
Functional Progression after Dose Suspension or Discontinuation of Nintedanib in Idiopathic Pulmonary Fibrosis: A Real-Life Multicentre Study

 ,
,  , ,
, ,  , ,
, ,  and
and
Abstract
1. Introduction
2. Results
3. Discussion
3.1. Main Results
3.2. Comparison with the Relevant Literature
3.3. Theoretical Implications
3.4. Limitations and Strengths
3.5. Recommendations for Future Research
4. Materials and Methods
4.1. Study Population and Design of the Study
4.2. Data Collection
4.3. Pulmonary Function Tests
4.4. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
| Baseline | n | 6 Months | n | p | Baseline | n | 12 Months | n | p |
|---|---|---|---|---|---|---|---|---|---|
| FVC | |||||||||
| 74.0 (62.5–82.5) | 27 | 72.5 (64.0–84.3) | 22 | 0.338 | 75.0 (63.0–83.0) | 25 | 76.0 (60.0–87.0) | 17 | 0.910 |
| FEV1 | |||||||||
| 81.0 (69.0–100.0) | 28 | 79.0 (67.5–91.0) | 19 | 0.736 | 81.0 (70.0–100.0) | 23 | 76.0 (74.0–87.0) | 13 | 0.317 |
| DLCO | |||||||||
| 44.0 (34.0–62.0) | 28 | 39.0 (28.0–49.5) | 19 | 0.056 | 47.0 (35.0–62.0) | 26 | 44.5 (35.0–59.8) | 18 | 0.099 |
References
- American Thoracic Society. Idiopathic pulmonary fibrosis: Diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am. J. Respir. Crit. Care Med. 2000, 161 Pt 1, 646–664. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef] [PubMed]
- Munchel, J.K.; Shea, B.S. Diagnosis and Management of Idiopathic Pulmonary Fibrosis. R. I. Med. J. (2013) 2021, 104, 26–29. [Google Scholar] [PubMed]
- Ruaro, B.; Gandin, I.; Pozzan, R.; Tavano, S.; Bozzi, C.; Hughes, M.; Kodric, M.; Cifaldi, R.; Lerda, S.; Confalonieri, M.; et al. Nintedanib in Idiopathic Pulmonary Fibrosis: Tolerability and Safety in a Real Life Experience in a Single Centre in Patients Also Treated with Oral Anticoagulant Therapy. Pharmaceuticals 2023, 16, 307. [Google Scholar] [CrossRef]
- Serra López-Matencio, J.M.; Gómez, M.; Vicente-Rabaneda, E.F.; González-Gay, M.A.; Ancochea, J.; Castañeda, S. Pharmacological Interactions of Nintedanib and Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis in Times of COVID-19 Pandemic. Pharmaceuticals 2021, 14, 819. [Google Scholar] [CrossRef] [PubMed]
- Ley, B.; Collard, H.R.; King, T.E. Clinical Course and Prediction of Survival in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2011, 183, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Luppi, F.; Kalluri, M.; Faverio, P.; Kreuter, M.; Ferrara, G. Idiopathic Pulmonary Fibrosis beyond the Lung: Understanding Disease Mechanisms to Improve Diagnosis and Management. Respir. Res. 2021, 22, 109. [Google Scholar] [CrossRef]
- Fujimoto, H.; Kobayashi, T.; Azuma, A. Idiopathic Pulmonary Fibrosis: Treatment and Prognosis. Clin. Med. Insights Circ. Respir. Pulm. Med. 2015, 9 (Suppl. S1), 179–185. [Google Scholar] [CrossRef]
- Cutolo, M.; Trombetta, A.C.; Melsens, K.; Pizzorni, C.; Sulli, A.; Ruaro, B.; Paolino, S.; Deschepper, E.; Smith, V. Automated assessment of absolute nailfold capillary number on videocapillaroscopic images: Proof of principle and validation in systemic sclerosis. Microcirculation 2018, 25, 12447. [Google Scholar] [CrossRef]
- Raghu, G.; Ley, B.; Brown, K.K.; Cottin, V.; Gibson, K.F.; Kaner, R.J.; Lederer, D.J.; Noble, P.W.; Song, J.W.; Wells, A.U.; et al. Risk factors for disease progression in idiopathic pulmonary fibrosis. Thorax 2020, 75, 78–80. [Google Scholar] [CrossRef]
- Baratella, E.; Ruaro, B.; Giudici, F.; Wade, B.; Santagiuliana, M.; Salton, F.; Confalonieri, P.; Simbolo, M.; Scarpa, A.; Tollot, S.; et al. Evaluation of Correlations between Genetic Variants and High-Resolution Computed Tomography Patterns in Idiopathic Pulmonary Fibrosis. Diagnostics 2021, 11, 762. [Google Scholar] [CrossRef]
- Sgalla, G.; Biffi, A.; Richeldi, L. Idiopathic Pulmonary Fibrosis: Diagnosis, Epidemiology and Natural History. Respirology 2016, 21, 427–437. [Google Scholar] [CrossRef]
- Salton, F.; Ruaro, B.; Confalonieri, P.; Confalonieri, M. Epithelial-Mesenchymal Transition: A Major Pathogenic Driver in Idiopathic Pulmonary Fibrosis? Medicina 2020, 56, 608. [Google Scholar] [CrossRef]
- Confalonieri, P.; Volpe, M.C.; Jacob, J.; Maiocchi, S.; Salton, F.; Ruaro, B.; Confalonieri, M.; Braga, L. Regeneration or Repair? The Role of Alveolar Epithelial Cells in the Pathogenesis of Idiopathic Pulmonary Fibrosis (IPF). Cells 2022, 11, 2095. [Google Scholar] [CrossRef]
- Neely, M.L.; Hellkamp, A.S.; Bender, S.; Todd, J.L.; Liesching, T.; Luckhardt, T.R.; Oldham, J.M.; Raj, R.; White, E.S.; Palmer, S.M. Lung Function Trajectories in Patients with Idiopathic Pulmonary Fibrosis. Respir. Res. 2023, 24, 209. [Google Scholar] [CrossRef]
- Denton, C.P.; Goh, N.S.; Humphries, S.M.; Maher, T.M.; Spiera, R.; Devaraj, A.; Ho, L.; Stock, C.; Erhardt, E.; Alves, M.; et al. Extent of Fibrosis and Lung Function Decline in Patients with Systemic Sclerosis and Interstitial Lung Disease: Data from the SENSCIS Trial. Rheumatology 2023, 62, 1870–1876. [Google Scholar] [CrossRef]
- Hilberg, F.; Roth, G.J.; Krssak, M.; Kautschitsch, S.; Sommergruber, W.; Tontsch-Grunt, U.; Garin-Chesa, P.; Bader, G.; Zoephel, A.; Quant, J.; et al. BIBF 1120: Triple Angiokinase Inhibitor with Sustained Receptor Blockade and Good Antitumor Efficacy. Cancer Res. 2008, 68, 4774–4782. [Google Scholar] [CrossRef]
- Flaherty, K.R.; Wells, A.U.; Cottin, V.; Devaraj, A.; Walsh, S.L.F.; Inoue, Y.; Richeldi, L.; Kolb, M.; Tetzlaff, K.; Stowasser, S.; et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N. Engl. J. Med. 2019, 381, 1718–1727. [Google Scholar] [CrossRef]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and Safety of Nintedanib in Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef]
- Cottin, V.; Martinez, F.J.; Jenkins, R.G.; Belperio, J.A.; Kitamura, H.; Molina-Molina, M.; Tschoepe, I.; Coeck, C.; Lievens, D.; Costabel, U. Safety and Tolerability of Nintedanib in Patients with Progressive Fibrosing Interstitial Lung Diseases: Data from the Randomized Controlled INBUILD Trial. Respir. Res. 2022, 23, 85. [Google Scholar] [CrossRef]
- Ikeda, S.; Sekine, A.; Baba, T.; Yamanaka, Y.; Sadoyama, S.; Yamakawa, H.; Oda, T.; Okuda, R.; Kitamura, H.; Okudela, K.; et al. Low Body Surface Area Predicts Hepatotoxicity of Nintedanib in Patients with Idiopathic Pulmonary Fibrosis. Sci. Rep. 2017, 7, 10811. [Google Scholar] [CrossRef] [PubMed]
- Cameli, P.; Alonzi, V.; d’Alessandro, M.; Bergantini, L.; Pordon, E.; Guerrieri, M.; Refini, R.M.; Sestini PBargagli, E. The Effectiveness of Nintedanib in Patients with Idiopathic Pulmonary Fibrosis, Familial Pulmonary Fibrosis and Progressive Fibrosing Interstitial Lung Diseases: A Real-World Study. Biomedicines 2022, 10, 1973. [Google Scholar] [CrossRef] [PubMed]
- Hostettler, K.E.; Zhong, J.; Papakonstantinou, E.; Karakiulakis, G.; Tamm, M.; Seidel, P.; Sun, Q.; Mandal, J.; Lardinois, D.; Lambers, C.; et al. Anti-Fibrotic Effects of Nintedanib in Lung Fibroblasts Derived from Patients with Idiopathic Pulmonary Fibrosis. Respir. Res. 2014, 15, 157. [Google Scholar] [CrossRef]
- Campochiaro, C.; De Luca, G.; Lazzaroni, M.G.; Armentaro, G.; Spinella, A.; Vigone, B.; Ruaro, B.; Stanziola, A.; Benfaremo, D.; De Lorenzis, E. Real-life efficacy and safety of nintedanib in systemic sclerosis-interstitial lung disease: Data from an Italian multicentre study. RMD Open 2023, 9, e002850. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Cottin, V.; Du Bois, R.M.; Selman, M.; Kimura, T.; Bailes, Z.; Schlenker-Herceg, R.; Stowasser, S.; Brown, K.K. Nintedanib in Patients with Idiopathic Pulmonary Fibrosis: Combined Evidence from the TOMORROW and INPULSIS® Trials. Respir. Med. 2016, 113, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Costabel, U.; Selman, M.; Kim, D.S.; Hansell, D.M.; Nicholson, A.G.; Brown, K.K.; Flaherty, K.R.; Noble, P.W.; Raghu, G.; et al. Efficacy of a Tyrosine Kinase Inhibitor in Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2011, 365, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Crestani, B.; Huggins, J.T.; Kaye, M.; Costabel, U.; Glaspole, I.; Ogura, T.; Song, J.W.; Stansen, W.; Quaresma, M.; Stowasser, S.; et al. Long-Term Safety and Tolerability of Nintedanib in Patients with Idiopathic Pulmonary Fibrosis: Results from the Open-Label Extension Study, INPULSIS-ON. Lancet Respir. Med. 2019, 7, 60–68. [Google Scholar] [CrossRef]
- Okamori, S.; Asakura, T.; Masuzawa, K.; Yasuda, H.; Kamata, H.; Ishii, M.; Betsuyaku, T. Suspected Accelerated Disease Progression after Discontinuation of Nintedanib in Patients with Idiopathic Pulmonary Fibrosis: Two Case Reports. Medicine 2017, 96, e9081. [Google Scholar] [CrossRef]
- Grimminger, F.; Günther, A.; Vancheri, C. The Role of Tyrosine Kinases in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Eur. Respir. J. 2015, 45, 1426–1433. [Google Scholar] [CrossRef]
- Iacovelli, R.; Massari, F.; Albiges, L.; Loriot, Y.; Massard, C.; Fizazi, K.; Escudier, B. Evidence and Clinical Relevance of Tumor Flare in Patients Who Discontinue Tyrosine Kinase Inhibitors for Treatment of Metastatic Renal Cell Carcinoma. Eur. Urol. 2015, 68, 154–160. [Google Scholar] [CrossRef]
- Chaft, J.E.; Oxnard, G.R.; Sima, C.S.; Kris, M.G.; Miller, V.A.; Riely, G.J. Disease Flare after Tyrosine Kinase Inhibitor Discontinuation in Patients with EGFR -Mutant Lung Cancer and Acquired Resistance to Erlotinib or Gefitinib: Implications for Clinical Trial Design. Clin. Cancer Res. 2011, 17, 6298–6303. [Google Scholar] [CrossRef] [PubMed]
- Stanojevic, S.; Kaminsky, D.A.; Miller, M.R.; Thompson, B.; Aliverti, A.; Barjaktarevic, I.; Cooper, B.G.; Culver, B.; Derom, E.; Hall, G.L.; et al. ERS/ATS Technical Standard on Interpretive Strategies for Routine Lung Function Tests. Eur. Respir. J. 2022, 60, 2101499. [Google Scholar] [CrossRef] [PubMed]
- Bonella, F.; Cottin, V.; Valenzuela, C.; Wijsenbeek, M.; Voss, F.; Rohr, K.B.; Stowasser, S.; Maher, T.M. Meta-Analysis of Effect of Nintedanib on Reducing FVC Decline Across Interstitial Lung Diseases. Adv Ther. 2022, 39, 3392–3402. [Google Scholar] [CrossRef] [PubMed]
- Hirasawa, Y.; Abe, M.; Terada, J.; Sakayori, M.; Suzuki, K.; Yoshioka, K.; Kawasaki, T.; Tsushima, K.; Tatsumi, K. Tolerability of Nintedanib-related diarrhea in patients with idiopathic pulmonary fibrosis. Pulm. Pharmacol. Ther. 2020, 62, 101917. [Google Scholar] [CrossRef]
- Fletcher, S.V.; Jones, M.G.; Renzoni, E.A.; Parfrey, H.; Hoyles, R.K.; Spinks, K.; Kokosi, M.; Kwok, A.; Warburton, C.; Titmuss, V.; et al. Safety and tolerability of nintedanib for the treatment of idiopathic pulmonary fibrosis in routine UK clinical practice. ERJ Open Res. 2018, 4, 00049–02018. [Google Scholar] [CrossRef]
- Galli, J.A.; Pandya, A.; Vega-Olivo, M.; Dass, C.; Zhao, H.; Criner, G.J. Pirfenidone and nintedanib for pulmonary fibrosis in clinical practice: Tolerability and adverse drug reactions. Respirology 2017, 22, 1171–1178. [Google Scholar] [CrossRef]
- Hughes, G.; Toellner, H.; Morris, H.; Leonard, C.; Chaudhuri, N. Real World Experiences: Pirfenidone and Nintedanib are Effective and Well Tolerated Treatments for Idiopathic Pulmonary Fibrosis. J. Clin. Med. 2016, 5, 78. [Google Scholar] [CrossRef]
| n = 54 | |
|---|---|
| Age at baseline (years), median (IQR) | 75.0 (69.0–79.0) |
| Age of onset (years), median (IQR) | 73.5 (68.0–77.0) |
| Gender | |
| Males | 38 (70.4) |
| Females | 16 (29.6) |
| Ever-smoker | 31 (57.4) |
| Previous coexisting disease | |
| GERD | 16 (29.6) |
| COPD | 5 (9.3) |
| Cancer | 7 (13.0) |
| Concomitant medication | |
| OCS | 17 (31.5) |
| Hydroxychloroquine | 0 (0.0) |
| Azathioprine | 1 (1.9) |
| Methotrexate | 0 (0.0) |
| Mycophenolate mofetil | 2 (3.7) |
| Rituximab | 0 (0.0) |
| Tocilizumab | 0 (0.0) |
| PPI | 35 (64.8) |
| HRCT pattern | |
| UIP | 38 (70.4) |
| NSIP | 9 (16.7) |
| n = 54 | |
|---|---|
| Completion of therapy # | |
| Full dose | 20 (37.0) |
| Reduced dose | 11 (20.4) |
| Suspended | 15 (27.8) |
| Mortality | |
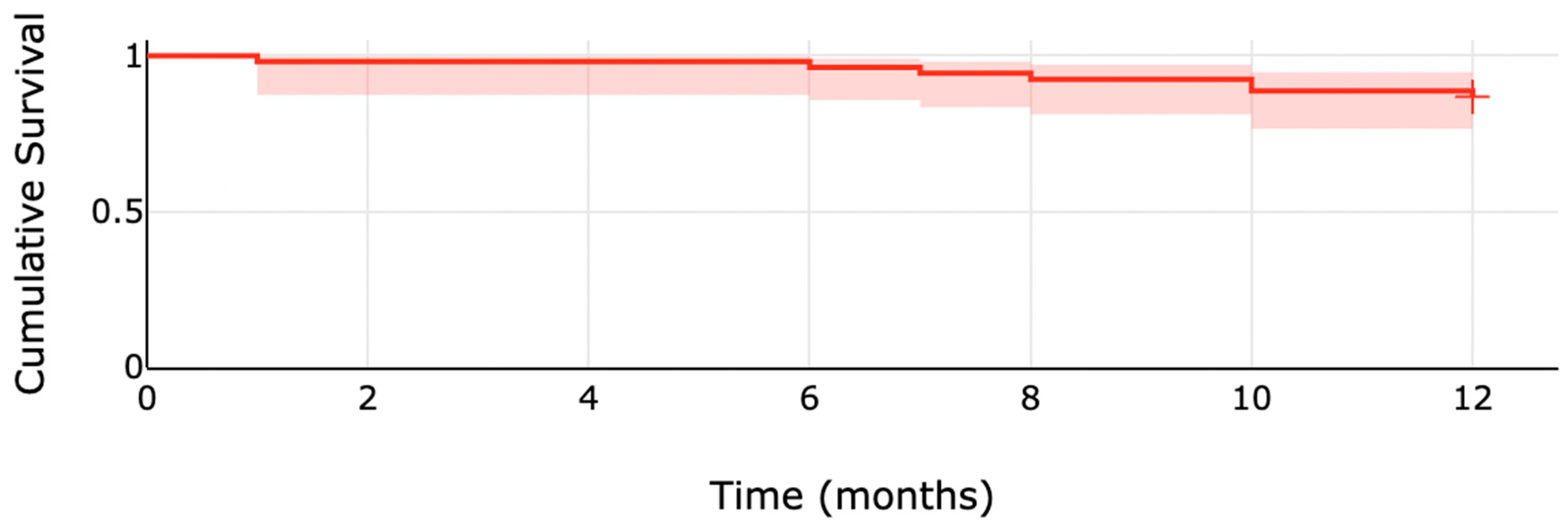
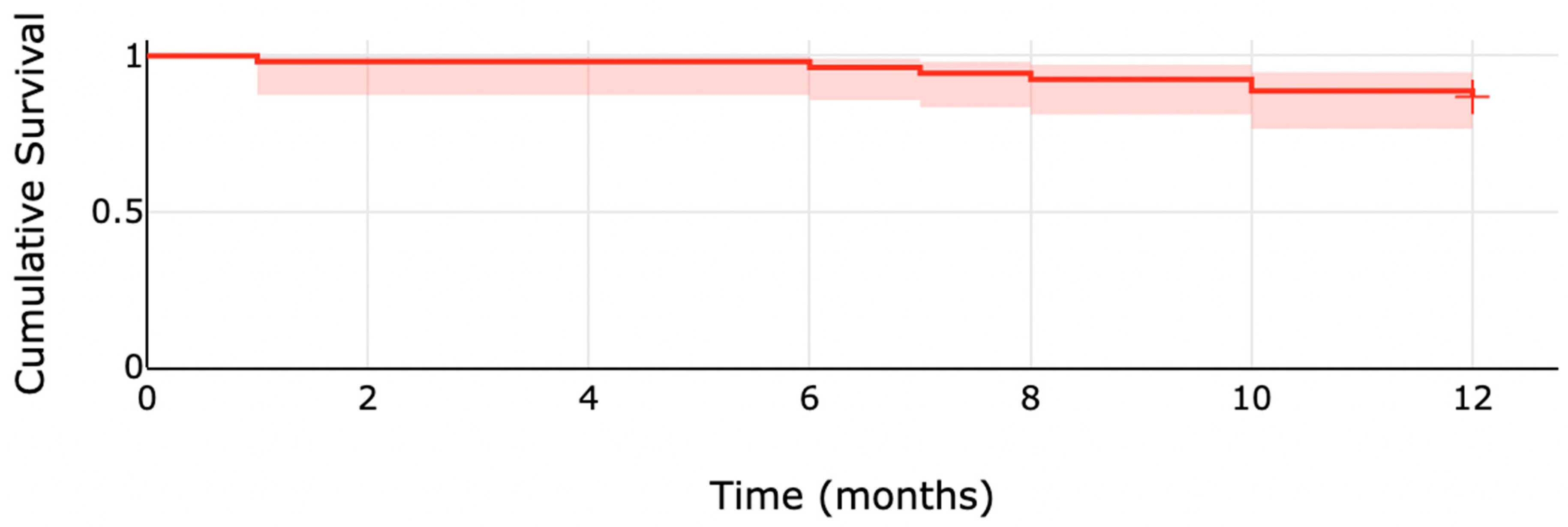
| 6 months | 2 (3.7) |
| 12 months | 7 (13.0) |
| Dose Reduction | n = 11 |
|---|---|
| Gastrointestinal intolerance | 7 (13.0) |
| Diarrhea | 7 (13.0) |
| Nausea/vomiting | 1 (1.9) |
| Abdominal pain | 1 (1.9) |
| Weight loss | 1 (1.9) |
| Allergic reaction | 0 (0.0) |
| Skin ulcer | 0 (0.0) |
| Cough | 0 (0.0) |
| Upper respiratory tract infection | 0 (0.0) |
| Fatigue | 0 (0.0) |
| Liver enzyme derangement | 6 (11.1) |
| Treatment suspension | n = 15 |
| Gastrointestinal intolerance | 11 (20.4) |
| Diarrhea | 8 (14.8) |
| Nausea/vomiting | 2 (3.7) |
| Abdominal pain | 1 (1.9) |
| Weight loss | 2 (3.7) |
| Allergic reaction | 0 (0.0) |
| Skin ulcer | 0 (0.0) |
| Cough | 0 (0.0) |
| Upper respiratory tract infection | 0 (0.0) |
| Fatigue | 0 (0.0) |
| Baseline | n | 6 Months | n | p | Baseline | n | 12 Months | n | p |
|---|---|---|---|---|---|---|---|---|---|
| FVC | |||||||||
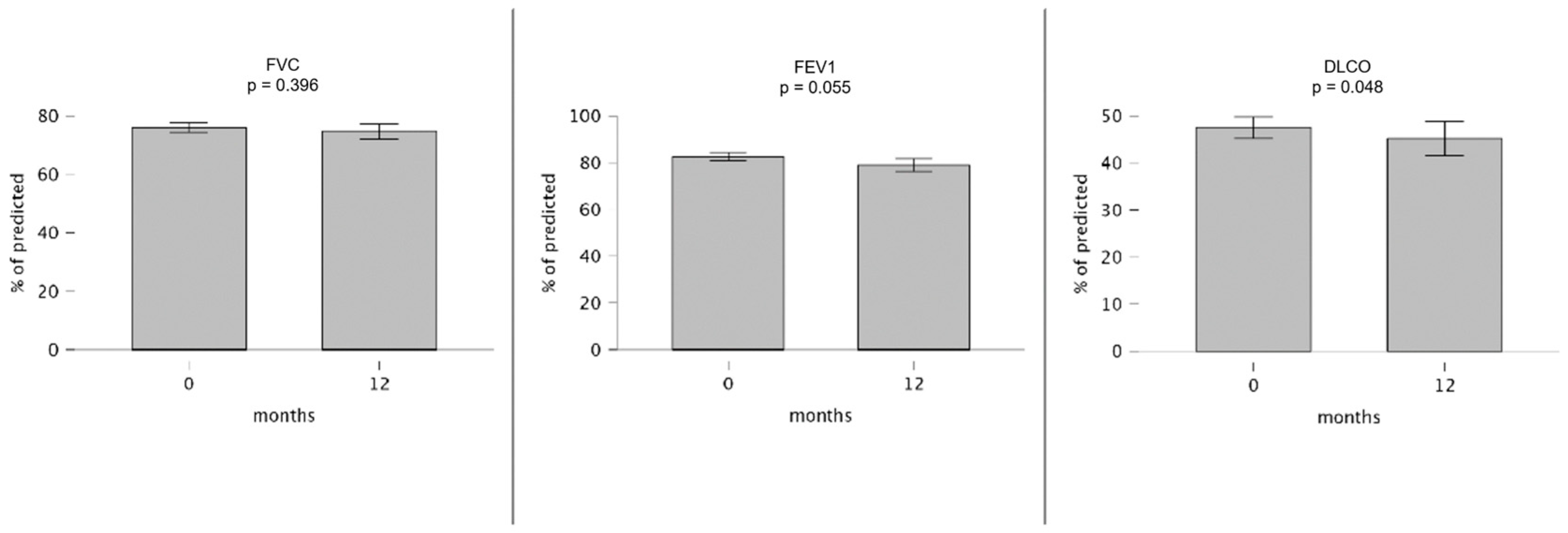
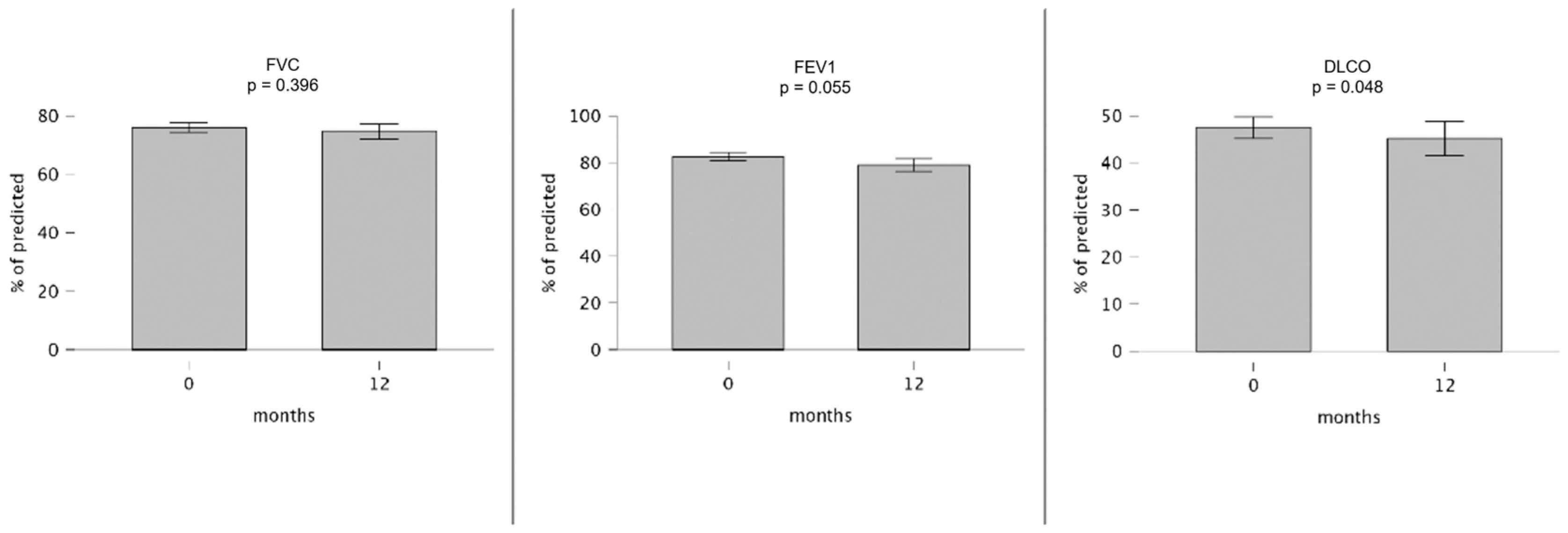
| 74.0 (62.5–82.5) | 39 | 73.0 (64.0–84.5) | 31 | 0.469 | 75.0 (64.0–82.5) | 35 | 76.0 (58.0–84.5) | 23 | 0.396 |
| FEV1 | |||||||||
| 81.0 (69.0–94.5) | 35 | 81.0 (67.5–90.5) | 27 | 0.949 | 81.0 (70.0–97.0) | 31 | 75.0 (69.5–86.8) | 18 | 0.055 |
| DLCO | |||||||||
| 43.0 (34.0–60.0) | 37 | 38.0 (31.0–48.8) | 26 | 0.041 | 44.0 (34.0–60.8) | 34 | 42.5 (32.5–57.8) | 22 | 0.048 |
| Baseline | n | 12 Months | n | p | |
|---|---|---|---|---|---|
| Full dose | |||||
| FVC | 74.0 (66.0–81.0) | 17 | 66.0 (61.0–82.0) | 11 | 0.975 |
| DLCO | 44.0 (35.0–58.5) | 18 | 44.0 (29.8–53.8) | 12 | 0.098 |
| Reduced dose | |||||
| FVC | 81.0 (68.0–87.5) | 10 | 81.0 (77.0–89.0) | 7 | 0.880 |
| DLCO | 46.0 (38.0–58.0) | 8 | 38.5 (37.3–50.3) | 6 | 0.084 |
| Suspended | |||||
| FVC | 70.0 (59.0–82.0) | 13 | 55.0 (52.8–66.3) | 8 | 0.035 |
| DLCO | 43.0 (31.8–50.5) | 12 | 37.5 (31.0–55.3) | 6 | 0.350 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruaro, B.; Salotti, A.; Reccardini, N.; Kette, S.; Da Re, B.; Nicolosi, S.; Zuccon, U.; Confalonieri, M.; Mondini, L.; Pozzan, R.; et al. Functional Progression after Dose Suspension or Discontinuation of Nintedanib in Idiopathic Pulmonary Fibrosis: A Real-Life Multicentre Study. Pharmaceuticals 2024, 17, 119. https://doi.org/10.3390/ph17010119
Ruaro B, Salotti A, Reccardini N, Kette S, Da Re B, Nicolosi S, Zuccon U, Confalonieri M, Mondini L, Pozzan R, et al. Functional Progression after Dose Suspension or Discontinuation of Nintedanib in Idiopathic Pulmonary Fibrosis: A Real-Life Multicentre Study. Pharmaceuticals. 2024; 17(1):119. https://doi.org/10.3390/ph17010119
Chicago/Turabian StyleRuaro, Barbara, Andrea Salotti, Nicolò Reccardini, Stefano Kette, Beatrice Da Re, Salvatore Nicolosi, Umberto Zuccon, Marco Confalonieri, Lucrezia Mondini, Riccardo Pozzan, and et al. 2024. "Functional Progression after Dose Suspension or Discontinuation of Nintedanib in Idiopathic Pulmonary Fibrosis: A Real-Life Multicentre Study" Pharmaceuticals 17, no. 1: 119. https://doi.org/10.3390/ph17010119
APA StyleRuaro, B., Salotti, A., Reccardini, N., Kette, S., Da Re, B., Nicolosi, S., Zuccon, U., Confalonieri, M., Mondini, L., Pozzan, R., Hughes, M., Confalonieri, P., & Salton, F. (2024). Functional Progression after Dose Suspension or Discontinuation of Nintedanib in Idiopathic Pulmonary Fibrosis: A Real-Life Multicentre Study. Pharmaceuticals, 17(1), 119. https://doi.org/10.3390/ph17010119