
Imidazopyridine-Based Thiazole Derivatives as Potential Antidiabetic Agents: Synthesis, In Vitro Bioactivity, and In Silico Molecular Modeling Approach


 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Results
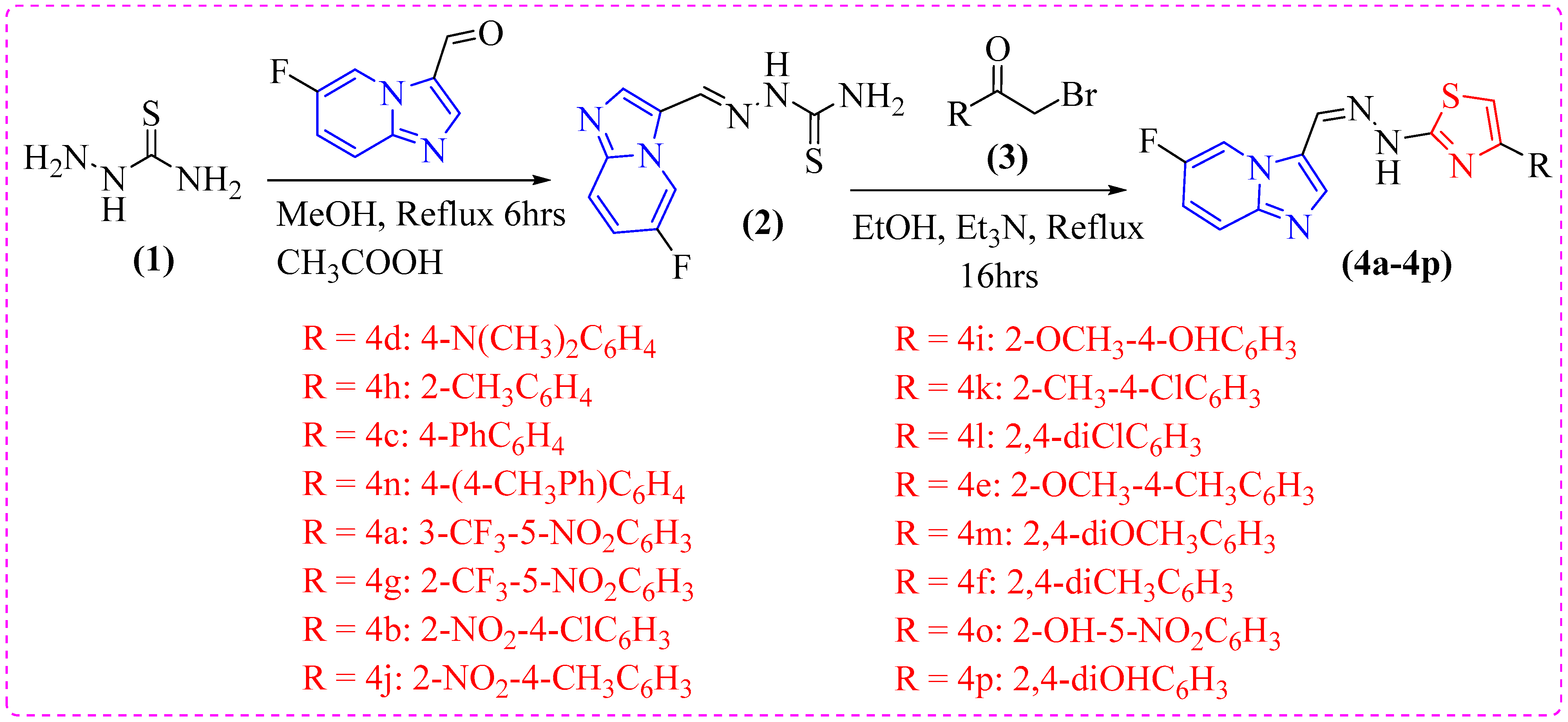
2.1. Chemistry
2.2. Biological Analysis (4a-p)
α-Glucosidase Inhibitory Activity
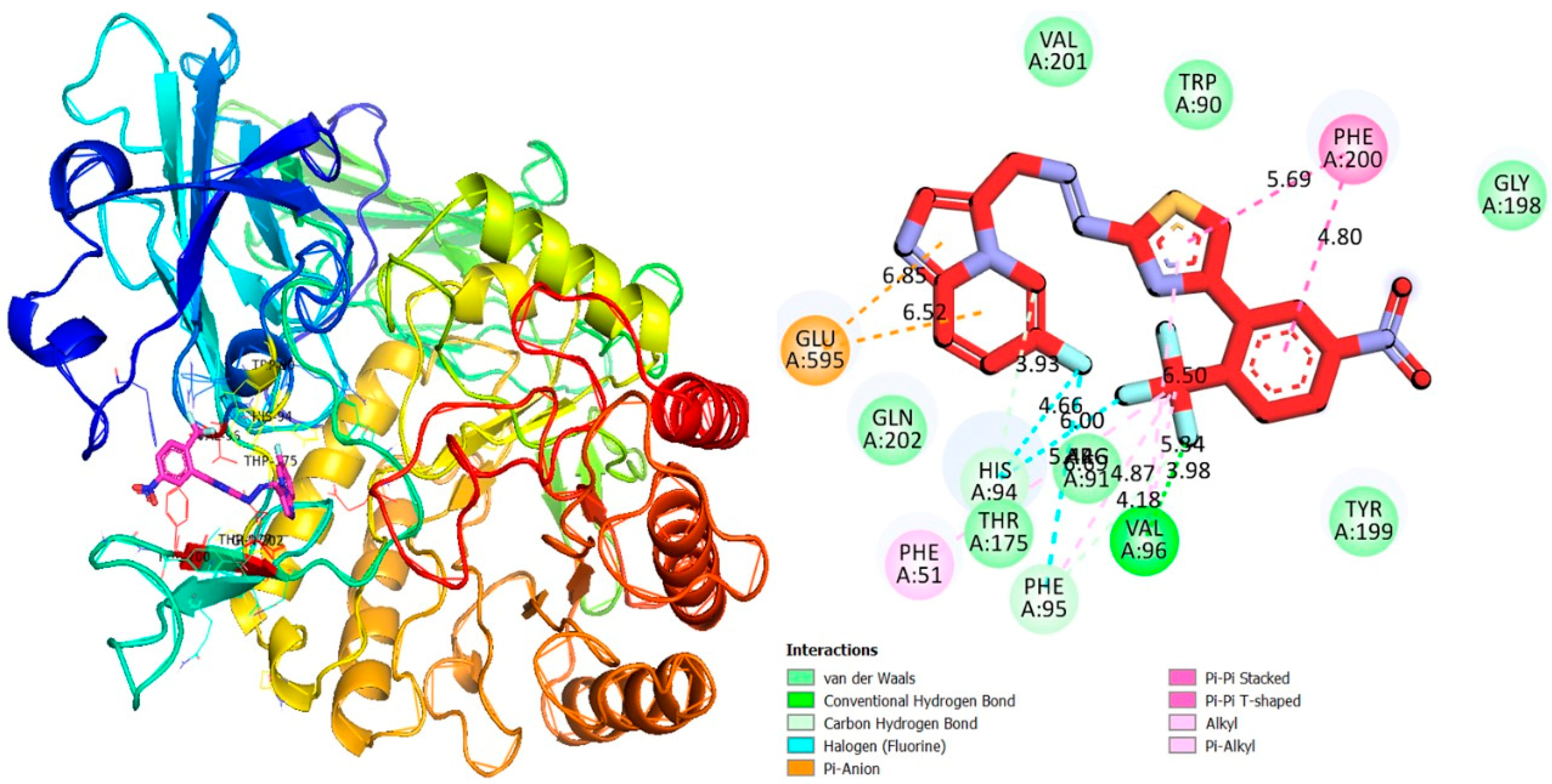
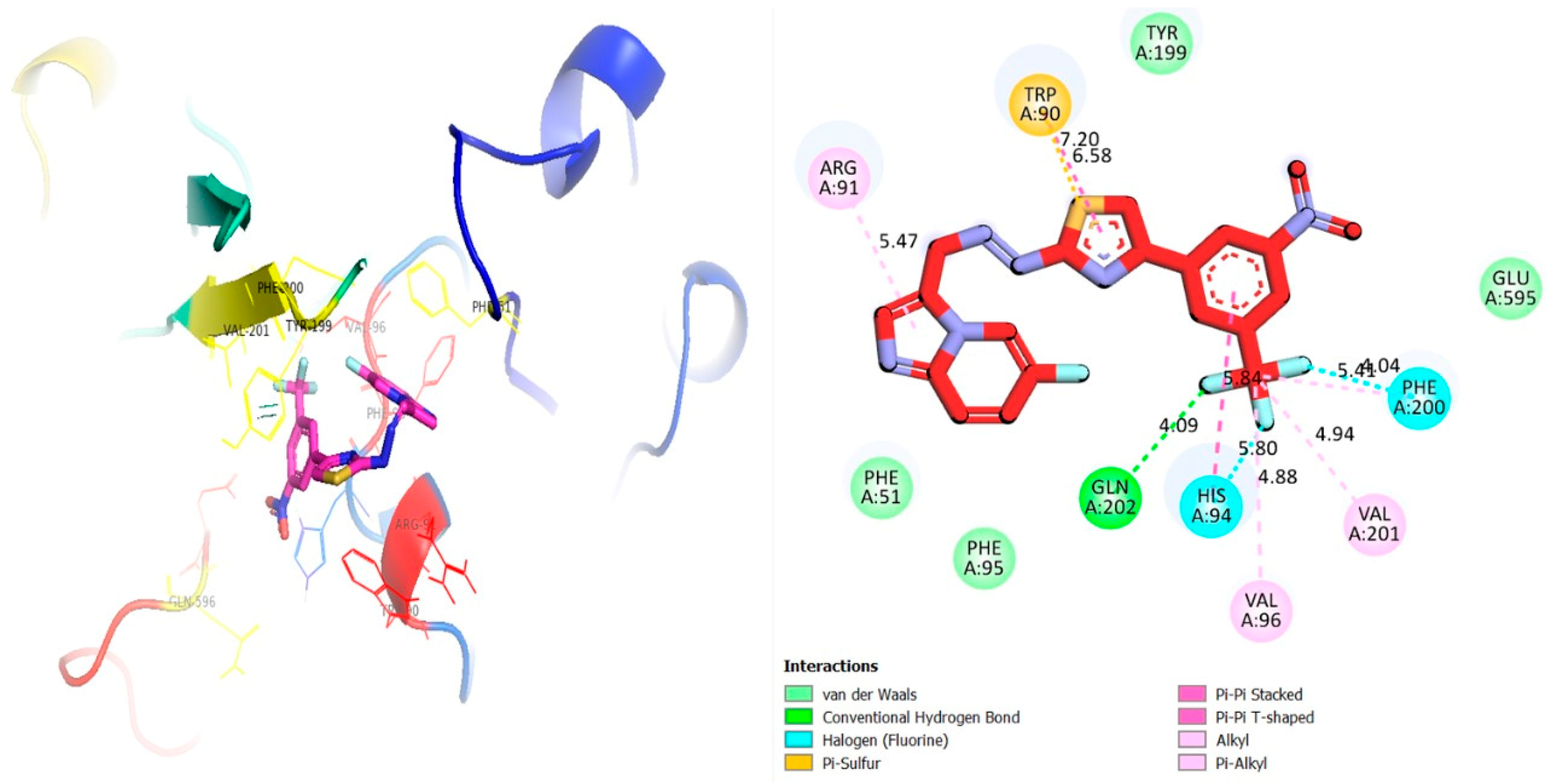
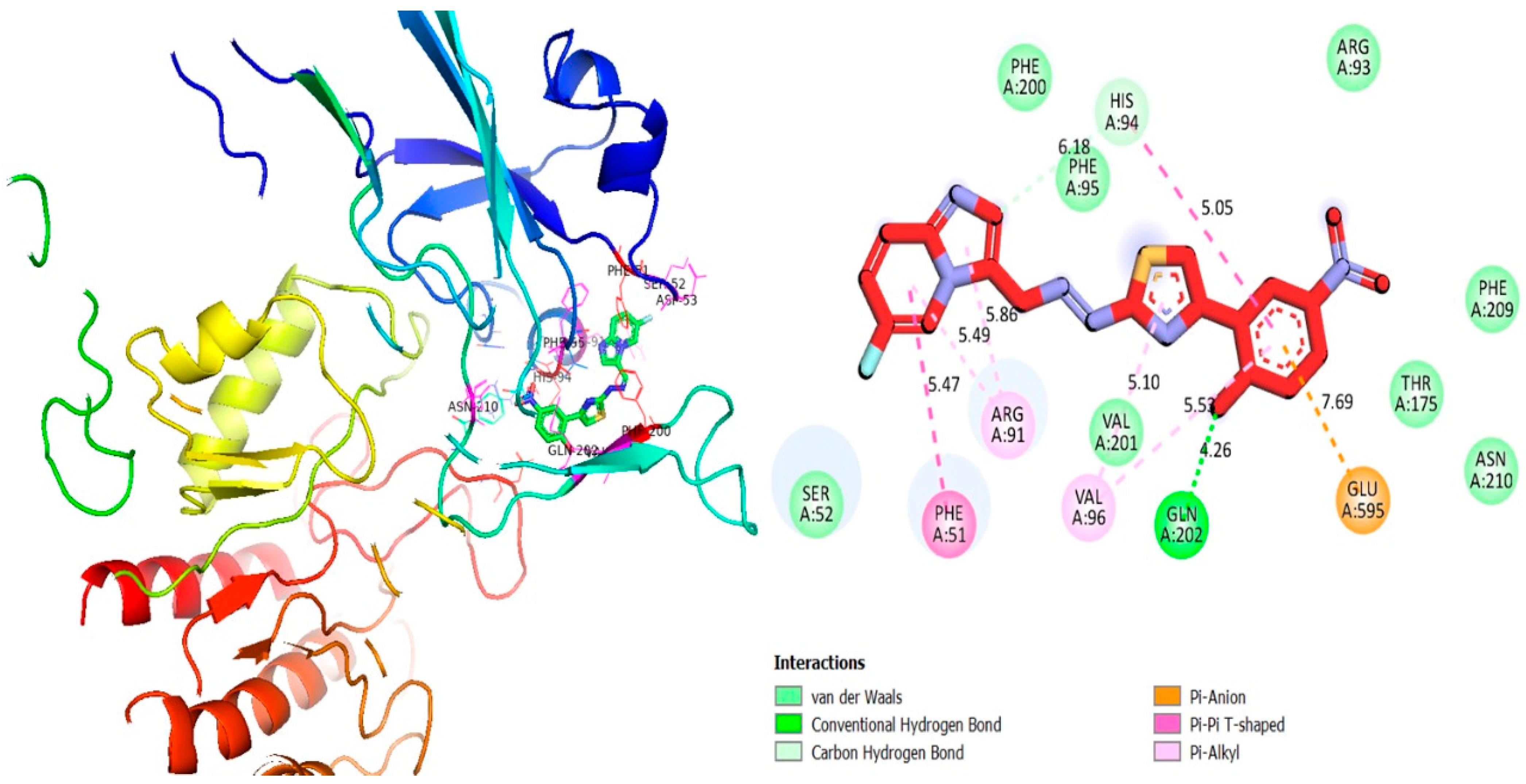
2.3. Molecular Docking Study
3. Discussion
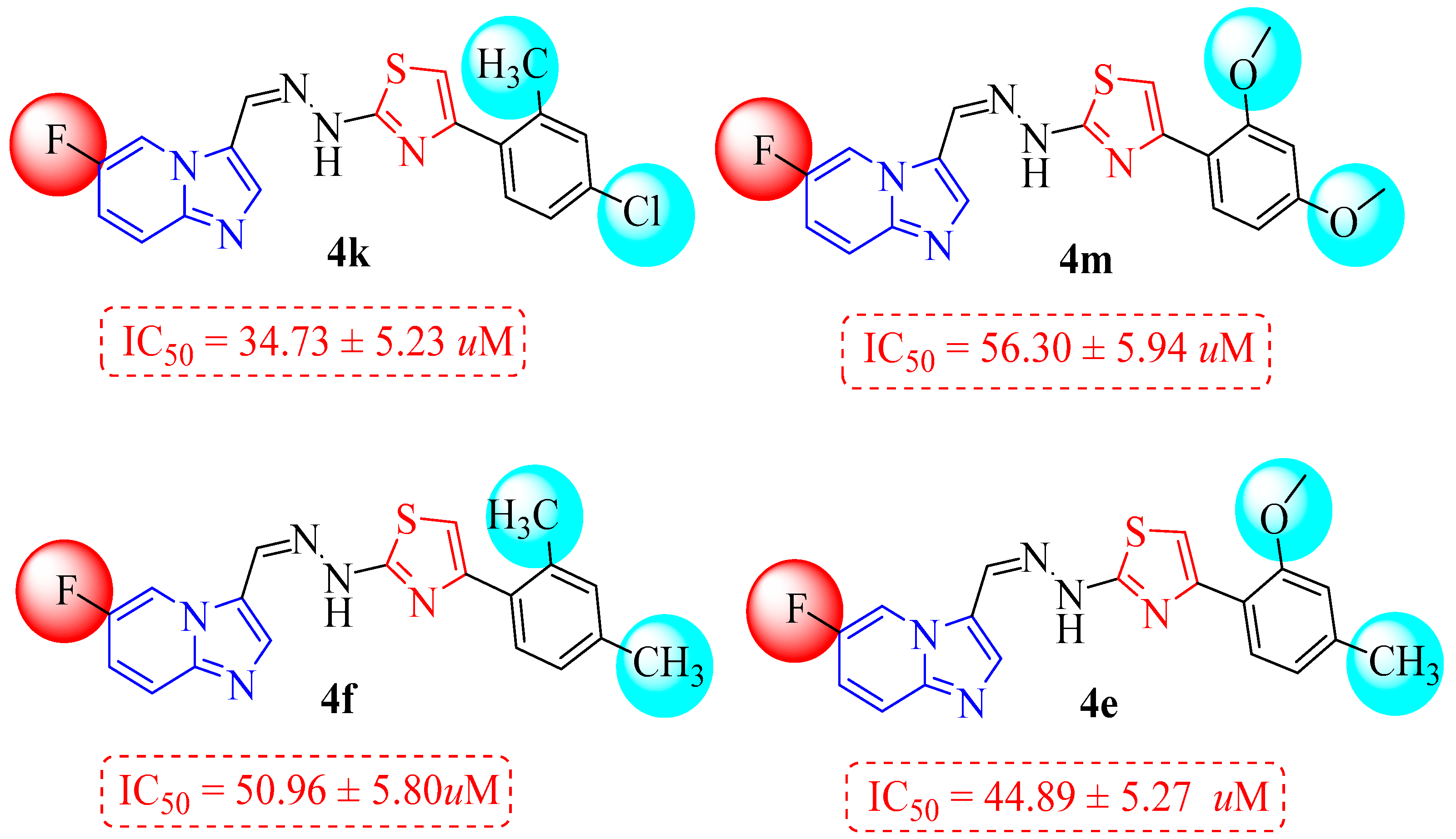
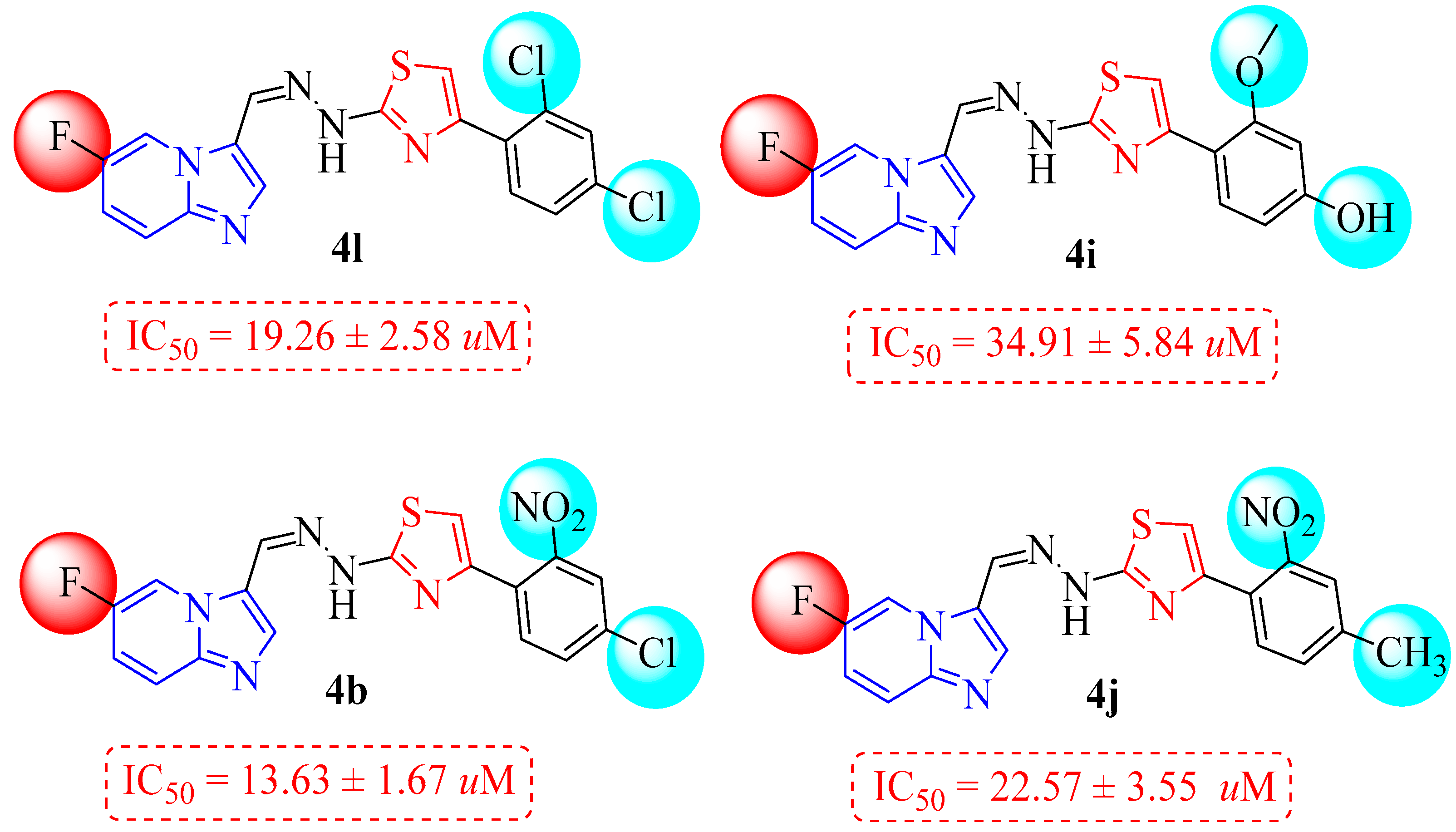
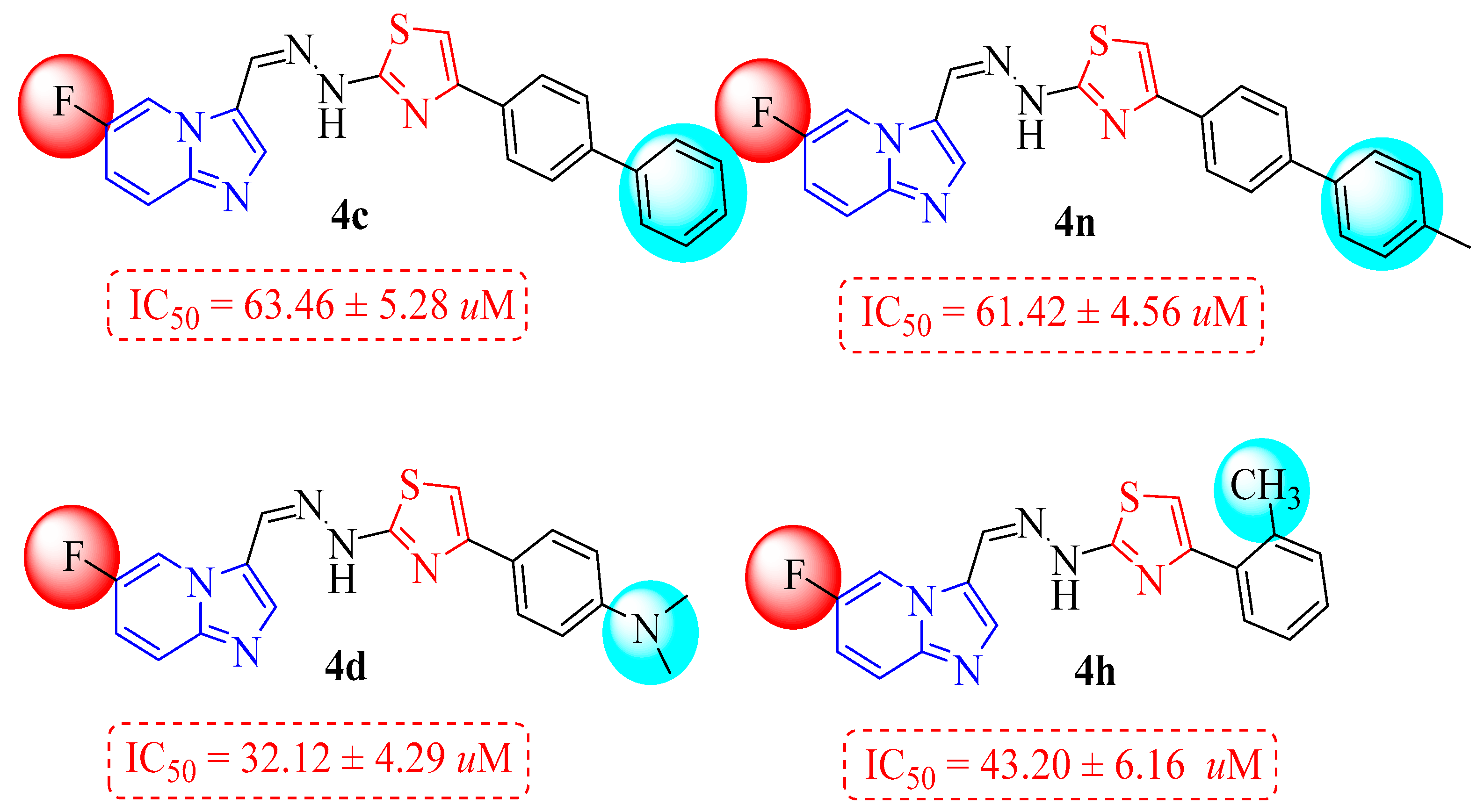
3.1. Structure–Activity Relationship (SAR) Study for α-Glucosidase Inhibitory Profile
3.2. Molecular Docking Study
3.2.1. Discussion
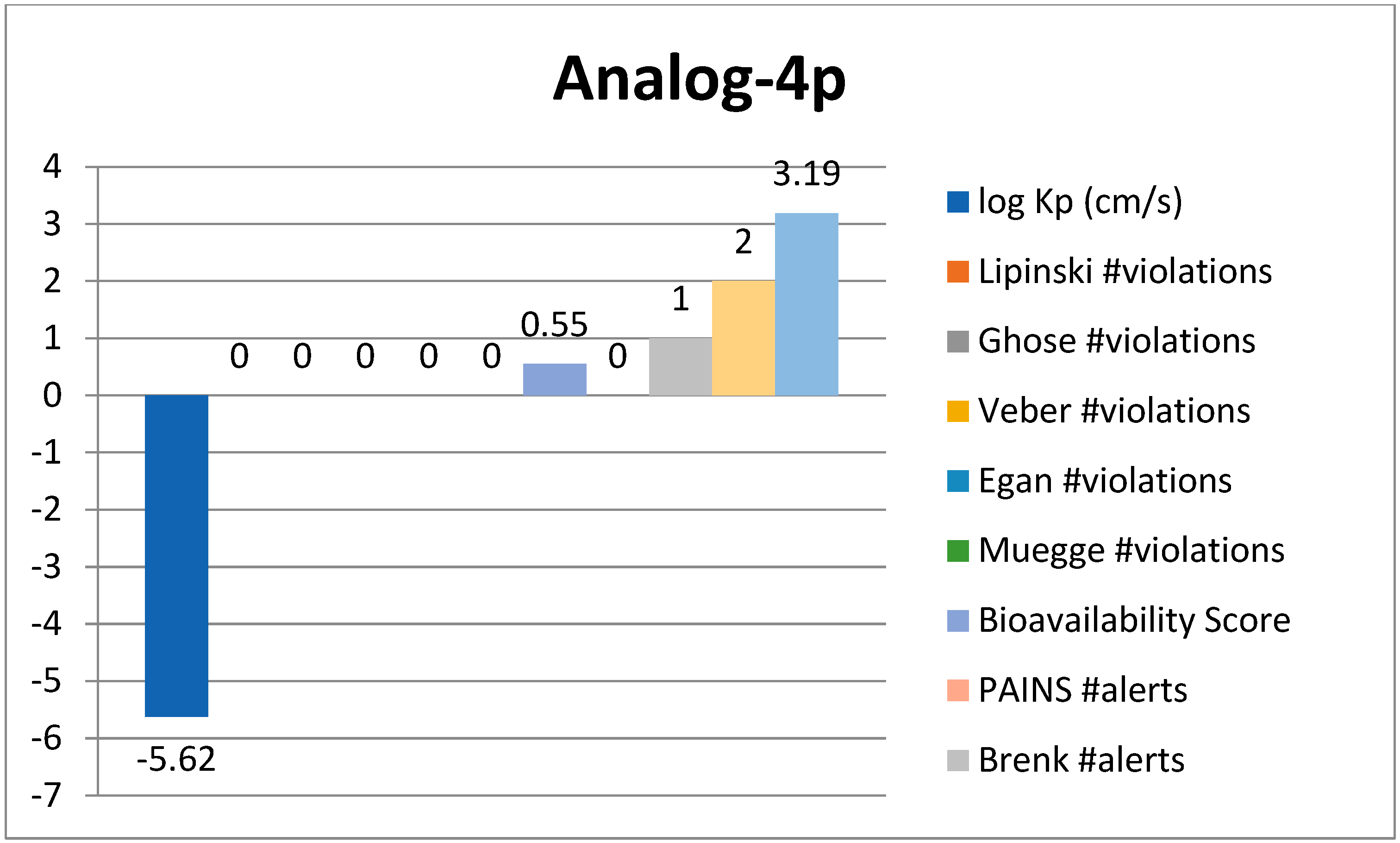
3.2.2. ADME Analysis
4. Materials and Methods
4.1. General Information
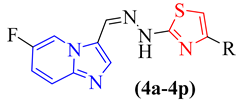
4.2. General Procedure for Synthesis of Imidazopyridine-Based Thiazole Analogs (4a-p)
4.3. Spectral Analysis (Provided in Supplementary Information)
4.3.1. Assay Protocol for α-Glucosidase Inhibition
4.3.2. Assay Protocol for Molecular Docking Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Association, A.D. 5. Prevention or delay of type 2 diabetes: Standards of medical care in diabetes-2018. Diabetes Care 2018, 41, S51–S54. [Google Scholar] [CrossRef]
- Javid, M.T.; Rahim, F.; Taha, M.; Rehman, H.U.; Nawaz, M.; Imran, S.; Uddin, I.; Mosaddik, A.; Khan, K.M. Synthesis, in vitro α-glucosidase inhibitory potential and molecular docking study of thiadiazole analogs. Bioorg. Chem. 2018, 78, 201–209. [Google Scholar] [CrossRef]
- Swaroopa, P.; Code, Q. Review on antidiabetic activity on medicinal plants. Int. J. Pharmacol. Res. 2017, 7, 230–235. [Google Scholar]
- Xu, X.; Wang, G.; Zhou, T.; Chen, L.; Chen, J.; Shen, X. Novel approaches to drug discovery for the treatment of type 2 diabetes. Expert Opin. Drug Discov. 2014, 9, 1047–1058. [Google Scholar] [CrossRef]
- Prabhakar, P.K.; Doble, M. Mechanism of action of natural products used in the treatment of diabetes mellitus. Chin. J. Integr. Med. 2011, 17, 563–574. [Google Scholar] [CrossRef]
- Bell, D.S. Type 2 diabetes mellitus: What is the optimal treatment regimen? Am. J. Med. 2004, 116, 23–29. [Google Scholar] [CrossRef]
- Rakesh, K.; Darshini, N.; Shubhavathi, T.; Mallesha, N. Biological Applications of Quinazolinone Analogues A Review. Org. Med. Chem. Int. J. 2017, 2, 41–45. [Google Scholar]
- Reddy, M.M.; Sivaramakrishna, A. Remarkably flexible quinazolinones—Synthesis and biological applications. J. Heterocycl. Chem. 2020, 57, 942–954. [Google Scholar] [CrossRef]
- Hussain, R.; Ullah, H.; Rahim, F.; Sarfraz, M.; Taha, M.; Iqbal, R.; Rehman, W.; Khan, S.; Shah, S.A.A.; Hyder, S. Multipotent Cholinesterase Inhibitors for the Treatment of Alzheimer’s Disease: Synthesis, Biological Analysis and Molecular Docking Study of Benzimidazole-Based Thiazole Derivatives. Molecules 2022, 27, 6087. [Google Scholar] [CrossRef]
- Parekh, N.M.; Juddhawala, K.V.; Rawal, B.M. Antimicrobial activity of thiazolylbenzenesulfonamide-condensed 2,4-thiazolidinediones derivatives. Med. Chem. Res. 2013, 22, 2737–2745. [Google Scholar] [CrossRef]
- Rostom, S.A.; El-Ashmawy, I.M.; Abd El Razik, H.A.; Badr, M.H.; Ashour, H.M. Design and synthesis of some thiazolyl and thiadiazolyl derivatives of antipyrine as potential non-acidic anti-inflammatory, analgesic and antimicrobial agents. Bioorg. Med. Chem. 2009, 17, 882–895. [Google Scholar] [CrossRef] [PubMed]
- Haroun, M.; Tratrat, C.; Tsolaki, E.; Geronikaki, A. Thiazole-based thiazolidinones as potent antimicrobial agents. Design, synthesis and biological evaluation. Comb. Chem. High Throughput Screen. 2016, 19, 51–57. [Google Scholar] [CrossRef]
- Mishra, I.; Mishra, R.; Mujwar, S.; Chandra, P.; Sachan, N. A retrospect on antimicrobial potential of thiazole scaffold. J. Heterocycl. Chem. 2020, 57, 2304–2329. [Google Scholar] [CrossRef]
- Luzina, E.L.; Popov, A.V. Synthesis and anticancer activity of N-bis (trifluoromethyl) alkyl-N′-thiazolyl and N-bis (trifluoromethyl) alkyl-N′-benzothiazolylureas. Eur. J. Med. Chem. 2009, 44, 4944–4953. [Google Scholar] [CrossRef] [PubMed]
- Zablotskaya, A.; Segal, I.; Germane, S.; Shestakova, I.; Domracheva, I.; Nesterova, A.; Geronikaki, A.; Lukevics, E. Silyl modification of biologically active compounds. 8. Trimethylsilyl ethers of hydroxyl-containing thiazole derivatives. Chem. Heterocycl. Compd. 2002, 38, 859–866. [Google Scholar] [CrossRef]
- Kalkhambkar, R.; Kulkarni, G.; Shivkumar, H.; Rao, R.N. Synthesis of novel triheterocyclicthiazoles as anti-inflammatory and analgesic agents. Eur. J. Med. Chem. 2007, 42, 1272–1276. [Google Scholar] [CrossRef] [PubMed]
- de Santana, T.I.; de Oliveira Barbosa, M.; de Moraes Gomes, P.A.T.; da Cruz, A.C.N.; da Silva, T.G.; Leite, A.C.L. Synthesis, anticancer activity and mechanism of action of new thiazole derivatives. Eur. J. Med. Chem. 2018, 144, 874–886. [Google Scholar] [CrossRef]
- Ayati, A.; Emami, S.; Moghimi, S.; Foroumadi, A. Thiazole in the targeted anticancer drug discovery. Future Med. Chem. 2019, 11, 1929–1952. [Google Scholar] [CrossRef]
- Britschgi, M.; Greyerz, S.; Burkhart, C.; Pichler, W.J. Molecular aspects of drug recognition by specific T cells. Curr. Drug Targets 2003, 4, 1–11. [Google Scholar] [CrossRef]
- Liaras, K.; Fesatidou, M.; Geronikaki, A. Thiazoles and thiazolidinones as COX/LOX inhibitors. Molecules 2018, 23, 685. [Google Scholar] [CrossRef]
- Turan-Zitouni, G.; Chevallet, P.; Kilic, F.S.; Erol, K. Synthesis of some thiazolyl-pyrazoline derivatives and preliminary investigation of their hypotensive activity. Eur. J. Med. Chem. 2000, 35, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Haroon, M.; Khalid, M.; Shahzadi, K.; Akhtar, T.; Saba, S.; Rafique, J.; Ali, S.; Irfan, M.; Alam, M.M.; Imran, M. Alkyl 2-(2-(arylidene) alkylhydrazinyl) thiazole-4-carboxylates: Synthesis, acetyl cholinesterase inhibition and docking studies. J. Mol. Struct. 2021, 1245, 131063. [Google Scholar] [CrossRef]
- Chhabria, M.T.; Patel, S.; Modi, P.; Brahmkshatriya, P.S. Thiazole: A review on chemistry, synthesis and therapeutic importance of its derivatives. Curr. Top. Med. Chem. 2016, 16, 2841–2862. [Google Scholar] [CrossRef] [PubMed]
- Orlemans, E.; Verboom, W.; Scheltinga, M.; Reinhoudt, D.; Lelieveld, P.; Fiebig, H.; Winterhalter, B.; Double, J.; Bibby, M. Synthesis, mechanism of action, and biological evaluation of mitosenes. J. Med. Chem. 1989, 32, 1612–1620. [Google Scholar] [CrossRef]
- Belohlavek, D.; Malfertheiner, P. The effect of zolimidine, imidazopyridine-derivate, on the duodenal ulcer healing. Scand. J. Gastroenterol. Suppl. 1979, 54, 44. [Google Scholar]
- Gali, R.; Banothu, J.; Bavantula, R. One-Pot Multicomponent Synthesis of Novel Substituted Imidazo [1,2-a] Pyridine Incorporated Thiazolyl Coumarins and their Antimicrobial Activity. J. Heterocycl. Chem. 2015, 52, 641–646. [Google Scholar] [CrossRef]
- Althagafi, I.; Abdel-Latif, E. Synthesis and antibacterial activity of new imidazo [1,2-a] pyridines festooned with pyridine, thiazole or pyrazole moiety. Polycycl. Aromat. Compd. 2022, 42, 4487–4500. [Google Scholar] [CrossRef]
- Rahim, F.; Javed, M.T.; Ullah, H.; Wadood, A.; Taha, M.; Ashraf, M.; Khan, M.A.; Khan, F.; Mirza, S.; Khan, K.M. Synthesis, molecular docking, acetylcholinesterase and butyrylcholinesterase inhibitory potential of thiazole analogs as new inhibitors for Alzheimer disease. Bioorg. Chem. 2015, 62, 106–116. [Google Scholar] [CrossRef]
- Mekky, A.E.; Sanad, S.M.; El-Idreesy, T.T. New thiazole and thiazole-chromene hybrids possessing morpholine units: Piperazine-mediated one-pot synthesis of potential acetylcholinesterase inhibitors. Synth. Commun. 2021, 51, 3332–3344. [Google Scholar] [CrossRef]
- Hemaida, A.Y.; Hassan, G.S.; Maarouf, A.R.; Joubert, J.; El-Emam, A.A. Synthesis and biological evaluation of thiazole-based derivatives as potential acetylcholinesterase inhibitors. ACS Omega 2021, 6, 19202–19211. [Google Scholar] [CrossRef]
- Park, S.; Kim, H.; Son, J.-Y.; Um, K.; Lee, S.; Baek, Y.; Seo, B.; Lee, P.H. Synthesis of Imidazopyridines via Copper-Catalyzed, Formal Aza-[3 + 2] Cycloaddition Reaction of Pyridine Derivatives with α-Diazo Oxime Ethers. J. Org. Chem. 2017, 82, 10209–10218. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.P.; Mohapatra, S.; Sahoo, C.R.; Raiguru, B.P.; Nayak, S.; Jena, S.; Padhy, R.N. Design, one-pot synthesis, molecular docking study, and antibacterial evaluation of novel 2H-chromene based imidazo [1,2-a] pyridine derivatives as potent peptide deformylase inhibitors. J. Mol. Struct. 2021, 1246, 131183. [Google Scholar] [CrossRef]
- Thakur, A.; Pereira, G.; Patel, C.; Chauhan, V.; Dhaked, R.K.; Sharma, A. Design, one-pot green synthesis and antimicrobial evaluation of novel imidazopyridine bearing pyran bis-heterocycles. J. Mol. Struct. 2020, 1206, 127686. [Google Scholar] [CrossRef]
- Eskandani, M.; Babak Bahadori, M.; Zengin, G.; Dinparast, L.; Bahadori, S. Novel natural agents from Lamiaceae family: An evaluation on toxicity and enzyme inhibitory potential linked to diabetes mellitus. Curr. Bioact. Compd. 2016, 12, 34–38. [Google Scholar] [CrossRef]
- Khan, S.; Iqbal, S.; Rahim, F.; Shah, M.; Hussain, R.; Alrbyawi, H.; Rehman, W.; Dera, A.A.; Rasheed, L.; Somaily, H. New Biologically Hybrid Pharmacophore Thiazolidinone-Based Indole Derivatives: Synthesis, In Vitro Alpha-Amylase and Alpha-Glucosidase along with Molecular Docking Investigations. Molecules 2022, 27, 6564. [Google Scholar] [CrossRef]
- Khan, S.; Iqbal, S.; Shah, M.; Rehman, W.; Hussain, R.; Rasheed, L.; Alrbyawi, H.; Dera, A.A.; Alahmdi, M.I.; Pashameah, R.A. Synthesis, In Vitro Anti-Microbial Analysis and Molecular Docking Study of Aliphatic Hydrazide-Based Benzene Sulphonamide Derivatives as Potent Inhibitors of α-Glucosidase and Urease. Molecules 2022, 27, 7129. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
| S.NO | R | IC50 ± SEM a [μM] | S.NO | R | IC50 ± SEM a [μM] |
|---|---|---|---|---|---|
| 4a |  | 6.85 ± 2.18 | 4i |  | 34.91 ± 5.84 |
| 4b | 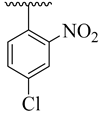 | 13.63 ± 1.67 | 4j | 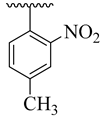 | 22.57 ± 3.55 |
| 4c |  | 63.46 ± 5.28 | 4k | 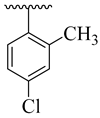 | 34.73 ± 5.23 |
| 4d | 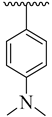 | 32.12 ± 4.29 | 4l | 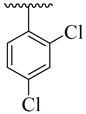 | 19.26 ± 2.58 |
| 4e | 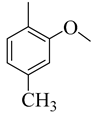 | 44.89 ± 5.27 | 4m | 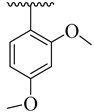 | 56.30 ± 5.94 |
| 4f | 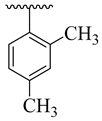 | 50.96 ± 5.80 | 4n | 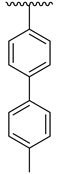 | 61.42 ± 4.56 |
| 4g |  | 5.57 ± 3.45 | 4o | 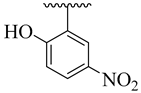 | 7.16 ± 1.40 |
| 4h | 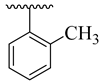 | 43.20 ± 6.16 | 4p |  | 10.48 ± 2.20 |
| a Standard Acarbose drug | 48.71 ± 2.65 | ||||
| Active Analogs | Targeted Enzyme | Receptors | Types of Interactions | Distance (oA) | Docking Score |
|---|---|---|---|---|---|
| 4g | α-glucosidase | GLU-A-595 | Pi-Anion | 6.52 | −13.45 |
| GLU-A-595 | Pi-Anion | 6.85 | |||
| PHE-A-200 | Pi-Pi stacked | 5.69 | |||
| PHE-A-200 | Pi-Pi stacked | 4.80 | |||
| VAL-A-96 | H-B | 3.98 | |||
| VAL-A-96 | Pi-R | 5.34 | |||
| PHE-A-95 | Pi-R | 6.69 | |||
| PHE-A-95 | Pi-R | 4.18 | |||
| PHE-A-95 | H-F | 4.87 | |||
| HIS-A-94 | Pi-R | 5.48 | |||
| HIS-A-94 | H-F | 6.00 | |||
| HIS-A-94 | H-F | 4.66 | |||
| PHE-A-95 | C-H | 3.93 | |||
| 4a | α-glucosidase | ARG-A-91 | Pi-R | 5.47 | −12.87 |
| TRP-A-90 | Pi-S | 7.20 | |||
| TRP-A-90 | Pi-R | 6.58 | |||
| PHE-A-200 | H-F | 4.04 | |||
| PHE-A-200 | Pi-R | 5.41 | |||
| VAL-A-201 | Pi-R | 4.94 | |||
| VAL-A-96 | Pi-R | 4.88 | |||
| HIS-A-94 | H-F | 5.80 | |||
| HIS-A-94 | Pi-R | 5.84 | |||
| GLN-A-202 | H-B | 4.09 | |||
| 4o | α-glucosidase | GLU-A-595 | Pi-anion | 7.69 | −12.15 |
| GLN-A-202 | H-B | 4.26 | |||
| VAL-A-96 | Pi-R | 5.53 | |||
| VAL-A-96 | Pi-R | 5.10 | |||
| ARG-A-91 | Pi-R | 5.86 | |||
| ARG-A-91 | Pi-R | 5.49 | |||
| PHE-A-51 | Pi-Pi T-shaped | 5.47 | |||
| HIS-A-94 | C-H | 6.18 | |||
| HIS-A-94 | Pi-Pi T-shaped | 5.05 | |||
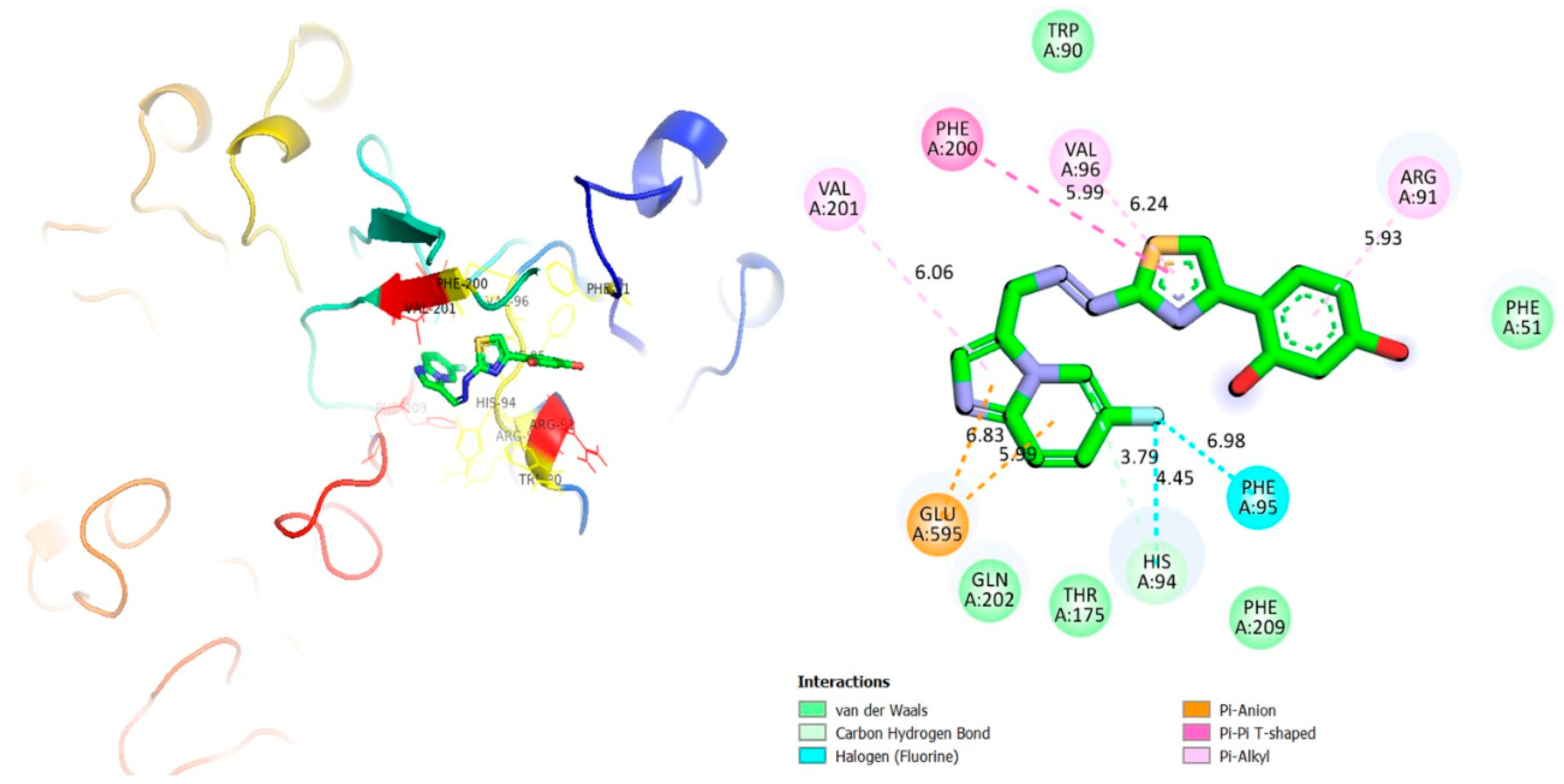
| 4p | α-glucosidase | PHE-A-51 | H-F | 6.98 | −11.25 |
| HIS-A-94 | H-F | 4.45 | |||
| HIS-A-94 | C-H | 3.79 | |||
| GLU-A-595 | Pi-Anion | 5.99 | |||
| GLU-A-595 | Pi-Anion | 6.83 | |||
| VAL-A-201 | Pi-R | 6.06 | |||
| PHE-A-200 | Pi-Pi T-shaped | 5.99 | |||
| VAL-A-96 | Pi-R | 6.24 | |||
| ARG-A-91 | Pi-R | 5.93 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussain, R.; Rehman, W.; Khan, S.; Maalik, A.; Hefnawy, M.; Alanazi, A.S.; Khan, Y.; Rasheed, L. Imidazopyridine-Based Thiazole Derivatives as Potential Antidiabetic Agents: Synthesis, In Vitro Bioactivity, and In Silico Molecular Modeling Approach. Pharmaceuticals 2023, 16, 1288. https://doi.org/10.3390/ph16091288
Hussain R, Rehman W, Khan S, Maalik A, Hefnawy M, Alanazi AS, Khan Y, Rasheed L. Imidazopyridine-Based Thiazole Derivatives as Potential Antidiabetic Agents: Synthesis, In Vitro Bioactivity, and In Silico Molecular Modeling Approach. Pharmaceuticals. 2023; 16(9):1288. https://doi.org/10.3390/ph16091288
Chicago/Turabian StyleHussain, Rafaqat, Wajid Rehman, Shoaib Khan, Aneela Maalik, Mohamed Hefnawy, Ashwag S. Alanazi, Yousaf Khan, and Liaqat Rasheed. 2023. "Imidazopyridine-Based Thiazole Derivatives as Potential Antidiabetic Agents: Synthesis, In Vitro Bioactivity, and In Silico Molecular Modeling Approach" Pharmaceuticals 16, no. 9: 1288. https://doi.org/10.3390/ph16091288
APA StyleHussain, R., Rehman, W., Khan, S., Maalik, A., Hefnawy, M., Alanazi, A. S., Khan, Y., & Rasheed, L. (2023). Imidazopyridine-Based Thiazole Derivatives as Potential Antidiabetic Agents: Synthesis, In Vitro Bioactivity, and In Silico Molecular Modeling Approach. Pharmaceuticals, 16(9), 1288. https://doi.org/10.3390/ph16091288