biogeographic origin | European | 51 | 28 (22–33) $ | 1.72 (1.66–1.8) $ | 65.50 (59.50–74.80) | 23.05 (2.85) * |
| Other # | 127 | 30 (25–36) | 1.70 (1.6–1.75) | 69.70 (62.60–78.20) | 24.84 (2.44) | |
| Clinical trial | A | 36 | 28 (24–34) | 1.70 (1.62–1.76) | 67.40 (63.50–74.33) | 23.86 (2.51) |
| B | 36 | 29 (25–33) | 1.70 (1.60–1.70) | 67.55 (62.30–77.80) | 24.25 (2.34) | |
| C | 34 | 31.5 (27–38) | 1.70 (1.70–1.80) | 68.70 (61.00–82.38) | 24.34 (2.41) | |
| D | 36 | 28.5 (25–36) | 1.70 (1.61–1.77) | 66.85 (61.00–76.75) | 24.32 (2.88) | |
| E | 36 | 28.5 (22–36) | 1.70 (1.61–1.75) | 68.75 (60.00–82.00) | 24.88 (3.23) | |
| Total (Mean (SD)) | 178 | 30.60 (8.07) | 1.70 (0.09) | 70.17 (11.66) | 24.33 (2.69) | |
| Variable | AUC∞ (ng·h/mL) | AUC∞/D (ng·h/mL*mg) | AUC∞/DW (kg*ng·h/mL*mg) | Cmax (ng/mL) | Cmax/D (ng/mL*mg) | Cmax/DW (kg*ng/mL*mg) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | Median (Q1–Q3) | N | Median (Q1–Q3) | N | Median (Q1–Q3) | N | Median (Q1–Q3) | N | Median (Q1–Q3) | N | Median (Q1–Q3) | ||
| Sex | Male | 190 | 4601.77 (2981.92–5988.57) | 190 | 57.95 (49.56–65.32) | 190 | 4361.7 (3821.39–4807.32) | 191 | 179.62 (131.35–248.41) | 191 | 2.34 (1.94–2.92) | 191 | 178.06 (147.91–222.31) |
| Female | 157 | 6136.57 (3998.87–7254.42) * | 157 | 69.75 (61.89–79.35) * | 157 | 4374.77 (3871.71–4869.57) | 159 | 257.39 (186.18–343.11) * | 159 | 3.21 (2.53–3.85) * | 159 | 201.45 (161.33–237.33) * | |
| Biogeographic origin | European | 99 | 5829.78 (4212.25–7032.24) | 99 | 65.07 (56.92–78.06) $ | 99 | 4350.45 (3821.05–4981.42) | 101 | 214.54 (172.41–396.81) | 101 | 2.66 (2.03–3.39) | 101 | 168.15 (140.84–223.85) |
| Other | 248 | 4770.84 (3094.42–6378.03) | 248 | 61.95 (52.68–71.79) | 248 | 4373.02 (3846.55–4810.85) | 249 | 210.54 (136.53–297.74) | 249 | 2.76 (2.16–3.39) | 249 | 188.18 (156.19–233.56) $ | |
| Feeding conditions | Fasting | 208 | 5297.56 (3223.13–6752.41) | 208 | 61.61 (52.7–71.51) | 208 | 4300.33 (3737.77–4702.98) | 211 | 198.94 (123.2–270.45) | 211 | 2.32 (1.97–2.87) | 211 | 160.95 (140.17–183.72) |
| Fed | 139 | 4415.57 (3390.96–6356.21) | 139 | 64.5 (54.85–75.22) | 139 | 4575.56 (4040.28–5052.86) * | 139 | 244.42 (165.75–339.04) * | 139 | 3.32 (2.78–4.02) * | 139 | 233.88 (208.16–264.09) * | |
| Total | 347 | 4956.22 (3319.66–6549.21) | 347 | 62.68 (53.83–72.61) | 347 | 4371.66 (3831.29–4834.93) | 350 | 212.29 (146.39–296.42) | 350 | 2.73 (2.13–3.39) | 350 | 185.75 (154.32–231.69) | |
| SEFV Score | N | AUC∞ (ng·h/mL) | Cmax (ng/mL) |
|---|---|---|---|
| Median (Q1–Q3) | Median (Q1–Q3) | ||
| 1 or 2 | 12 | 4344.94 (3471.56–6177.11) | 177.19 (138.11–233.21) |
| 3 | 19 | 5046.77 (3620.79–6385.53) | 243.52 (175.60–330.11) |
| Total | 31 | 5008.30 (3547.24–6305.36) | 201.06 (147.89–279.71) |
| MedDRA SOC | MedDRA PT | AUC∞ (ng·h/mL) | Cmax (ng/mL) | Initial Score | New Score |
|---|---|---|---|---|---|
| Nervous system disorders | Headache | 5792.34 | 178.66 | 3 | 3 |
| Headache | 2138.86 | 67.39 | 3 | 3 | |
| Presyncope | 3620.8 | 208.05 | 3 | 3 | |
| Headache | 6305.37 | 243.52 | 3 | 3 | |
| Dizziness | 7514.45 * | 455.74 * | 3 | 4 | |
| Headache | 6284.71 | 330.11 * | 3 | 4 | |
| Headache | 4705.14 | 200.32 | 3 | 3 | |
| Psychomotor hyperactivity | 6481.22 | 179.62 | 3 | 3 | |
| Presyncope | 5008.3 | 279.71 | 3 | 3 | |
| Headache | 5008.3 | 279.71 | 3 | 3 | |
| Headache | 4597.18 | 256.68 | 3 | 3 | |
| Gastrointestinal disorders | Dry mouth | 3547.24 | 175.6 | 3 | 3 |
| Vomiting | 6385.53 | 330.58 * | 3 | 4 | |
| Nausea | 7440.14 * | 380.9 * | 3 | 4 | |
| Respiratory, thoracic and mediastinal disorders | Epistaxis | 3099.63 | 116.37 | 3 | 3 |
| Metabolism and nutrition disorders | Decreased appetite | 6284.71 | 330.11 * | 3 | 4 |
| General disorders and administration site conditions | Asthenia | 5046.78 | 147.89 | 3 | 3 |
| Eye disorders | Vision blurred | 1258.01 | 67.78 | 3 | 3 |
| Musculoskeletal and connective tissue disorders | Muscle spasms | 7521.95 * | 357.21 * | 3 | 4 |
| Internal Code | EudraCT Code | Dose | Type of Study | Sample Size |
|---|---|---|---|---|
| A | 2019-000628-17 | 100 mg | Single dose—fasting | 36 |
| B | 2019-002739-26 | 50 mg | Single dose—fasting | 36 |
| C | 2019-004289-16 | 50 mg | Single dose—fed | 36 |
| D | 2019-004882-41 | 100 mg | Single dose—fasting | 36 |
| E | 2020-003002-31 | 100 mg | Single dose—fed | 36 |
| Total | 180 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Lopez, A.; Mejía-Abril, G.; Zubiaur, P.; Calleja, S.; Román, M.; Abad-Santos, F.; Ochoa, D. Use of Exposure Data to Establish Causality in Drug–Adverse Event Relationships: An Example with Desvenlafaxine. Pharmaceuticals 2024, 17, 69. https://doi.org/10.3390/ph17010069
Rodríguez-Lopez A, Mejía-Abril G, Zubiaur P, Calleja S, Román M, Abad-Santos F, Ochoa D. Use of Exposure Data to Establish Causality in Drug–Adverse Event Relationships: An Example with Desvenlafaxine. Pharmaceuticals. 2024; 17(1):69. https://doi.org/10.3390/ph17010069
Chicago/Turabian StyleRodríguez-Lopez, Andrea, Gina Mejía-Abril, Pablo Zubiaur, Sofía Calleja, Manuel Román, Francisco Abad-Santos, and Dolores Ochoa. 2024. "Use of Exposure Data to Establish Causality in Drug–Adverse Event Relationships: An Example with Desvenlafaxine" Pharmaceuticals 17, no. 1: 69. https://doi.org/10.3390/ph17010069
APA StyleRodríguez-Lopez, A., Mejía-Abril, G., Zubiaur, P., Calleja, S., Román, M., Abad-Santos, F., & Ochoa, D. (2024). Use of Exposure Data to Establish Causality in Drug–Adverse Event Relationships: An Example with Desvenlafaxine. Pharmaceuticals, 17(1), 69. https://doi.org/10.3390/ph17010069