

Expanding the Analytical Toolbox: Developing New Lys-C Peptide Mapping Methods with Minimized Assay-Induced Artifacts to Fully Characterize Antibodies

Abstract
: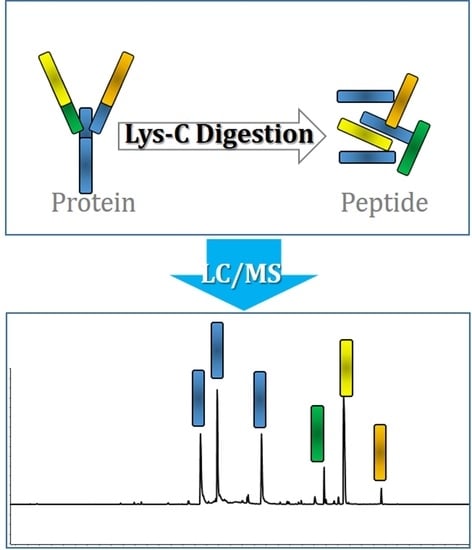
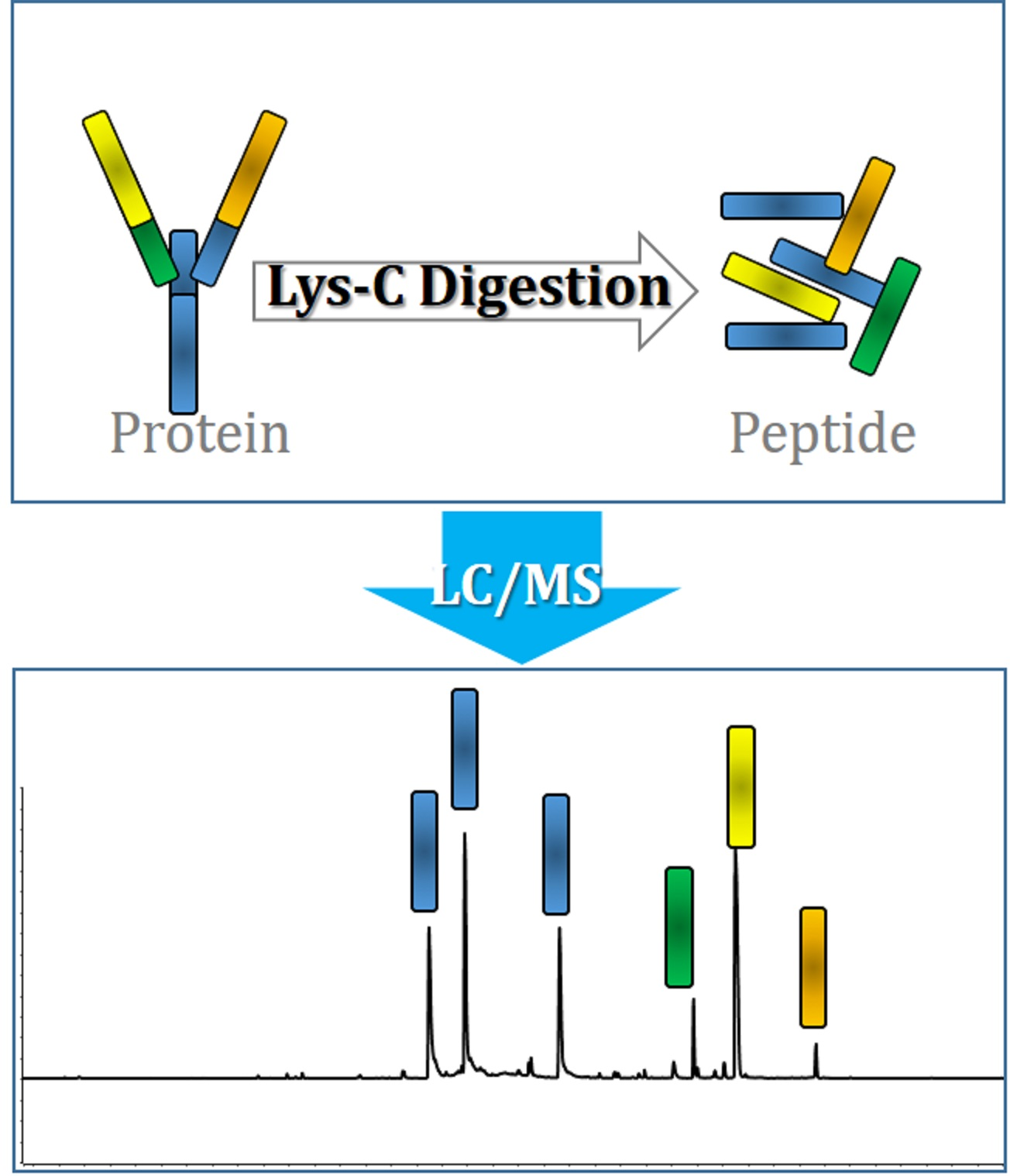
1. Introduction
2. Results and Discussion
2.1. Method Development: Lys-C Digestion Method 1
2.2. Method Development: Lys-C Digestion Method 2
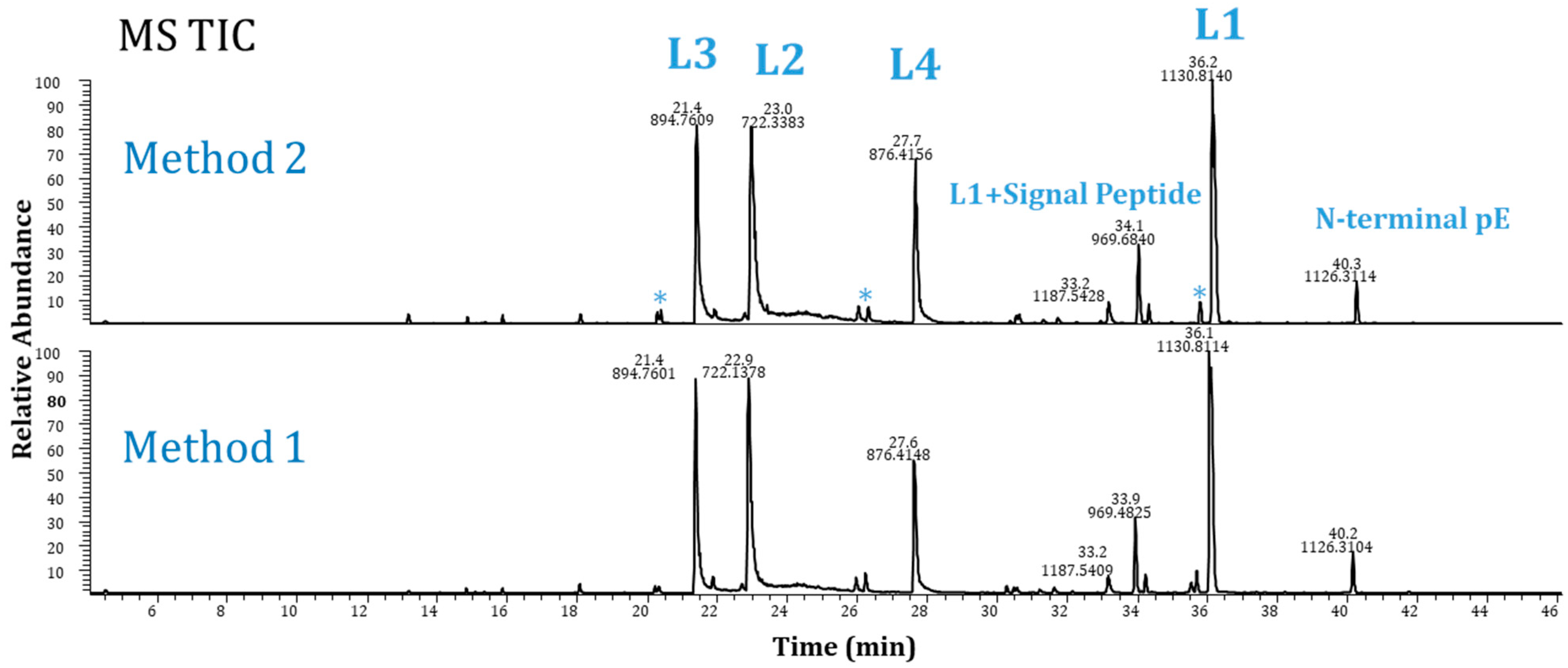
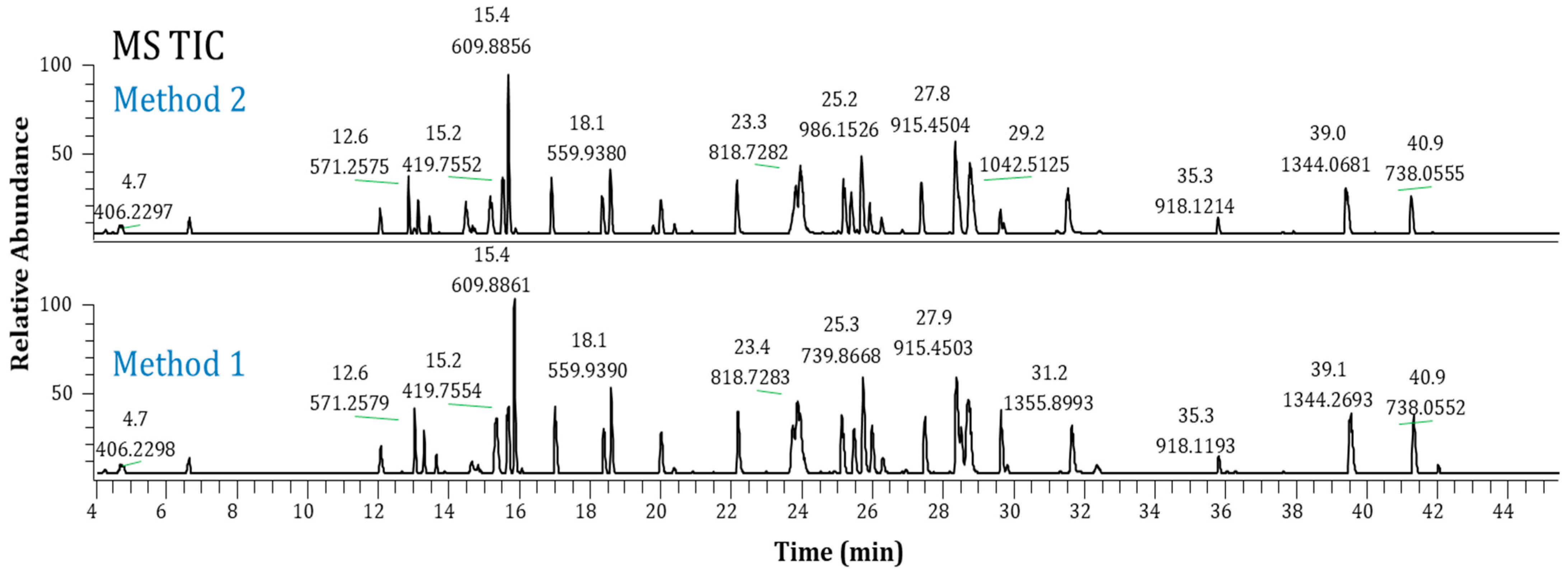
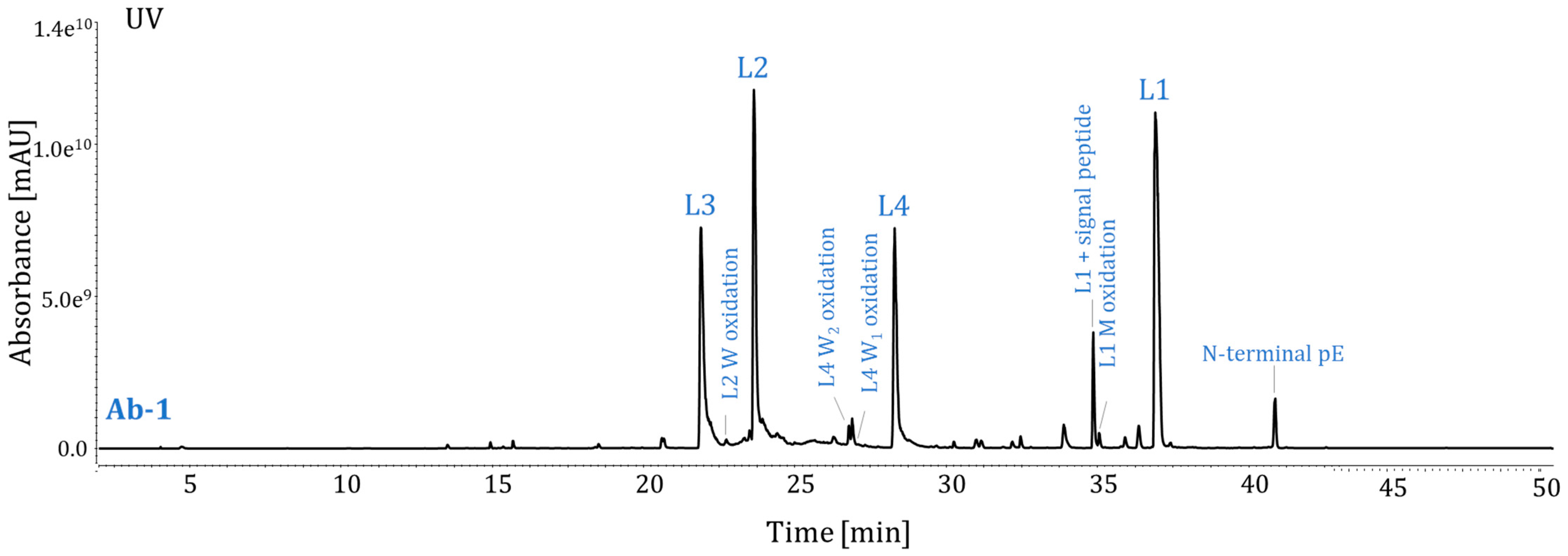
2.3. Method Performance Evaluation
2.4. Method Robustness
2.5. Suitability for Ab-1 Stability Study
3. Materials and Methods
3.1. Materials
3.2. Protein Digestion
3.2.1. Method 1: Lys-C Digestion for High-Concentration Samples (≥50 mg/mL)
3.2.2. Method 2: Lys-C Digestion for Low-Concentration Samples (≥3 mg/mL)
3.3. LC/MS Analysis
3.4. MS Data Analysis
3.5. Tryptic Peptide Map
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mullard, A. FDA approves 100th monoclonal antibody product. Nat. Rev. Drug Discov. 2021, 20, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Robotham, A.C.; Kelly, F.J. LC-MS characterization of antibody-based therapeutics: Recent highlights and future prospects, Approaches to the Purification. In Analysis and Characterization of Antibody-Based Therapeutics; Elsevier Limited: Amsterdam, The Netherlands, 2020; pp. 1–33. [Google Scholar]
- Grilo, A.L.; Mantalaris, A. The increasingly human and profitable monoclonal antibody market. Trends Biotechnol. 2019, 37, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Kaplon, H.; Muralidharan, M.; Schneider, Z.; Reichert, J.M. Antibodies to watch in 2020. mAbs 2020, 12, 1703531. [Google Scholar] [CrossRef] [PubMed]
- Kansy, M.; Caron, G. New therapeutic modalities in drug discovery and development: Insights & opportunities. ADMET DMPK 2021, 9, 227–230. [Google Scholar] [PubMed]
- Kaplon, H.; Chenoweth, A.; Crescioli, S.; Reichert, J.M. Antibodies to watch in 2022. mAbs 2022, 14, 2014296. [Google Scholar] [CrossRef]
- Vlasak, J.; Bussat, M.C.; Wang, S.; Wagner-Rousset, E.; Schaefer, M.; Klinguer-Hamour, C.; Kirchmeier, M.; Corvaïa, N.; Ionescu, R.; Beck, A. Identification and characterization of asparagine deamidation in the light chain CDR1 of a humanized IgG1 antibody. Anal. Biochem. 2009, 392, 145–154. [Google Scholar] [CrossRef]
- Bertolotti-Ciarlet, A.; Wang, W.; Lownes, R.; Pristatsky, P.; Fang, Y.; McKelvey, T.; Li, Y.; Li, Y.; Drummond, J.; Prueksaritanont, T.; et al. Impact of methionine oxidation on the binding of human IgG1 to Fc Rn and Fc gamma receptors. Mol. Immunol. 2009, 46, 1878–1882. [Google Scholar] [CrossRef]
- Pan, H.; Chen, K.; Chu, L.; Kinderman, F.; Apostol, I.; Huang, G. Methionine oxidation in human IgG2 Fc decreases binding affinities to protein A and FcRn. Protein Sci. 2009, 18, 424–433. [Google Scholar] [CrossRef]
- Liu, Y.D.; Chen, X.; Enk, J.Z.; Plant, M.; Dillon, T.M.; Flynn, G.C. Human IgG2 antibody disulfide rearrangement in vivo. J. Biol. Chem. 2008, 283, 29266–29272. [Google Scholar] [CrossRef]
- Wei, Z.; Feng, J.; Lin, H.Y.; Mullapudi, S.; Bishop, E.; Tous, G.I.; Casas-Finet, J.; Hakki, F.; Strouse, R.; Schenerman, M.A. Identification of a single tryptophan residue as critical for binding activity in a humanized monoclonal antibody against respiratory syncytial virus. Anal. Chem. 2007, 79, 2797–2805. [Google Scholar] [CrossRef]
- Wei, B.; Berning, K.; Quan, C.; Zhang, Y.T. Glycation of antibodies: Modification, methods and potential effects on biological functions. mAbs 2017, 9, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Goetze, A.M.; Liu, Y.D.; Zhang, Z.Q.; Shah, B.; Lee, E.; Bondarenko, P.V.; Flynn, G.C. High-mannose glycans on the Fc region of therapeutic IgG antibodies increase serum clearance in humans. Glycobiology 2011, 21, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, Y.D.; Flynn, G.C. The effect of Fc Glycan forms on human IgG2 antibody clearance in humans. Glycobiology 2009, 19, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.D.; Flynn, G.C. Effect of high mannose glycan pairing on antibody clearance. Biologicals 2016, 44, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, D.; Mason, B.; Rossomando, T.; Li, N.; Liu, D.; Cheung, J.K.; Xu, W.; Raghava, S.; Katiyar, A.; et al. Structure, heterogeneity and developability assessment of therapeutic antibodies. mAbs 2019, 11, 239–264. [Google Scholar] [CrossRef]
- Liu, Y.D.; Enk, J.Z.; Flynn, G.C. Human antibody Fc deamidation in vivo. Biologicals 2009, 37, 313–322. [Google Scholar] [CrossRef]
- Goetze, A.M.; Liu, Y.D.; Arroll, T.; Chu, L.; Flynn, G.C. Rates and impact of human antibody glycation in vivo. Glycobiology 2012, 22, 221–234. [Google Scholar] [CrossRef]
- Liu, Y.D.; Goetze, A.M.; Bass, R.B.; Flynn, G.C. N-terminal glutamate to pyroglutamate conversion in vivo. JBC 2011, 286, 11211–11217. [Google Scholar] [CrossRef]
- Liu, H.; Ponniah, G.; Zhang, H.-M.; Nowak, C.; Neill, A.; Gonzalez-Lopez, N.; Patel, R.; Cheng, G.; Kita, A.Z.; Andrien, B. In vitro and in vivo modifications of recombinant and human IgG antibodies. mAbs 2014, 6, 1145–1154. [Google Scholar] [CrossRef]
- Liu, Y.D.; Cadang, L.; Bol, K.; Pan, X.; Tschudi, K.; Jazayri, M.; Camperi, J.; Michels, D.; Stults, J.; Harris, R.J.; et al. Challenges and strategies for a thorough characterization of antibody acidic charge variants. Bioengineering 2022, 9, 641. [Google Scholar] [CrossRef]
- Goetze, A.M.; Schenauer, M.R.; Flynn, G.C. Assessing monoclonal antibody product quality attribute criticality through clinical studies. mAbs 2010, 2, 500–507. [Google Scholar] [CrossRef]
- Rogstad, S.; Yan, H.; Wang, X.; Powers, D.; Brorson, K.; Damdinsuren, B.; Lee, S. Multi-attribute method for quality control of therapeutic proteins. Anal. Chem. 2019, 91, 14170–14177. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.E.; Dubois, H.; Hoffmann, M.; Eri, S.D.; Fromentin, Y.; Wiesner, J.; Pfenninger, A.; Clavier, S.; Pieper, A.; Duhau, L.; et al. Automated mass spectrometry multi-attribute method analyses for process development and characterization of mAbs. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2021, 1166, 122540. [Google Scholar] [CrossRef] [PubMed]
- Tajiri-Tsukada, M.; Hashii, N.; Ishii-Watabe, A. Establishment of a highly precise multi-attribute method for the characterization and quality control of therapeutic monoclonal antibodies. Bioengineered 2020, 11, 984–1000. [Google Scholar] [CrossRef] [PubMed]
- Ren, D. An improved trypsin digestion method minimizes digestion-induced modifications on proteins. Anal. Biochem. 2009, 392, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Sadek, M.; Moore, B.N.; Yu, C.; Ruppe, N.; Abdun-Nabi, A.; Hao, Z.; Alvarez, M.; Dahotre, S.; Deperalta, G. A robust purity method for biotherapeutics using new peak detection in an LC–MS-based multi-attribute method. J. Am. Soc. Mass Spectrom. 2023, 34, 484–492. [Google Scholar] [CrossRef]
- Giansanti, P.; Tsiatsiani, L.; Low, T.Y.; Heck, A.J. Six alternative proteases for mass spectrometry-based proteomics beyond trypsin. Nat. Protoc. 2016, 11, 993–1006. [Google Scholar] [CrossRef]
- Buettner, A.; Maier, M.; Bonnington, L.; Bulau, P.; Reusch, D. Multi-attribute monitoring of complex erythropoietin beta glycosylation by GluC liquid chromatography–mass spectrometry peptide mapping. Anal. Chem. 2020, 92, 7574–7580. [Google Scholar] [CrossRef]
- Li, W.; Yang, B.; Zhou, D.; Xu, J.; Ke, Z.; Suen, W.C. Discovery and characterization of antibody variants using mass spectrometry-based comparative analysis for biosimilar candidates of monoclonal antibody drugs. J. Chromatogr. B 2016, 1025, 57–67. [Google Scholar] [CrossRef]
- Li, X.; Rawal, B.; Rivera, S.; Letarte, S.; Richardson, D.D. Improvements on sample preparation and peptide separation for reduced peptide mapping based multi-attribute method analysis of therapeutic monoclonal antibodies using lysyl endopeptidase digestion. J Chromatogr. A 2022, 1675, 463161. [Google Scholar] [CrossRef]
- Li, X.; Pierson, N.A.; Hua, X.; Patel, B.A.; Olma, M.H.; Strulson, C.A.; Letarte, S.; Richardson, D.D. Analytical Performance Evaluation of Identity, Quality-Attribute Monitoring and new Peak Detection in a Platform Multi-Attribute Method Using Lys-C Digestion for Characterization and Quality Control of Therapeutic Monoclonal Antibodies. J. Pharm. Sci. 2023, 112, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Lundell, N.; Schreitmuller, T. Sample preparation for peptide mapping—A pharmaceutical quality-control perspective. Anal. Biochem. 1999, 266, 31–47. [Google Scholar] [CrossRef] [PubMed]
- Suttapitugsakul, S.; Xiao, H.; Smeekens, J.; Wu, R. Evaluation and optimization of reduction and alkylation methods to maximize peptide identification with MS-based proteomics. Mol. Biosyst. 2017, 13, 2574–2582. [Google Scholar] [CrossRef] [PubMed]
- Hutterer, K.M.; Hong, R.W.; Lull, J.; Zhao, X.; Wang, T.; Pei, R.; Le, M.E.; Borisov, O.; Piper, R.; Liu, Y.D.; et al. Monoclonal antibody disulfide reduction during manufacturing: Untangling process effects from product effects. mAbs 2013, 5, 608–613. [Google Scholar] [CrossRef]
- Jekel, P.A.; Weijer, W.J.; Beintema, J.J. Use of endoproteinase Lys-C from Lysobacter enzymogenes in protein sequence analysis. Anal. Biochem. 1983, 134, 347–354. [Google Scholar] [CrossRef]
- Poulsen, J.W.; Madsen, C.T.; Young, C.; Poulsen, F.M.; Nielsen, M.L. Using guanidine-hydrochloride for fast and efficient protein digestion and single-step affinity-purification mass spectrometry. J. Proteome Res. 2013, 12, 1020–1030. [Google Scholar] [CrossRef]
- Liu, Y.D.; Chen, Y.; Tsui, G.; Wei, B.; Yang, F.; Yu, C.; Cornell, C. Predictive in vitro vitreous and serum models to assess thiol-related quality attributes in protein therapeutics. Anal. Chem. 2020, 92, 6869–6876. [Google Scholar] [CrossRef]
- Yang, F.; Zhang, J.; Buettner, A.; Vosika, E.; Sadek, M.; Hao, Z.; Reusch, D.; Koenig, M.; Chan, W.; Bathke, A.; et al. Mass spectrometry-based multi-attribute method in protein therapeutics product quality monitoring and quality control. mAbs 2023, 15, 2197668. [Google Scholar] [CrossRef]
- Hao, Z.; Moore, B.; Ren, C.; Sadek, M.; Macchi, F.; Yang, L.; Harris, J.; Yee, L.; Liu, E.; Tran, V.; et al. Multi-Attribute Method Performance Profile for Quality Control of Monoclonal Antibody Therapeutics. J. Pharm. Biomed. Anal. 2021, 205, 114330. [Google Scholar] [CrossRef]

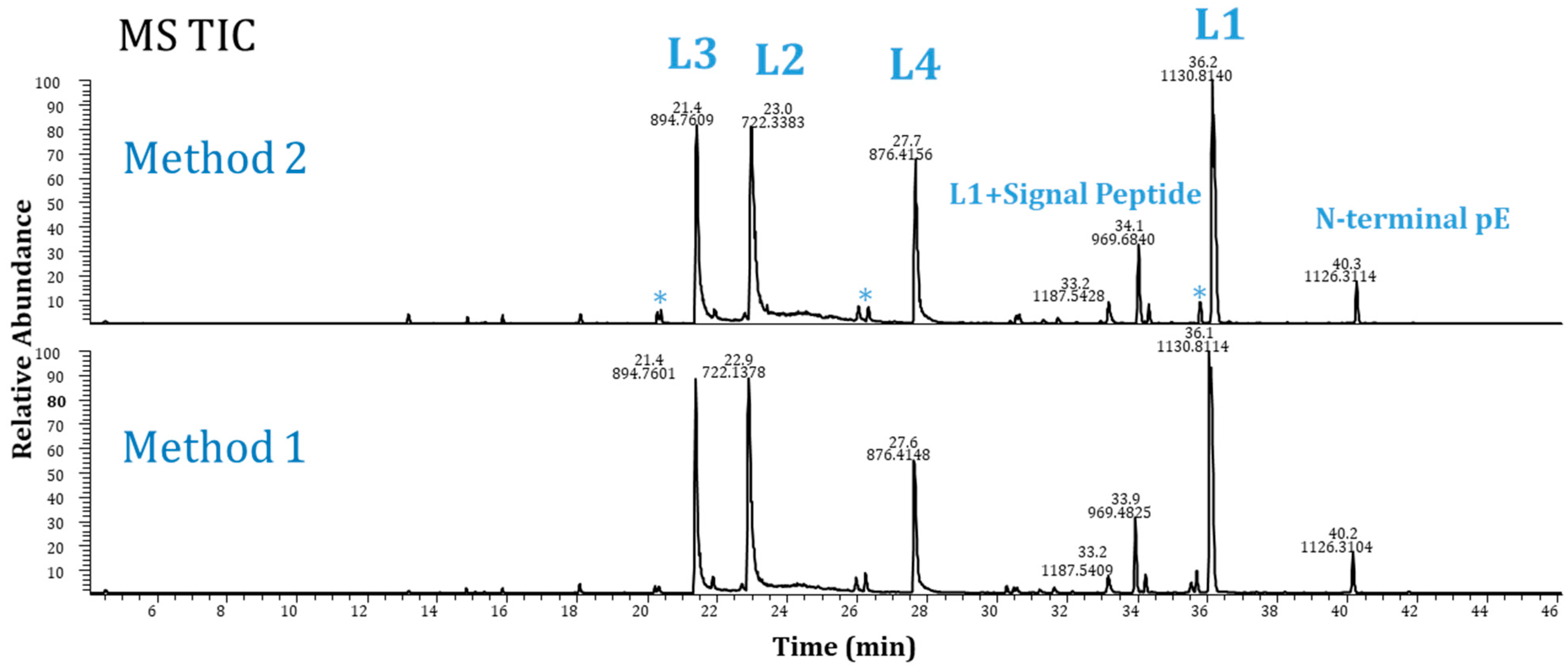
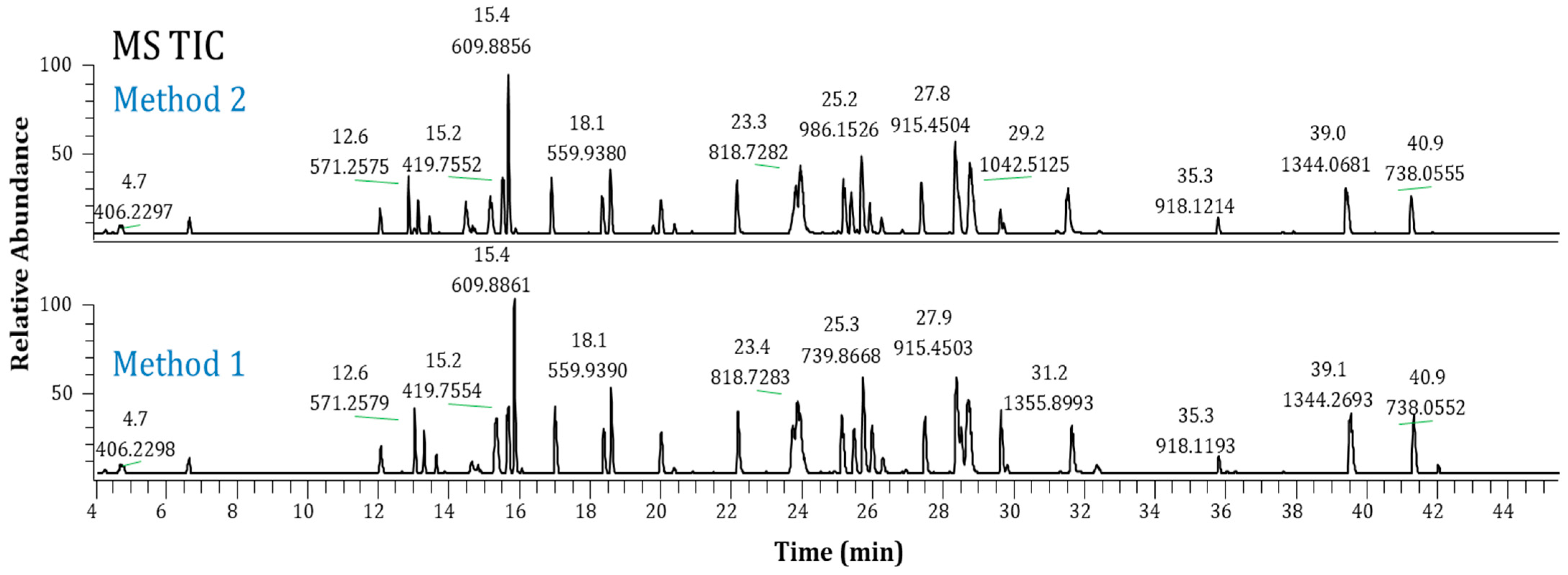
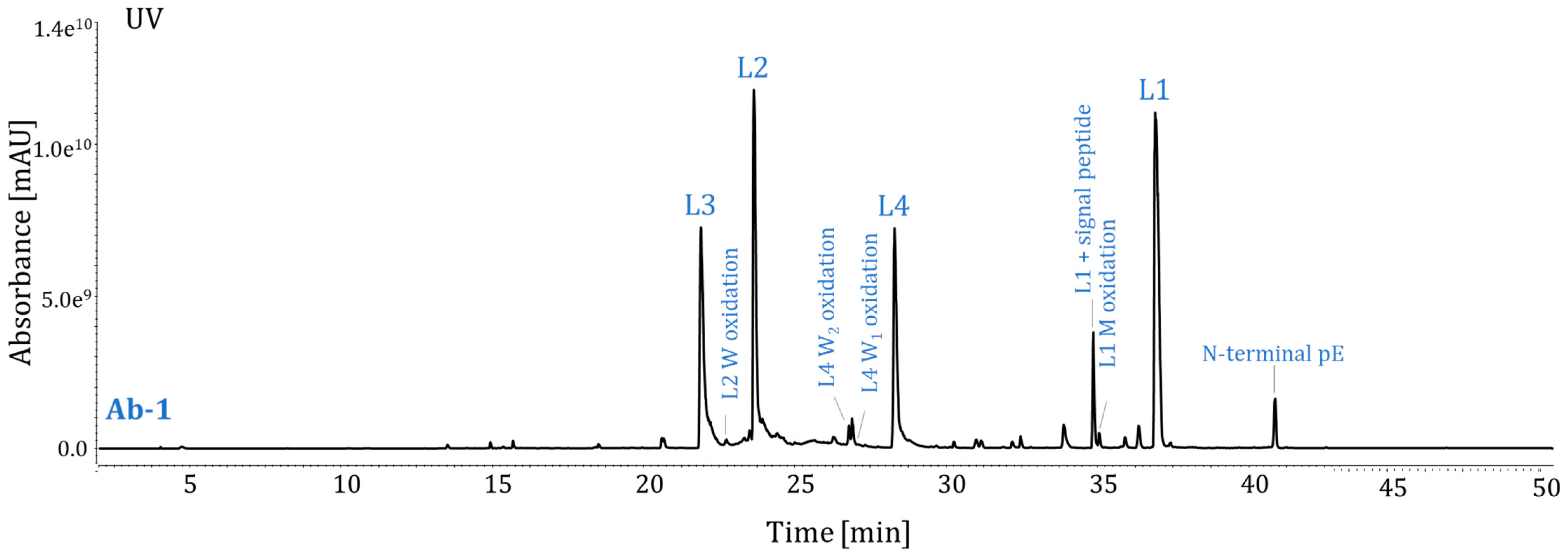

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Test Condition a | Note | |
|---|---|---|---|
| Reduction | DTT (mM) | 3.5, 5 | 6 M GuHCl for protein denaturation at 37 °C. Use Met (10–20 mM) as a scavenger throughout the procedure to reduce artificial oxidation |
| Buffer pH | 7, 7.5, 8 | ||
| Incubation time (min) | 30, 45, 60, 90 | ||
| Alkylation | IAM (mM) | 6.8, 8.5 | Alkylation at 37 °C in the dark |
| Incubation time (min) | 15, 30 | ||
| Dilution |
| Dilution to lower GuHCl level (≤2 M) in digestion and more dilution (<1 M) before LC/MS injection | |
| Digestion | Lys-C enzyme-to-protein ratio | 1:20, 1:10 | Digestion at pH 7 to minimize artificial deamidation Digestion temperature: 37 °C |
| Incubation time | 30 min, 1 h, 2 h | ||
| Method 1 | Reagent | Concentration | Method 2 | Reagent | Concentration | |
|---|---|---|---|---|---|---|
| Reduction | 8 M GuHCl 85 µL | GuHCl | 6.0 M | 8 M GuHCl 67 µL | GuHCl | 4.7 M |
| 1 M Tris pH 7.5/0.2 M Met 6 µL | Tris HCl | 0.05 M | 1 M Tris HCl pH 7.5/0.2 M Met/65 mM DTT 6 µL | Tris HCl | 0.05 M | |
| Met | 11 mM | Met | 11 mM | |||
| 40 mM DTT 10 µL | DTT | 3.5 mM | DTT | 3.5 mM | ||
| 50 mg/mL Ab–1 12 µL | protein | 5.1 mg/mL | 3 mg/mL Ab–1 40 µL | protein | 1.0 mg/mL | |
| Incubate at 37 °C for 1 h | Incubate at 37 °C for 1 h | |||||
| Alkylation | 200 mM IAM 5 µL | IAM | 8.5 mM | 200 mM IAM 5 µL | IAM | 8.5 mM |
| Incubate at 37 °C for 15 min in the dark | Incubate at 37 °C for 15 min in the dark | |||||
| Digestion | Take 25 µLof the alkylated sample (125 µg) into a new tube | Take 68 µL of the reduced sample (68 µg) into a new tube | ||||
| Reduced sample 25 µL | protein | 1.5 mg/mL | Reduced sample 68 µL | protein | 0.4 mg/mL | |
| Add H2O 39 µL (GuHCl conc. < 2 M) | GuHCl | 1.8 M | Add H2O 74 µL (GuHCl conc. < 2 M) | GuHCl | 2.0 M | |
| 1 M Tris pH 7/0.2 M Met 6 µL | Tris HCl | 0.1 M | 1 M Tris HCl pH 7/0.2 M Met 12 µL | Tris HCl | 0.1 M | |
| Met | 18 mM | Met | 19 mM | |||
| 0.5 mg/mL Lys-C solution 13 µL | Lys-C: protein | 1:20 (w:w) | 0.5 mg/mL Lys-C solution 7 µL | Lys-C: protein | 1:20 (w:w) | |
| Incubate at 37 °C for 2 h | Incubate at 37 °C for 2 h | |||||
| Dilution & Quench | Dilution (3×) | protein | 0.5 mg/mL | Pipette 79 µL digested sample, dilute 3× | protein | 0.2 mg/mL |
| Add H2O 140 µL (GuHCl conc. < 1 M) | GuHCl | 0.6 M | Add H2O 120 µL (GuHCl conc. < 1 M) | GuHCl | 0.7 M | |
| Add 20 µL 0.2 M Met | Met | 16 mM | Add20 µL 0.2 M Met | Met | 18 mM | |
| Add 10% TFA 5 µL to quench digestion | TFA | 0.2% | Add 10%TFA 5 µL to quench digestion | TFA | 0.2% |
| DoE Factor | Condition Specifications | DoE Level (Low/High) |
|---|---|---|
| Digest buffer (pH) a | 7.0 ± 0.1 | 6.9/7.1 |
| Digest time (min) a | 120 ± 10 | 110/130 |
| Digest temperature (°C) a | 37 ± 2 | 35/39 |
| Reduction buffer (pH) a | 7.5 ± 0.1 | 7.4/7.6 |
| Injection volume (µL) b | 5 for Method 1 | 4/6 |
| 17 for Method 2 | 15/19 |
| Statistics | Method 1 | Method 2 | ||
|---|---|---|---|---|
| Signal Peptide (%) | N-Terminal pE (%) | Signal Peptide (%) | N-Terminal pE (%) | |
| Max | 13.2 | 7.1 | 13.9 | 7.3 |
| Min | 11.7 | 6.3 | 11.9 | 6.1 |
| Difference (Max-Min) | 1.5 | 0.8 | 2.0 | 1.2 |
| Average | 12.5 | 6.7 | 12.8 | 6.7 |
| SD | 0.4 | 0.2 | 0.5 | 0.3 |
| % RSD | 3.3% | 2.8% | 4.2% | 5.1% |
| Attribute | Relative Abundance (%) | ||||||
|---|---|---|---|---|---|---|---|
| t = 0 | 4 Weeks at 40 °C | 2 Weeks at 50 °C | pH Control | pH 3.25 | AAPH Control | 10 mM AAPH | |
| L1 + Signal peptide | 9.6 | 9.0 | 9.3 | 10.3 | 10.2 | 10.6 | 10.0 |
| N-terminal pE | 5.7 | 7.3 | 9.5 | 6.4 | 7.0 | 7.3 | 6.7 |
| L1 M oxidation | 2.3 | 2.3 | 2.2 | 2.2 | 2.1 | 2.2 | 2.1 |
| L1 W oxidation | <0.1 | <0.1 | <0.1 | <0.1 | <0.1 | <0.1 | <0.1 |
| L2 W oxidation | <0.1 | ND | ND | 0.1 | 0.2 | 0.1 | 1.1 |
| L3 N1 deamidation | ND | ND | ND | ND | ND | ND | ND |
| L3 N2 deamidation | ND | ND | ND | ND | ND | ND | ND |
| L4 W1 oxidation | 0.3 | 0.3 | 0.4 | 0.3 | 0.2 | 0.3 | 3.7 |
| L4 W2 oxidation | 1.3 | 1.0 | 1.3 | 0.9 | 0.8 | 0.9 | 11.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.D.; Beardsley, M.I.; Yang, F. Expanding the Analytical Toolbox: Developing New Lys-C Peptide Mapping Methods with Minimized Assay-Induced Artifacts to Fully Characterize Antibodies. Pharmaceuticals 2023, 16, 1327. https://doi.org/10.3390/ph16091327
Liu YD, Beardsley MI, Yang F. Expanding the Analytical Toolbox: Developing New Lys-C Peptide Mapping Methods with Minimized Assay-Induced Artifacts to Fully Characterize Antibodies. Pharmaceuticals. 2023; 16(9):1327. https://doi.org/10.3390/ph16091327
Chicago/Turabian StyleLiu, Y. Diana, Michelle Irwin Beardsley, and Feng Yang. 2023. "Expanding the Analytical Toolbox: Developing New Lys-C Peptide Mapping Methods with Minimized Assay-Induced Artifacts to Fully Characterize Antibodies" Pharmaceuticals 16, no. 9: 1327. https://doi.org/10.3390/ph16091327
APA StyleLiu, Y. D., Beardsley, M. I., & Yang, F. (2023). Expanding the Analytical Toolbox: Developing New Lys-C Peptide Mapping Methods with Minimized Assay-Induced Artifacts to Fully Characterize Antibodies. Pharmaceuticals, 16(9), 1327. https://doi.org/10.3390/ph16091327