
Synthesis and Biological Evaluation of Chalconesulfonamides: En Route to Proapoptotic Agents with Antiestrogenic Potency

 , ,
, ,  ,
,  and
and
Abstract
:
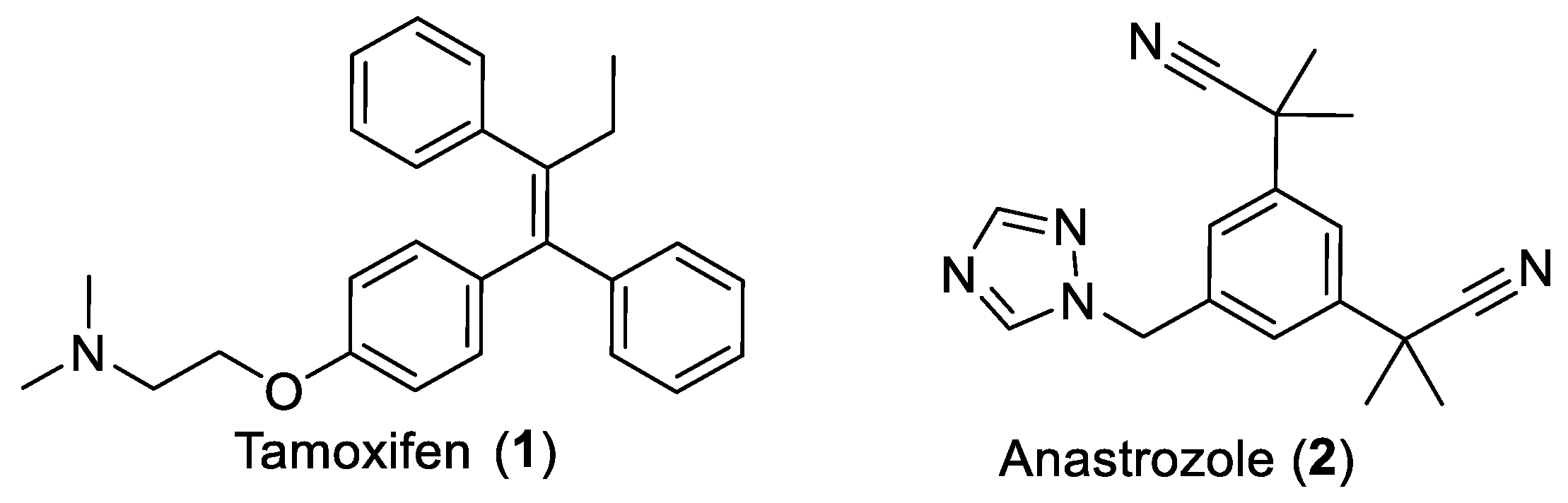
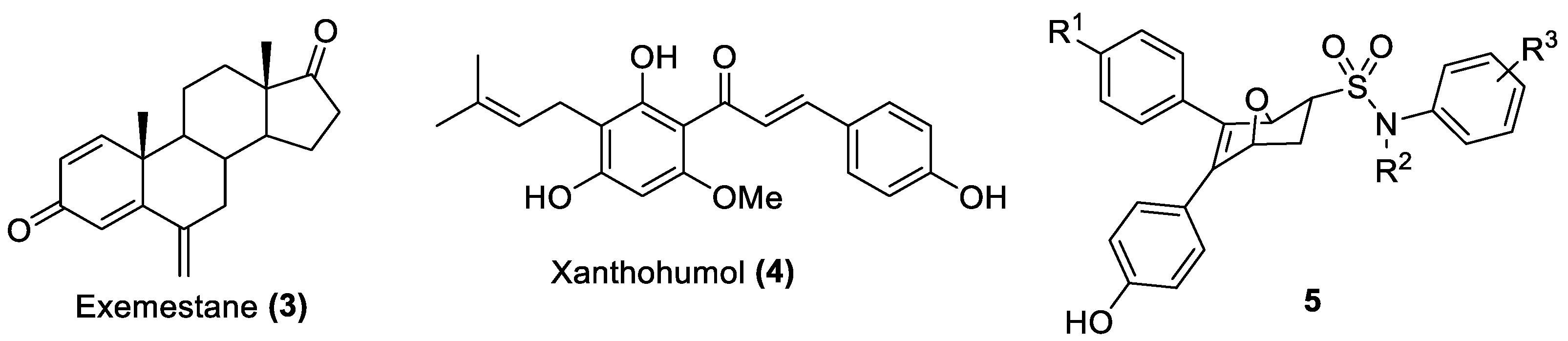
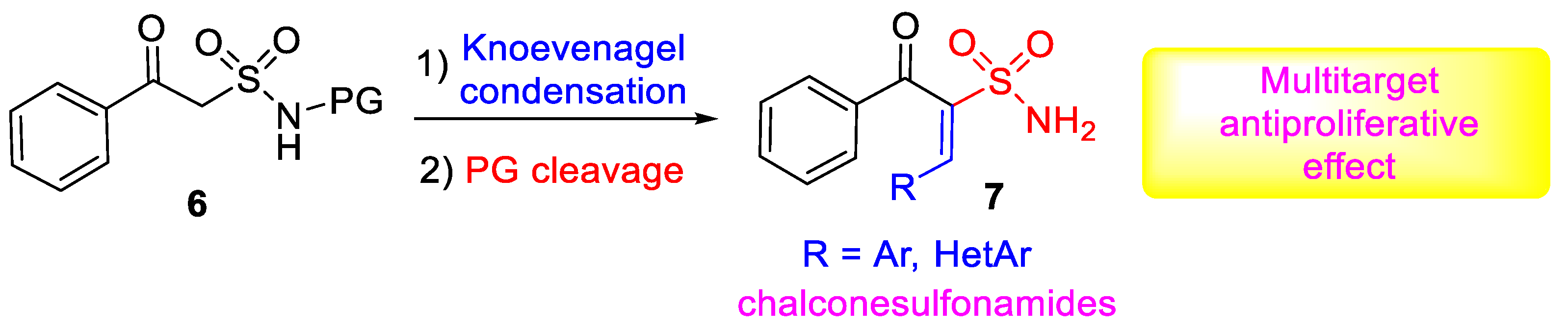
1. Introduction
2. Results
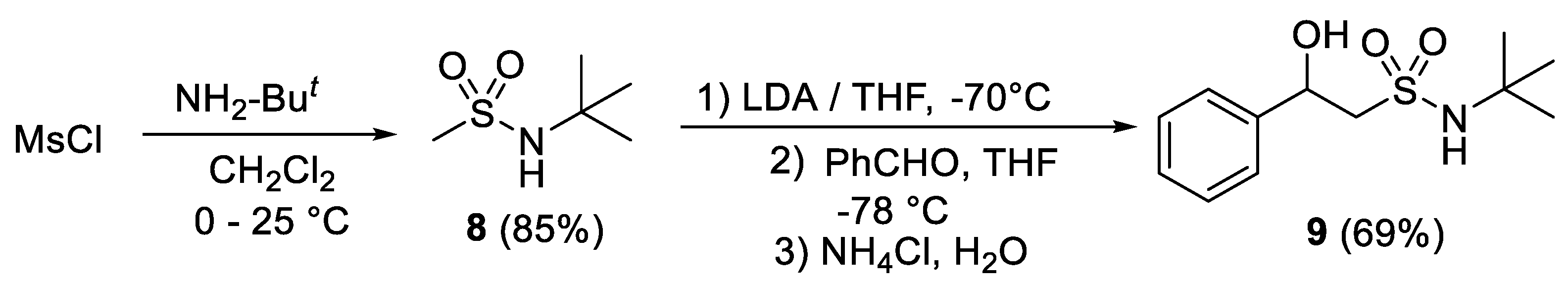
2.1. Chemistry
2.2. Antiproliferative Effects and Selectivity
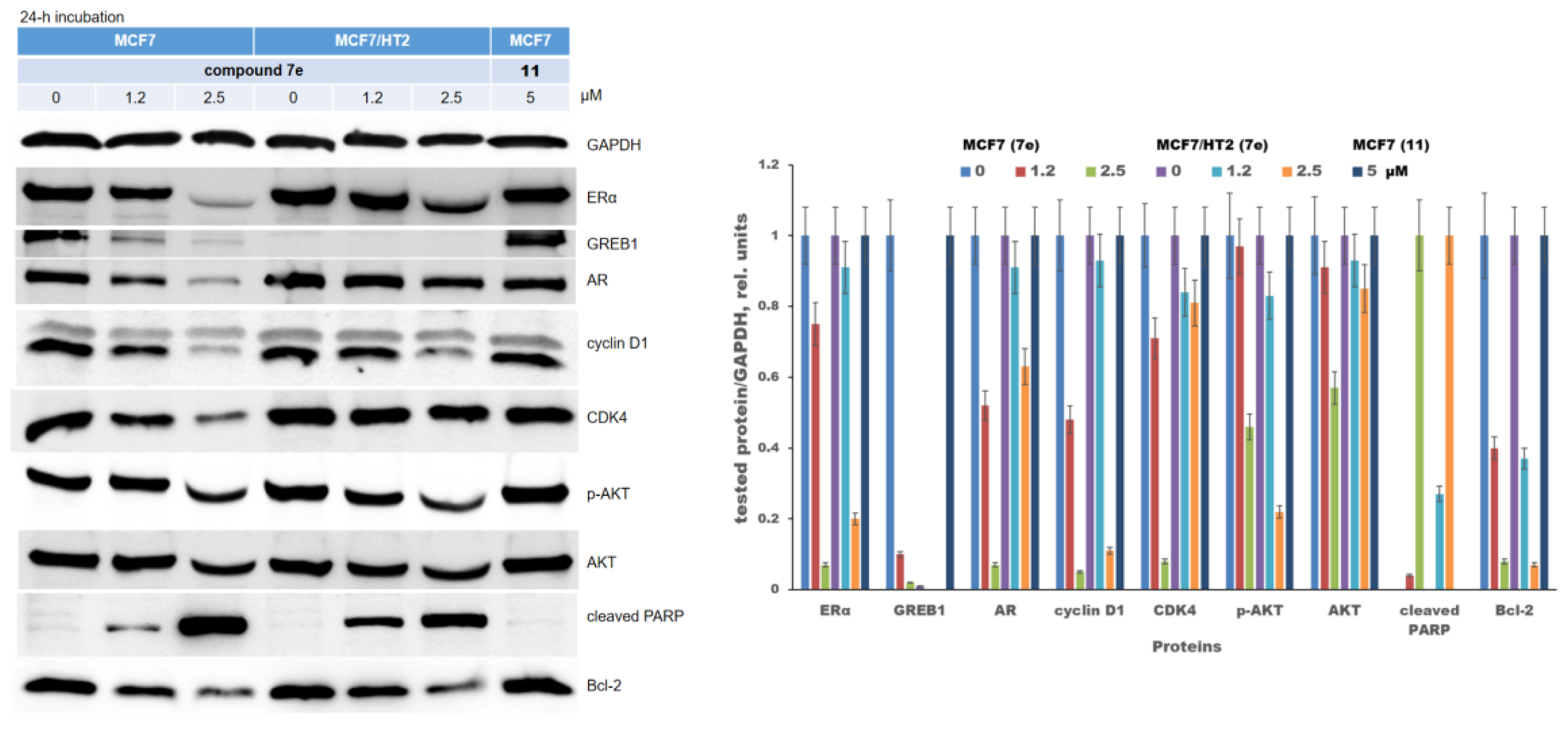
2.3. Signaling Pathways Regulated by Chalconesulfonamide 7e
3. Discussion
4. Materials and Methods
4.1. Synthesis
- N-(tert-Butyl)-2-oxo-2-phenylethane-1-sulfonamide (6)
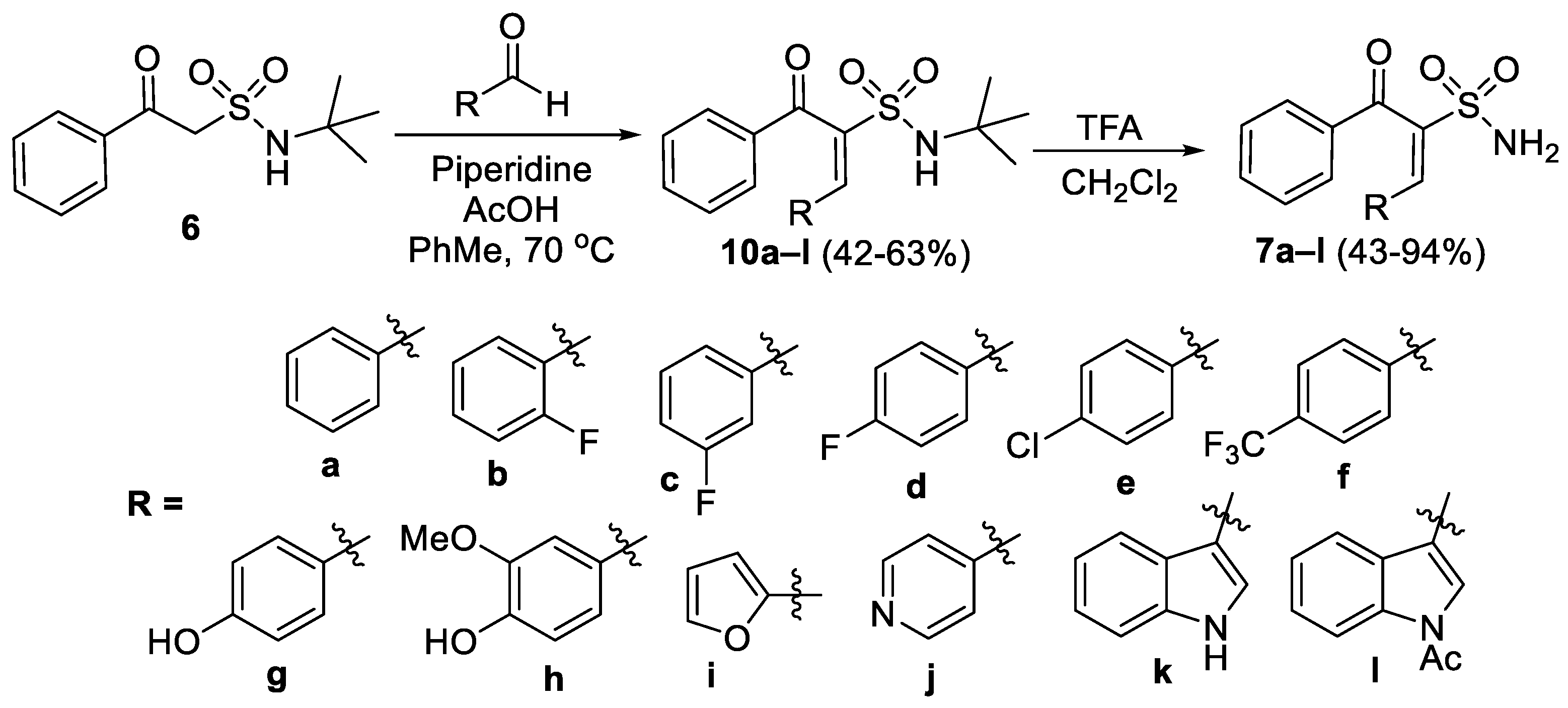
- (E)-N-(tert-Butyl)-3-oxo-1,3-diphenylprop-1-ene-2-sulfonamide (10a)
- (E)-N-(tert-Butyl)-1-(2-fluorophenyl)-3-oxo-3-phenylprop-1-ene-2-sulfonamide (10b)
- (E)-N-(tert-Butyl)-1-(3-fluorophenyl)-3-oxo-3-phenylprop-1-ene-2-sulfonamide (10c)
- (E)-N-(tert-Butyl)-1-(4-fluorophenyl)-3-oxo-3-phenylprop-1-ene-2-sulfonamide (10d)
- (E)-N-(tert-Butyl)-1-(4-chlorophenyl)-3-oxo-3-phenylprop-1-ene-2-sulfonamide (10e)
- (E)-N-(tert-Butyl)-3-oxo-3-phenyl-1-(4-(trifluoromethyl)phenyl)prop-1-ene-2-sulfonamide (10f)
- (E)-N-(tert-Butyl)-1-(4-hydroxyphenyl)-3-oxo-3-phenylprop-1-ene-2-sulfonamide (10g)
- (E)-N-(tert-Butyl)-1-(4-hydroxy-3-methoxyphenyl)-3-oxo-3-phenylprop-1-ene-2-sulfonamide (10h)
- (E)-N-(tert-Butyl)-1-(furan-2-yl)-3-oxo-3-phenylprop-1-ene-2-sulfonamide (10i).
- (E)-N-(tert-Butyl)-3-oxo-3-phenyl-1-(pyridin-4-yl)prop-1-ene-2-sulfonamide (10j)
- (E)-N-(tert-Butyl)-1-(1H-indol-3-yl)-3-oxo-3-phenylprop-1-ene-2-sulfonamide (10k)
- (E)-1-(1-Acetyl-1H-indol-3-yl)-N-(tert-butyl)-3-oxo-3-phenylprop-1-ene-2-sulfonamide (10l)
- (E)-3-Oxo-1,3-diphenylprop-1-ene-2-sulfonamide (7a)
- (E)-1-(2-Fluorophenyl)-3-oxo-3-phenylprop-1-ene-2-sulfonamide (7b)
- (E)-1-(3-Fluorophenyl)-3-oxo-3-phenylprop-1-ene-2-sulfonamide (7c)
- (E)-1-(4-Fluorophenyl)-3-oxo-3-phenylprop-1-ene-2-sulfonamide (7d)
- (E)-1-(4-Chlorophenyl)-3-oxo-3-phenylprop-1-ene-2-sulfonamide (7e)
- (E)-3-Oxo-3-phenyl-1-(4-(trifluoromethyl)phenyl)prop-1-ene-2-sulfonamide (7f)
- (E)-1-(4-Hydroxyphenyl)-3-oxo-3-phenylprop-1-ene-2-sulfonamide (7g)
- (E)-1-(4-Hydroxy-3-methoxyphenyl)-3-oxo-3-phenylprop-1-ene-2-sulfonamide (7h)
- (E)-1-(Furan-2-yl)-3-oxo-3-phenylprop-1-ene-2-sulfonamide (7i)
- (E)-3-Oxo-3-phenyl-1-(pyridin-4-yl)prop-1-ene-2-sulfonamide (7j)
- (E)-1-(1H-Indol-3-yl)-3-oxo-3-phenylprop-1-ene-2-sulfonamide (7k)
- (E)-1-(1-Acetyl-1H-indol-3-yl)-3-oxo-3-phenylprop-1-ene-2-sulfonamide (7l)
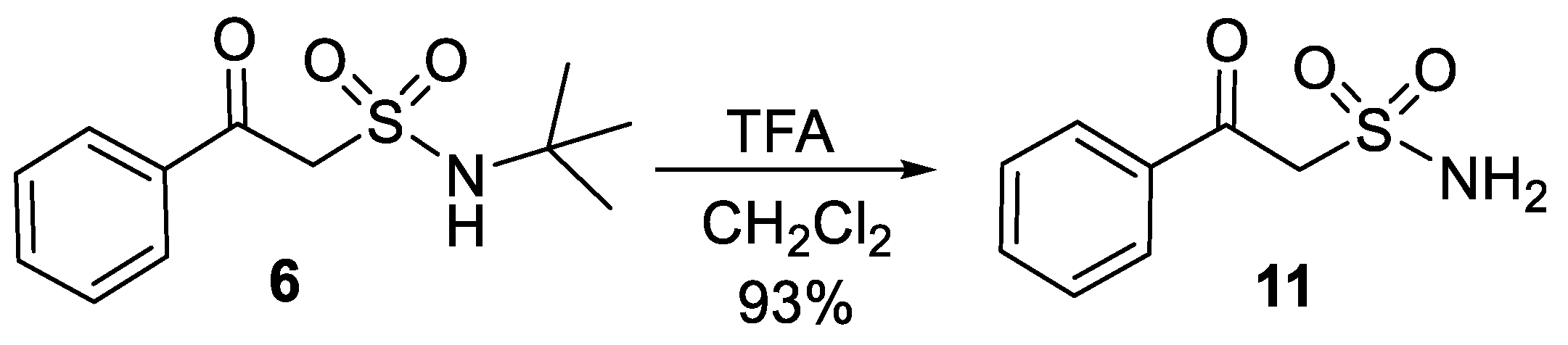
- 2-Oxo-2-phenylethan-1-sulfonamide (11)
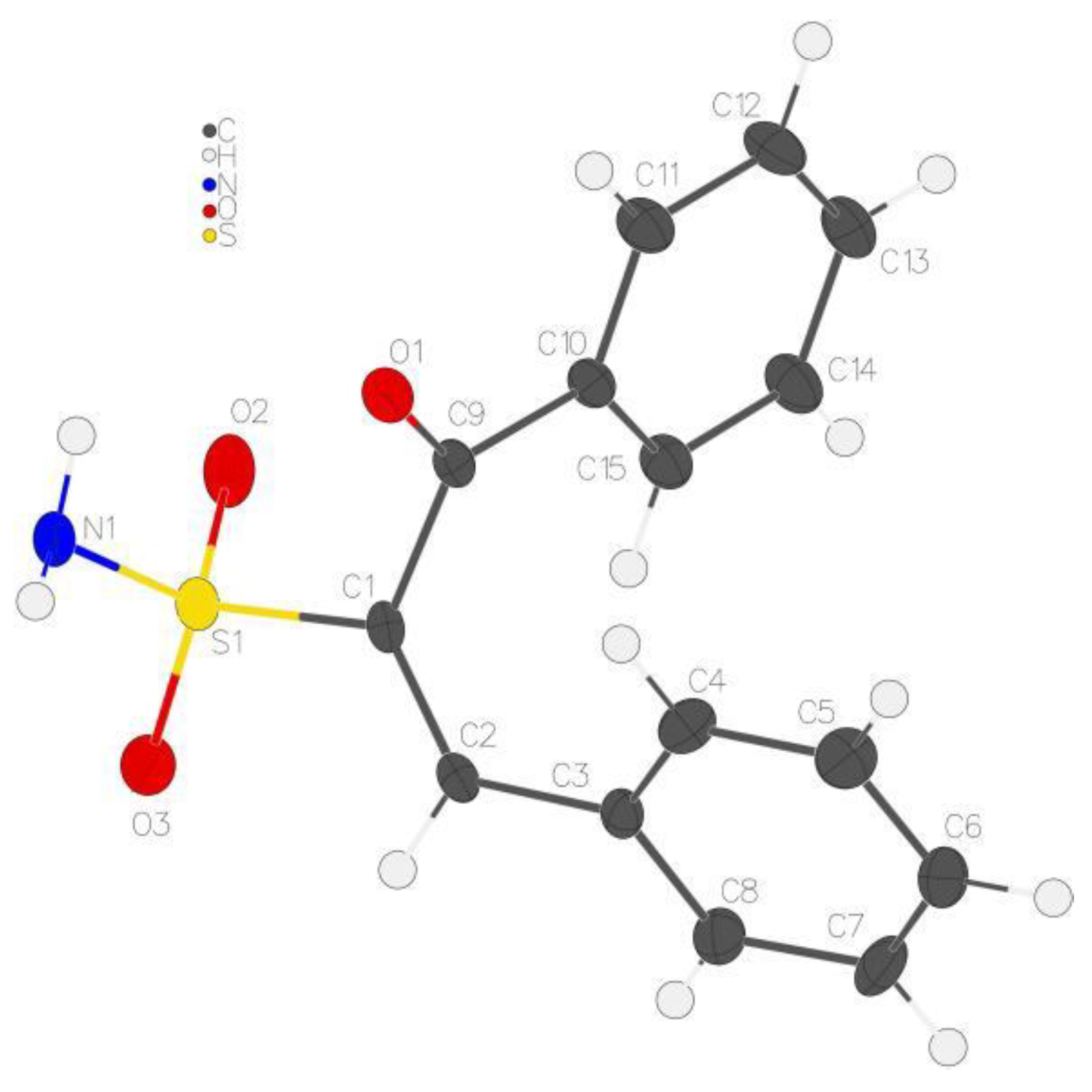
4.2. X-ray Study of 7a
4.3. Antiproliferative Activity and Selectivity
4.4. Immunoblotting
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HRMS | High-resolution mass spectrometry; |
| HPLC | High-performance liquid chromatography; |
| LDA | Lithium diisopropylamide; |
| NMR | Nuclear magnetic resonance; |
| PG | Protecting group; |
| TFA | Trifluoroacetic acid. |
| ER α | Estrogen receptor α |
| AI | Aromatase inhibitor |
| SERM | Selective estrogen receptor modulator |
| SI | Selectivity index |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| GREB1 | Gene regulated in breast cancer 1 protein |
| AR | Androgen receptor |
| CDK4 | Cyclin-dependent kinase 4 |
| PARP | Poly (ADP-ribose) polymerase 1 |
| Bcl-2 | B-cell CLL/lymphoma 2 |
| HT | 4-hydroxytamoxifen |
| MDR | Multidrug-resistant |
References
- Hanker, A.B.; Sudhan, D.R.; Arteaga, C.L. Overcoming endocrine resistance in breast cancer. Cancer Cell 2020, 37, 496–513. [Google Scholar] [CrossRef] [PubMed]
- Härkönen, P.L.; Mäkelä, S.I. Role of estrogens in development of prostate cancer. J. Steroid Biochem. Mol. Biol. 2004, 92, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Mungenast, F.; Thalhammer, T. Estrogen biosynthesis and action in ovarian cancer. Front. Endocrinol. 2014, 5, 192. [Google Scholar] [CrossRef] [PubMed]
- Martinkovich, S.; Shah, D.; Planey, S.L.; Arnott, J.A. Selective estrogen receptor modulators: Tissue specificity and clinical utility. Clin. Interv. Aging 2014, 2014, 1437–1452. [Google Scholar] [CrossRef]
- Ma, C.X.; Reinert, T.; Chmielewska, I.; Ellis, M.J. Mechanisms of aromatase inhibitor resistance. Nat. Rev. Cancer 2015, 15, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, R.; Ward, R.A. Structure-based design of targeted covalent inhibitors. Chem. Soc. Rev. 2018, 47, 3816–3830. [Google Scholar] [CrossRef] [PubMed]
- Sutanto, F.; Konstantinidou, M.; Dömling, A. Covalent inhibitors: A rational approach to drug discovery. RSC Med. Chem. 2020, 11, 876–884. [Google Scholar] [CrossRef]
- Van Asten, K.; Neven, P.; Lintermans, A.; Wildiers, H.; Paridaens, R. Aromatase inhibitors in the breast cancer clinic: Focus on exemestane. Endocr. Relat. Cancer 2014, 21, 31–49. [Google Scholar] [CrossRef]
- Hong, Y.; Yu, B.; Sherman, M.; Yuan, Y.C.; Zhou, D.; Chen, S. Molecular basis for the aromatization reaction and exemestane-mediated irreversible inhibition of human aromatase. Mol. Endocrinol. 2007, 21, 401–414. [Google Scholar] [CrossRef]
- Coombes, R.C.; Hall, E.; Gibson, L.J.; Paridaens, R.; Jassem, J.; Delozier, T.; Bliss, J.M. A randomized trial of exemestane after two to three years of tamoxifen therapy in postmenopausal women with primary breast cancer. New Engl. J. Med. 2004, 350, 1081–1092. [Google Scholar] [CrossRef]
- Sobral, A.F.; Amaral, C.; Correia-da-Silva, G.; Teixeira, N. Unravelling exemestane: From biology to clinical prospects. J. Steroid Biochem. Mol. Biol. 2016, 163, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Park, I.H.; Lee, H.W.; Lee, K.S.; Nam, B.H.; Ro, J.S. Efficacy of exemestane after nonsteroidal aromatase inhibitor use in metastatic breast cancer patients. Asian Pac. J. Cancer Prev. 2012, 13, 979–983. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A privileged structure in medicinal chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef] [PubMed]
- Salehi, B.; Quispe, C.; Chamkhi, I.; El Omari, N.; Balahbib, A.; Sharifi-Rad, J.; Les, F. Pharmacological properties of chalcones: A review of preclinical including molecular mechanisms and clinical evidence. Front. Pharmacol. 2021, 11, 592654. [Google Scholar] [CrossRef] [PubMed]
- Harikumar, K.B.; Kunnumakkara, A.B.; Ahn, K.S.; Anand, P.; Krishnan, S.; Guha, S.; Aggarwal, B.B. Modification of the cysteine residues in IkappaBalpha kinase and NF-kappaB (p65) by xanthohumol leads to suppression of NF-kappaB-regulated gene products and potentiation of apoptosis in leukemia cells. Blood 2009, 113, 2003–2013. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Duan, D.; Ge, C.; Yao, J.; Liu, Y.; Li, X.; Fang, J. Synthesis of xanthohumol analogues and discovery of potent thioredoxin reductase inhibitor as potential anticancer agent. J. Med. Chem. 2015, 58, 1795–1805. [Google Scholar] [CrossRef] [PubMed]
- Hegazy, M.E.F.; Mohamed, A.E.H.H.; El-Halawany, A.M.; Djemgou, P.C.; Shahat, A.A.; Paré, P.W. Estrogenic activity of chemical constituents from Tephrosia candida. J. Nat. Prod. 2011, 74, 937–942. [Google Scholar] [CrossRef]
- Kommidi, D.R.; Pagadala, R.; Rana, S.; Singh, P.; Shintre, S.A.; Koorbanally, N.A.; Moodley, B. Novel carbapenem chalcone derivatives: Synthesis, cytotoxicity and molecular docking studies. Org. Biomol. Chem. 2015, 13, 4344–4350. [Google Scholar] [CrossRef]
- Min, J.; Nwachukwu, J.C.; Min, C.K.; Njeri, J.W.; Srinivasan, S.; Rangarajan, E.S.; Nettles, K.W. Dual-mechanism estrogen receptor inhibitors. Biophys. Comput. Biol. 2021, 118, e2101657118. [Google Scholar] [CrossRef]
- Zhu, M.; Zhang, C.; Nwachukwu, J.C.; Srinivasan, S.; Cavett, V.; Zheng, Y.; Zhou, H.B. Bicyclic core estrogens as full antagonists: Synthesis, biological evaluation and structure-activity relationships of estrogen receptor ligands based on bridged oxabicyclic core arylsulfonamides. Org. Biomol. Chem. 2012, 10, 8692–8700. [Google Scholar] [CrossRef]
- Lefebvre, C.A.; Forcellini, E.; Boutin, S.; Côté, M.F.; René, C.; Mathieu, P.; Paquin, J.F. Synthesis of novel substituted pyrimidine derivatives bearing a sulfamide group and their in vitro cancer growth inhibition activity. Bioorg. Med. Chem. Lett. 2017, 27, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Spevak, W.; Zhang, Y.; Burton, E.A.; Ma, Y.; Habets, G.; Bollag, G. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature 2015, 526, 583–586. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Lee, S.; Kim, B.; Lee, H.; Hong, S.S.; Hong, S. Discovery of new azaindole-based PI3Kα inhibitors: Apoptotic and antiangiogenic effect on cancer cells. Bioorg. Med. Chem. Lett. 2010, 20, 7212–7215. [Google Scholar] [CrossRef] [PubMed]
- Krymov, S.K.; Scherbakov, A.M.; Salnikova, D.I.; Sorokin, D.V.; Dezhenkova, L.G.; Ivanov, I.V.; Shchekotikhin, A.E. Synthesis, biological evaluation, and in silico studies of potential activators of apoptosis and carbonic anhydrase inhibitors on isatin-5-sulfonamide scaffold. Eur. J. Med. Chem. 2022, 228, 113997. [Google Scholar] [CrossRef] [PubMed]
- Krymov, S.K.; Scherbakov, A.M.; Dezhenkova, L.G.; Salnikova, D.I.; Solov’eva, S.E.; Sorokin, D.V.; Shchekotikhin, A.E. Indoline-5-Sulfonamides: A Role of the Core in Inhibition of Cancer-Related Carbonic Anhydrases, Antiproliferative Activity and Circumventing of Multidrug Resistance. Pharmaceuticals 2022, 15, 1453. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.E. N-Alkanesulfonamide dianions: Formation and chemoselective C-alkylation. J. Org. Chem. 1984, 49, 1700–1703. [Google Scholar] [CrossRef]
- Reddy, M.R.; Mallireddigari, M.R.; Pallela, V.R.; Cosenza, S.C.; Billa, V.K.; Akula, B.; Reddy, E.P. Design, synthesis, and biological evaluation of (E)-N-aryl-2-arylethenesulfonamide analogues as potent and orally bioavailable microtubule-targeted anticancer agents. J. Med. Chem. 2013, 56, 5562–5586. [Google Scholar] [CrossRef]
- Korolev, A.M.; Shchekotikhin, A.E.; Lysenkova, L.N.; Preobrazhenskaya, M.N. Synthesis of (Indol-3-yl) methanesulfonamide and its 5-Methoxy Derivative. Synthesis 2003, 3, 383–388. [Google Scholar] [CrossRef]
- Dauben, W.G.; Ponaras, A.A.; Chollet, A. An approach to angularly functionalized methylhydrindan systems. J. Org. Chem. 1980, 45, 4413–4417. [Google Scholar] [CrossRef]
- Corey, E.J.; Barrette, E.P.; Magriotis, P.A. A new Cr (VI) reagent for the catalytic oxidation of secondary alcohols to ketones. Tetrahedron. Lett. 1985, 26, 5855–5858. [Google Scholar] [CrossRef]
- Burke, S.D.; Danheiser, R.L. Oxidizing and Reducing Agents, 1st ed.; John Wiley & Sons: Chichester, UK, 1999; pp. 100–102. [Google Scholar]
- Allen, C.F.H. Ethyl benzalmalonate. Org. Synth. 1945, 25, 42. [Google Scholar] [CrossRef]
- Magee, D.I.; Ratshonka, S.; McConaghy, J.; Hood, M. Synthesis of β-and β, β-substituted Morita–Baylis–Hillman adducts using a two-step protocol. Can. J. Chem. 2012, 90, 450–463. [Google Scholar] [CrossRef]
- Scherbakov, A.M.; Basharina, A.A.; Sorokin, D.V.; Mikhaevich, E.I.; Mizaeva, I.E.; Mikhaylova, A.L.; Krasil’nikov, M.A. Targeting hormone-resistant breast cancer cells with docetaxel: A look inside the resistance. Cancer Drug Resist. 2023, 6, 103. [Google Scholar] [CrossRef] [PubMed]
- Tikhomirov, A.S.; Tsvetkov, V.B.; Volodina, Y.L.; Litvinova, V.A.; Andreeva, D.V.; Dezhenkova, L.G.; Kaluzhny, D.N.; Treshalin, I.D.; Shtil, A.A.; Shchekotikhin, A.E. Heterocyclic ring expansion yields anthraquinone derivatives potent against multidrug resistant tumor cells. Bioorg. Chem. 2022, 127, 105925. [Google Scholar] [CrossRef] [PubMed]
- Sagnou, M.; Novikov, F.N.; Ivanova, E.S.; Alexiou, P.; Stroylov, V.S.; Titov, I.Y.; Tatarskiy, V.V.; Vagida, M.S.; Pelecanou, M.; Shtil, A.A.; et al. Novel curcumin derivatives as P-glycoprotein inhibitors: Molecular modeling, synthesis and sensitization of multidrug resistant cells to doxorubicin. Eur. J. Med. Chem. 2020, 198, 112331. [Google Scholar] [CrossRef] [PubMed]
- Zorin, V.; Zorina, A.; Smetanina, N.; Kopnin, P.; Ozerov, I.V.; Leonov, S.; Isaev, A.; Klokov, D.; Osipov, A.N. Diffuse colonies of human skin fibroblasts in relation to cellular senescence and proliferation. Aging 2017, 9, 1404. [Google Scholar] [CrossRef]
- Zorin, V.; Grekhova, A.; Pustovalova, M.; Zorina, A.; Smetanina, N.; Vorobyeva, N.; Kopnin, P.; Gilmutdinova, I.; Moskalev, A.; Osipov, A.N.; et al. Spontaneous γH2AX foci in human dermal fibroblasts in relation to proliferation activity and aging. Aging 2019, 11, 4536. [Google Scholar] [CrossRef]
- Kamiloglu, S.; Sari, G.; Ozdal, T.; Capanoglu, E. Guidelines for cell viability assays. Food Front. 2020, 1, 332–349. [Google Scholar] [CrossRef]
- Larsson, P.; Engqvist, H.; Biermann, J.; Ronnerman, E.W.; Forssell-Aronsson, E.; Kovacs, A.; Karlsson, P.; Helou, K.; Parris, T.Z. Optimization of cell viability assays to improve replicability and reproducibility of cancer drug sensitivity screens. Sci. Rep. 2020, 10, 5798. [Google Scholar] [CrossRef]
- Lavogina, D.; Lust, H.; Tahk, M.J.; Laasfeld, T.; Vellama, H.; Nasirova, N.; Vardja, M.; Eskla, K.-L.; Salumets, A.; Rinken, A.; et al. Revisiting the resazurin-based sensing of cellular viability: Widening the application horizon. Biosensors 2022, 12, 196. [Google Scholar] [CrossRef]
- Welboren, W.J.; HStunnenberg, G.; Sweep, F.C.; Span, P.N. Identifying estrogen receptor target genes. Mol. Oncol. 2007, 1, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Clusan, L.; Ferrière, F.; Flouriot, G.; Pakdel, F. A Basic Review on Estrogen Receptor Signaling Pathways in Breast Cancer. Int. J. Mol. Sci. 2023, 24, 6834. [Google Scholar] [CrossRef] [PubMed]
- Anestis, A.; Zoi, I.; Papavassiliou, A.G.; Karamouzis, M.V. Androgen Receptor in Breast Cancer-Clinical and Preclinical Research Insights. Molecules 2020, 25, 358. [Google Scholar] [CrossRef] [PubMed]
- Kolyvas, E.A.; Caldas, C.; Kelly, K.; Ahmad, S.S. Androgen receptor function and targeted therapeutics across breast cancer subtypes. Breast Cancer Res. 2022, 24, 79. [Google Scholar] [CrossRef] [PubMed]
- Qie, S.; Diehl, J.A. Cyclin D1, cancer progression, and opportunities in cancer treatment. J. Mol. Med. 2016, 94, 1313–1326. [Google Scholar] [CrossRef]
- Montalto, F.I.; De Amicis, F. Cyclin D1 in Cancer: A Molecular Connection for Cell Cycle Control, Adhesion and Invasion in Tumor and Stroma. Cells 2020, 9, 2648. [Google Scholar] [CrossRef]
- Barnes, D.M.; Gillett, C.E. Cyclin D1 in breast cancer. Breast Cancer Res. Treat. 1998, 52, 1–15. [Google Scholar] [CrossRef]
- Shi, X.; Wang, J.; Lei, Y.; Cong, C.; Tan, D.; Zhou, X. Research progress on the PI3K/AKT signaling pathway in gynecological cancer. Mol. Med. Rep. 2019, 19, 4529–4535. [Google Scholar] [CrossRef]
- Rascio, F.; Spadaccino, F.; Rocchetti, M.T.; Castellano, G.; Stallone, G.; Netti, G.S.; Ranieri, E. The Pathogenic Role of PI3K/AKT Pathway in Cancer Onset and Drug Resistance: An Updated Review. Cancers 2021, 13, 3949. [Google Scholar] [CrossRef]
- Coppola, G.M.; Hardtmann, G.E. The chemistry of 2H-3,1-benzoxazine-2,4(1H)dione (isatoic anhydride). 7. Reactions with anions of active methylenes to form quinolines. J. Heterocycl. Chem. 1979, 16, 1605–1610. [Google Scholar] [CrossRef]
- Soley, J. Synthesis of ß-Keto-and ß-Hydroxy-α, α-Difluorosulfonamides. Ph.D. Thesis, University of Waterloo, Waterloo, ON, Canada, 2021. Available online: https://uwspace.uwaterloo.ca/handle/10012/17044 (accessed on 4 March 2023).
- Moreira, J.; Loureiro, J.B.; Correia, D.; Palmeira, A.; Pinto, M.M.; Saraiva, L.; Cidade, H. Structure–Activity Relationship Studies of Chalcones and Diarylpentanoids with Antitumor Activity: Potency and Selectivity Optimization. Pharmaceuticals 2023, 16, 1354. [Google Scholar] [CrossRef] [PubMed]
- Lai, W.; Chen, J.; Gao, X.; Jin, X.; Chen, G.; Ye, L. Design and Synthesis of Novel Chalcone Derivatives: Anti-Breast Cancer Activity Evaluation and Docking Study. Int. J. Mol. Sci. 2023, 24, 15549. [Google Scholar] [CrossRef] [PubMed]
- Scherbakov, A.M.; Zavarzin, I.V.; Vorontsova, S.K.; Hajra, A.; Andreeva, O.E.; Yadykov, A.V.; Levina, I.S.; Volkova, Y.A.; Shirinian, V.Z. Synthesis and evaluation of the antiproliferative activity of benzylidenes of 16-dehydroprogesterone series. Steroids 2018, 138, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Bruker. APEX3, RLATT, CELL_NOW, TWINABS, SAINT-Plus and SADABS, Version 2019.11-0; Bruker AXS Inc.: Madison, WI, USA, 2016. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Volodina, Y.L.; Dezhenkova, L.G.; Tikhomirov, A.S.; Tatarskiy, V.V.; Kaluzhny, D.N.; Moisenovich, A.M.; Isagulieva, A.K.; Shtil, A.A.; Tsvetkov, V.B.; Shchekotikhin, A.E. New anthra[2,3-b]furancarboxamides: A role of positioning of the carboxamide moiety in antitumor properties. Eur. J. Med. Chem. 2019, 165, 31–45. [Google Scholar] [CrossRef]
- Komendantova, A.S.; Scherbakov, A.M.; Komkov, A.V.; Chertkova, V.V.; Gudovanniy, A.O.; Chernoburova, E.I.; Sorokin, D.V.; Dzichenka, Y.U.; Shirinian, V.Z.; Volkova, Y.A.; et al. Novel steroidal 1,3,4-thiadiazines: Synthesis and biological evaluation in androgen receptor-positive prostate cancer 22Rv1 cells. Bioorg. Chem. 2019, 91, 103142. [Google Scholar] [CrossRef]
- Zapevalova, M.V.; Shchegravina, E.S.; Fonareva, I.P.; Salnikova, D.I.; Sorokin, D.V.; Scherbakov, A.M.; Maleev, A.A.; Ignatov, S.K.; Grishin, I.D.; Kuimov, A.N.; et al. Synthesis, Molecular Docking, In Vitro and In Vivo Studies of Novel Dimorpholinoquinazoline-Based Potential Inhibitors of PI3K/Akt/mTOR Pathway. Int. J. Mol. Sci. 2022, 23, 10854. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Mruk, D.D.; Cheng, C.Y. Enhanced chemiluminescence (ECL) for routine immunoblotting: An inexpensive alternative to commercially available kits. Spermatogenesis 2011, 1, 121–122. [Google Scholar] [CrossRef] [PubMed]
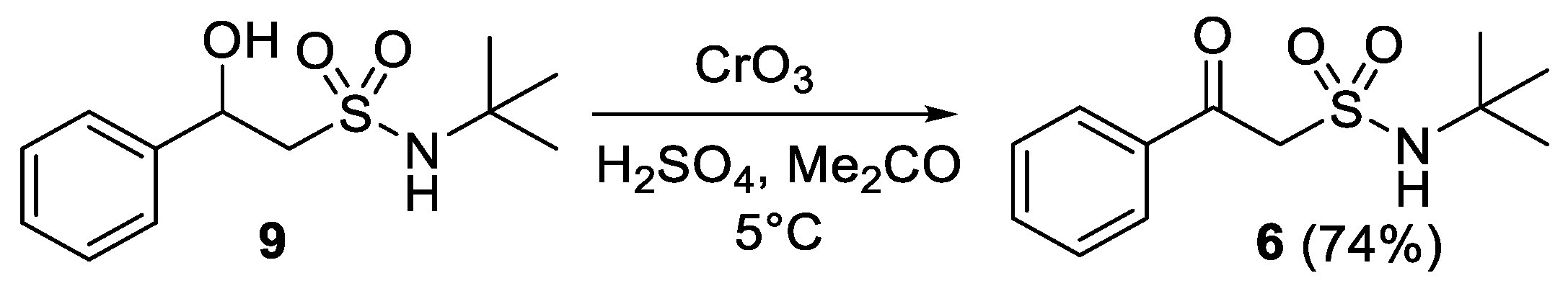

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
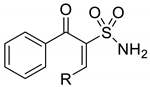
| Cmpd | R | IC50 (µM) a | ||||
| MCF7 | MCF7/HT2 | K-562 | K-562/4 | HCT-116 | ||
| 7a | 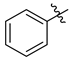 | 3.6 ± 0.2 | 3.5 ± 0.2 | 3.7 ± 0.3 | 12.5 ± 1.2 | 10.3 ± 1.1 |
| 7b | 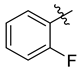 | 4.0 ± 0.3 | 4.5 ± 0.4 | 2.5 ± 0.3 | 6.4 ± 0.5 | 8.2 ± 0.8 |
| 7c | 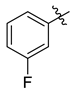 | 4.0 ± 0.3 | 3.9 ± 0.4 | 2.2 ± 0.1 | 11.5 ± 1.5 | 10.3 ± 1.1 |
| 7d | 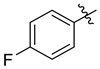 | 3.9 ± 0.2 | 4.8 ± 0.4 | 2.0 ± 0.1 | 15.3 ± 1.1 | 9.8 ± 1.1 |
| 7e | 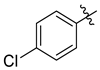 | 2.0 ± 0.1 | 3.8 ± 0.2 | 1.3 ± 0.1 | 2.8 ± 0.2 | 5.1 ± 0.4 |
| 7f | 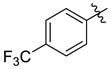 | 2.2 ± 0.3 | 2.5 ± 0.2 | 1.8 ± 0.2 | 2.8 ± 0.3 | 3.6 ± 0.3 |
| 7g | 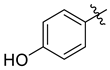 | 5.5 ± 0.7 | 7.3 ± 0.7 | 3.2 ± 0.2 | 18.5 ± 1.4 | 15.0 ± 1.4 |
| 7h | 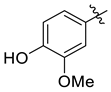 | 6.8 ± 0.3 | 8.8 ± 0.8 | 4.3 ± 0.6 | 32.0 ± 4.2 | 23.0 ± 3.0 |
| 7i | 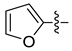 | 39.9 ± 3.3 | 35.9 ± 3.1 | 11.5 ± 1.2 | NT | 45.0 ± 7.0 |
| 7j | 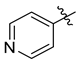 | 5.7 ± 0.3 | 5.1 ± 0.3 | 7.5 ± 0.8 | NT | 21.0 ± 2.5 |
| 7k | 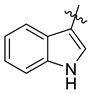 | 19.9 ± 1.4 | 14.1 ± 1.1 | 9.2 ± 1.1 | NT | 23.2 ± 2.5 |
| 7l | 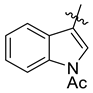 | 6.1 ± 0.4 | 6.7 ± 0.4 | 6.6 ± 0.7 | NT | 7.1 ± 0.8 |
| 11 | 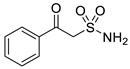 | >50 | >50 | >50 | >50 | >50 |
| Dox b | 0.31 ± 0.04 | NT | 0.12 ± 0.01 | 5.1 ± 0.5 | 0.32 ± 0.04 | |
| Cell Line | Compounds | ||
|---|---|---|---|
| 7e | 11 | ||
| IC50, µM | MCF7 cells | 2.6 ± 0.2 | >50 |
| Fibroblasts | 4.9 ± 0.3 | >50 | |
| SI * | 1.9 | - | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krymov, S.K.; Salnikova, D.I.; Dezhenkova, L.G.; Bogdanov, F.B.; Korlyukov, A.A.; Scherbakov, A.M.; Shchekotikhin, A.E. Synthesis and Biological Evaluation of Chalconesulfonamides: En Route to Proapoptotic Agents with Antiestrogenic Potency. Pharmaceuticals 2024, 17, 32. https://doi.org/10.3390/ph17010032
Krymov SK, Salnikova DI, Dezhenkova LG, Bogdanov FB, Korlyukov AA, Scherbakov AM, Shchekotikhin AE. Synthesis and Biological Evaluation of Chalconesulfonamides: En Route to Proapoptotic Agents with Antiestrogenic Potency. Pharmaceuticals. 2024; 17(1):32. https://doi.org/10.3390/ph17010032
Chicago/Turabian StyleKrymov, Stepan K., Diana I. Salnikova, Lyubov G. Dezhenkova, Fedor B. Bogdanov, Alexander A. Korlyukov, Alexander M. Scherbakov, and Andrey E. Shchekotikhin. 2024. "Synthesis and Biological Evaluation of Chalconesulfonamides: En Route to Proapoptotic Agents with Antiestrogenic Potency" Pharmaceuticals 17, no. 1: 32. https://doi.org/10.3390/ph17010032
APA StyleKrymov, S. K., Salnikova, D. I., Dezhenkova, L. G., Bogdanov, F. B., Korlyukov, A. A., Scherbakov, A. M., & Shchekotikhin, A. E. (2024). Synthesis and Biological Evaluation of Chalconesulfonamides: En Route to Proapoptotic Agents with Antiestrogenic Potency. Pharmaceuticals, 17(1), 32. https://doi.org/10.3390/ph17010032