
Chemistry and Pharmacology of Fluorinated Drugs Approved by the FDA (2016–2022)

 ,
, 
Abstract
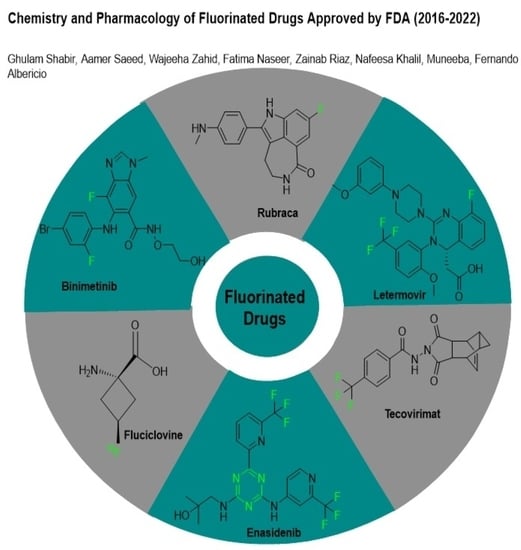
1. Introduction
2. FDA-Approved Fluorinated (F-18) Radiolabeled Drugs
2.1. Flortaucipir F-18
2.2. Fluciclovine F-18
2.3. Fluorodopa F-18
2.4. Fluoroestradiol F-18
2.5. Piflufolastat F-18
3. FDA-Approved Fluorinated Oligonucleotide Drugs
4. FDA-Approved Fluorinated Heterocyclic/Carbocyclic Drugs
5. Therapeutic Indications
5.1. FDA-Approved Fluorinated Anticancer Drugs
5.1.1. Abemaciclib
5.1.2. Alpelisib
5.1.3. Apalutamide
5.1.4. Avapritinib
5.1.5. Belzutifan
5.1.6. Binimetinib
5.1.7. Capmatinib
5.1.8. Dacomitinib
5.1.9. Encorafenib
5.1.10. Ivosidenib
5.1.11. Larotrectinib
5.1.12. Lorlatinib
5.1.13. Pexidartinib
5.1.14. Pralsetinib
5.1.15. Relugolix
5.1.16. Ripretinib
5.1.17. Rucaparib
5.1.18. Selumetinib
5.1.19. Sotorasib
5.1.20. Talazoparib
5.2. FDA-Approved Fluorinated Anti-Viral Drugs
5.2.1. Baloxavir Marboxil
5.2.2. Fostamatinib
5.2.3. Glecaprevir
5.2.4. Letermovir
5.2.5. Osilodrostat
5.2.6. Pibrentasvir
5.2.7. Siponimod
5.2.8. Sofosbuvir
5.2.9. Tecovirimat
5.2.10. Voxilaprevir
5.2.11. Umbralisib
5.3. FDA-Approved Fluorinated Anti-HIV Drugs
5.3.1. Bictegravir
5.3.2. Cabotegravir
5.3.3. Doravirine
5.3.4. Emtricitabine
5.4. FDA-Approved Fluorinated Antibacterial Drugs
5.4.1. Berotralstat
5.4.2. Elexacaftor
5.4.3. Eravacycline
5.4.4. Pretomanid
5.4.5. Telotristat Ethyl
5.5. FDA-Approved Miscellaneous Fluorinated Drugs
5.5.1. Asciminib
5.5.2. Atogepant
5.5.3. Avacopan
5.5.4. Delafloxacin
5.5.5. Elagolix
5.5.6. Enasidenib
5.5.7. Lasmiditan
5.5.8. Lemborexant
5.5.9. Lumateperone
5.5.10. Melphalan flufenamide
5.5.11. Netupitant
5.5.12. Pimavanserin
5.5.13. Selinexor
5.5.14. Tafenoquine
5.5.15. Ubrogepant
5.5.16. Upadacitinib
5.5.17. Vericiguat
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| S.NO. | Abbreviation | Explanation |
| 1 | 18F-DCFPyL | 2-(3-{1-carboxy-5-[(6-[(18)F]fluoro-pyridine-3-carbonyl)-amino]-pentyl}-ureido)-pentanedioic acid, |
| 2 | ANCA | Antineutrophilic Cytoplasmic Antibodies |
| 3 | ASCT1 | Alanine Serine Cysteine Transporter 1 |
| 4 | AUC | Area Under the Curve |
| 5 | CDK4 | Cyclin D and Cyclin-dependent Kinase 4 |
| 6 | CFTR | Cystic fibrosis transmembrane conductance regulator |
| 7 | CGRP | Calcitonin Gene-related Peptide |
| 8 | CML | Chronic Myelogenous Leukemia |
| 9 | CMV | Cytomegalovirus |
| 10 | DLBCL | Diffuse large B-cell lymphoma |
| 11 | EGFR | Epidermal Growth Factor Receptor |
| 12 | eGFR | Estimated Glomerular Filtration Rate |
| 13 | EMBRACA | Epidemiological Study of Familial Breast Cancer |
| 14 | ER | Estrogen Receptor |
| 15 | FDA | Food and Drug Administration |
| 16 | FDG | 18F-fluoro Deoxyglucose |
| 17 | GIST | Gastrointestinal Stromal tumors |
| 18 | GnRH | Gonadotropin-releasing Hormone |
| 19 | HCV | Hepatitis C Virus |
| 20 | HER1 | Human Epidermal Growth Factor Receptor 1 |
| 21 | HIF-2α | Hypoxia-Inducible Factor-2α |
| 22 | KRAS | Kirsten Rat Sarcoma Virus |
| 23 | KRAS | Kirsten Rat Sarcoma Viral Oncogene Homolog |
| 24 | LAT1, LAT3 | L-type Amino Acid Transporters |
| 25 | MAO | Monoamine Oxidase |
| 26 | MEK | Mitogen-activated Protein Kinase |
| 27 | MET | Mesenchymal Epithelial Transition |
| 28 | NF | Neurofibromas |
| 29 | NFTs | Neurofibrillary tangles |
| 30 | NO–sGC–cGMp | Nitric oxide (NO)–NO-sensitive (soluble) guanylyl cyclase (sGC)–cyclic guanosine monophosphate (cGMP) |
| 31 | NS3/4A | A non-structural protein 3 and 4a protease inhibitor |
| 32 | OX1R | Orexin Receptor Type 1 |
| 33 | PARP | Poly (ADP-ribose) Polymerase 1 |
| 34 | PARP | Poly Adenosine Diphosphate-ribose Polymerase |
| 35 | PDGFRA | Platelet-derived Growth Factor Receptor Alpha |
| 36 | PET | Positron Emission Tomography |
| 37 | PSMA | Prostate-specific Membrane Antigen |
| 38 | RET | Rational Emotive Therapy |
| 39 | TNF | Tumor Necrosis Factor |
References
- Banks, R.E.; Murtagh, V.; Marsden, H.M.; Syvret, R.G. Direct fluorination of bis (trifluoromethanesulfonyl) imide and its lithium salt and related studies. J. Fluor. Chem. 2001, 112, 271–275. [Google Scholar] [CrossRef]
- Yang, X.; Wu, T.; Phipps, R.J.; Toste, F.D. Advances in catalytic enantioselective fluorination, mono-, di-, and trifluoromethylation, and trifluoromethylthiolation reactions. Chem. Rev. 2015, 115, 826–870. [Google Scholar] [CrossRef]
- Syvret, R.G.; Butt, K.M.; Nguyen, T.P.; Bulleck, V.L.; Rieth, R.D. Novel process for generating useful electrophiles from common anions using Selectfluor® fluorination agent. J. Org. Chem. 2002, 67, 4487–4493. [Google Scholar] [CrossRef] [PubMed]
- Reichel, M.; Karaghiosoff, K. Reagents for selective fluoromethylation: A challenge in organofluorine chemistry. Angew. Chem. Int. Ed. 2020, 59, 12268–12281. [Google Scholar] [CrossRef] [PubMed]
- Bravo, P.; Guidetti, M.; Viani, F.; Zanda, M.; Markovsky, A.L.; Sorochinsky, A.E.; Soloshonok, I.V.; Soloshonok, V.A. Chiral sulfoxide controlled asymmetric additions to C=N double bond. An efficient approach to stereochemically defined α-fluoroalkyl amino compounds. Tetrahedron 1998, 54, 12789–12806. [Google Scholar] [CrossRef]
- Xu, X.H.; Matsuzaki, K.; Shibata, N. Synthetic methods for compounds having CF3–S units on carbon by trifluoromethylation, trifluoromethylthiolation, triflylation, and related reactions. Chem. Rev. 2015, 115, 731–764. [Google Scholar] [CrossRef] [PubMed]
- Ohkura, H.; Berbasov, D.O.; Soloshonok, V.A. Chemo-and regioselectivity in the reactions between highly electrophilic fluorine containing dicarbonyl compounds and amines. Improved synthesis of the corresponding imines/enamines. Tetrahedron 2003, 59, 1647–1656. [Google Scholar] [CrossRef]
- Gondo, K.; Kitamura, T. Reaction of iodonium ylides of 1,3-dicarbonyl compounds with HF reagents. Molecules 2012, 17, 6625–6632. [Google Scholar] [CrossRef]
- Han, J.; Sorochinsky, A.E.; Ono, T.; Soloshonok, V.A. Biomimetic transamination-a metal-free alternative to the reductive amination. Application for generalized preparation of fluorine-containing amines and amino acids. Cur. Org. Synth. 2011, 2, 281–294. [Google Scholar] [CrossRef]
- Syvret, R.G.; Casteel, W.J., Jr.; Lal, G.S.; Goudar, J.S. Selective fluorination of an aryl triazolinone herbicide intermediate. J. Fluor. Chem. 2004, 125, 33–35. [Google Scholar] [CrossRef]
- Röschenthaler, G.V.; Kukhar, V.P.; Kulik, I.B.; Belik, M.Y.; Sorochinsky, A.E.; Rusanov, E.B.; Soloshonok, V.A. Asymmetric synthesis of phosphonotrifluoroalanine and its derivatives using N-tert-butanesulfinyl imine derived from fluoral. Tetrahedron Lett. 2012, 53, 539–542. [Google Scholar] [CrossRef]
- Kirk, K.L. Fluorine in medicinal chemistry: Recent therapeutic applications of fluorinated small molecules. J. Fluor. Chem. 2006, 127, 1013–1029. [Google Scholar] [CrossRef]
- Ojima, I. Strategic incorporation of fluorine into taxoid anticancer agents for medicinal chemistry and chemical biology studies. J. Fluor. Chem. 2017, 198, 10–23. [Google Scholar] [CrossRef]
- Kowalczyk, D.; Wojciechowski, J.; Albrecht, L. Asymmetric Organocatalysis in the Synthesis of Pyrrolidine Derivatives Bearing a Benzofuran-3 (2H)-one Scaffold. Synthesis 2017, 49, 880–890. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J.L.; Soloshonok, V.A.; Izawa, K.; Liu, H. Next generation of fluorine-containing pharmaceuticals, compounds currently in phase II–III clinical trials of major pharmaceutical companies: New structural trends and therapeutic areas. Chem. Rev. 2016, 116, 422–518. [Google Scholar] [CrossRef]
- Hagmann, W.K. The many roles for fluorine in medicinal chemistry. J. Med. Chem. 2008, 51, 4359–4369. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Kiss, L.; Mei, H.; Remete, A.M.; Ponikvar-Svet, M.; Sedgwick, D.M.; Roman, R.; Fustero, S.; Moriwaki, H.; Soloshonok, V.A. Chemical aspects of human and environmental overload with fluorine. Chem. Rev. 2021, 121, 4678–4742. [Google Scholar] [CrossRef]
- Mei, H.; Han, J.; Klika, K.D.; Izawa, K.; Sato, T.; Meanwell, N.A.; Soloshonok, V.A. Applications of fluorine-containing amino acids for drug design. Eur. J. Med. Chem. 2020, 186, 111826. [Google Scholar] [CrossRef] [PubMed]
- Mei, H.; Han, J.; Klika, K.D.; Izawa, K.; Sato, T.; Meanwell, N.A.; Soloshonok, V.A.; White, S.; Graham, D.J.; Fustero, S. Tailor-made amino acids and fluorinated motifs as prominent traits in modern pharmaceuticals. Chem. Eur. J. 2020, 26, 11349–11390. [Google Scholar] [CrossRef]
- Burgey, C.S.; Robinson, K.A.; Lyle, T.A.; Sanderson, P.E.; Lewis, S.D.; Lucas, B.J.; Vacca, J.P. Metabolism-directed optimization of 3-aminopyrazinone acetamide thrombin inhibitors. Development of an orally bioavailable series containing P1 and P3 pyridines. J. Med. Chem. 2003, 46, 461–473. [Google Scholar] [CrossRef]
- Mei, H.; Han, J.; Soloshonok, V.A.; Remete, A.M.; Zou, Y.; Moriwaki, H.; Fustero, S.; Kiss, L. Fluorine-containing drugs approved by the FDA in 2019. Chin. Chem. Lett. 2020, 31, 2401–2413. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, A.; Dhawan, G.; Mei, H.; Zhang, W.; Izawa, K.; Soloshonok, V.A.; Han, J. Fluorine-containing pharmaceuticals approved by the FDA in 2020: Synthesis and biological activity. Chin. Chem. Lett. 2021, 32, 3342–3354. [Google Scholar] [CrossRef]
- Han, J.; Remete, A.M.; Dobson, L.S.; Kiss, L.; Izawa, K.; Moriwaki, H.; Soloshonok, V.A.; O’Hagan, D. Next generation organofluorine containing blockbuster drugs. J. Fluor. Chem. 2020, 239, 109639. [Google Scholar] [CrossRef]
- Böhm, H.J.; Banner, D.; Bendels, S.; Kansy, M.; Kuhn, B.; Müller, K.; Obst-Sander, U.; Stahl, M. Fluorine in medicinal chemistry. ChemBioChem 2004, 5, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Stoller, A.D. Synthesis of new substituted 1λ4-1, 2, 4, 6-thiatriazines. J. Heterocycl. Chem. 2000, 37, 583–595. [Google Scholar] [CrossRef]
- Wang, Q.; Song, H.; Wang, Q. Fluorine-containing agrochemicals in the last decade and approaches for fluorine incorporation. Chin. Chem. Lett. 2022, 32, 626–642. [Google Scholar] [CrossRef]
- Ogawa, Y.; Tokunaga, E.; Kobayashi, O.; Hirai, K.; Shibata, N. Current contributions of organofluorine compounds to the agrochemical industry. iScience 2020, 23, 101467. [Google Scholar] [CrossRef]
- Fujiwara, T.; O’Hagan, D. Successful fluorine-containing herbicide agrochemicals. J. Fluor. Chem. 2014, 167, 16–29. [Google Scholar] [CrossRef]
- Chen, P.; Bai, W.; Bao, Y. Fluorescent chemodosimeters for fluoride ions via silicon-fluorine chemistry: 20 years of progress. J. Mater. Chem. 2019, 7, 11731–11746. [Google Scholar] [CrossRef]
- Ilardi, E.A.; Vitaku, E.; Njardarson, J.T. Data-mining for sulfur and fluorine: An evaluation of pharmaceuticals to reveal opportunities for drug design and discovery: Miniperspective. J. Fluor. Chem. 2014, 57, 2832–2842. [Google Scholar] [CrossRef]
- Hop, C.E.; Kalgutkar, A.S.; Soglia, J.R. Importance of early assessment of bioactivation in drug discovery. Ann. Rep. Med. Chem. 2006, 41, 369–381. [Google Scholar] [CrossRef]
- Wang, J.; Sánchez-Roselló, M.; Aceña, J.L.; Del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef] [PubMed]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef]
- Shah, P.; Westwell, A.D. The role of fluorine in medicinal chemistry. J. Enzym. Inhib. Med. Chem. 2007, 22, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Swallow, S. Fluorine in Medicinal Chemistry. Progress in Medicinal Chemistry. Prog. Med. Chem. 2015, 54, 65–133. [Google Scholar] [CrossRef] [PubMed]
- Véliz, E.A.; Stephens, O.M.; Beal, P.A. Synthesis and analysis of RNA containing 6-trifluoromethylpurine ribonucleoside. Org. Lett. 2001, 3, 2969–2972. [Google Scholar] [CrossRef]
- Filler, R.; Saha, R. Fluorine in medicinal chemistry: A century of progress and a 60-year retrospective of selected highlights. Future Med. Chem. 2009, 1, 777–791. [Google Scholar] [CrossRef]
- Jarvis, L.M. Drug hunters explore allostery’s advantages. Chem. Eng. News. 2019, 97, 39–42. [Google Scholar] [CrossRef]
- Glasspool, R.M.; Evans, T.R.J. Clinical imaging of cancer metastasis. Eur. J. Cancer 2000, 36, 1661–1670. [Google Scholar] [CrossRef]
- Westera, G.; August, S.P. Functional imaging of physiological processes by positron emission tomography. Physiology 2003, 18, 175–178. [Google Scholar] [CrossRef][Green Version]
- Olivier, P. Nuclear oncology, a fast growing field of nuclear medicine. Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrometers Detect. Assoc. Equip. 2004, 527, 4–8. [Google Scholar] [CrossRef]
- McGoron, A.J.; Maob, X.; Georgiou, M.F.; Kuluz, J.W. Computer phantom study of brain PET glucose metabolism imaging using a rotating SPECT/PET camera. Comput. Biol. Med. 2005, 35, 511–531. [Google Scholar] [CrossRef] [PubMed]
- Herholz, K.; Heiss, W.D. Positron emission tomography in clinical neurology. Mol. Imaging Biol. 2004, 6, 239–269. [Google Scholar] [CrossRef]
- Wooten, D.W.; Guehl, N.J.; Verwer, E.E.; Shoup, T.M.; Yokell, D.L.; Zubcevik, N.; Vasdev, N. Pharmacokinetic evaluation of the tau PET radiotracer 18F-T807 (18F-AV-1451) in human subjects. J. Nucl. Med. 2017, 3, 484–491. [Google Scholar] [CrossRef]
- Nye, J.A.; Schuster, D.M.; Yu, W.; Camp, V.M.; Goodman, M.M.; Votaw, J.R. Biodistribution and radiation dosimetry of the synthetic nonmetabolized amino acid analogue anti-18F-FACBC in humans. J. Nucl. Med. 2007, 4, 1017–1020. [Google Scholar] [CrossRef]
- Deng, W.P.; Wong, K.A.; Kirk, K. Convenient syntheses of 2-, 5-and 6-fluoro-and 2, 6-difluoro-l-DOPA. Tetrahedron Asymm. 2002, 11, 1135–1140. [Google Scholar] [CrossRef]
- Liao, G.J.; Clark, A.S.; Schubert, E.K.; Mankoff, D.A. 18F-fluoroestradiol PET: Current status and potential future clinical applications. J. Nucl. Med. 2016, 8, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Osborne, J.R.; Akhtar, N.H.; Vallabhajosula, S.; Anand, A.; Deh, K.; Tagawa, S.T. Prostate-specific membrane antigen-based imaging. Urol. Oncol. Semin. Orig. Investig. 2013, 2, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Basetty, V.; Deruiter, J.; Pathak, S.; Dua, K.; Dhanasekaran, M. Advanced drug delivery systems targeting to improve therapeutic outcomes in porphyria. In Drug Delivery Systems for Metabolic Disorders; Academic Press: Cambridge, MA, USA, 2022; pp. 65–76. [Google Scholar] [CrossRef]
- Khvorova, A. Oligonucleotide therapeutics—A new class of cholesterol-lowering drugs. N. Engl. J. Med. 2017, 376, 4–7. [Google Scholar] [CrossRef]
- Scott, L.J.; Keam, S.J. Lumasiran: First approval. Drugs 2021, 81, 277–282. [Google Scholar] [CrossRef]
- de la Torre, B.G.; Albericio, F. The Pharmaceutical Industry in 2022. An Analysis of FDA Drug Approvals from the Perspective of Molecules. Molecules 2023, 28, 1038. [Google Scholar] [CrossRef]
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications. Molecules 2020, 25, 1909. [Google Scholar] [CrossRef] [PubMed]
- Martorana, A.; Giacalone, V.; Bonsignore, R.; Pace, A.; Gentile, C.; Pibiri, I.; Buscemi, S.; Lauria, A.; Piccionello, P.A. Heterocyclic Scaffolds for the Treatment of Alzheimer’s Disease. Curr. Pharm. Des. 2016, 22, 39713995. [Google Scholar] [CrossRef] [PubMed]
- Pathania, S.; Narang, R.K.; Rawal, R.K. Role of sulphur-heterocycles in medicinal chemistry: An update. Eur. J. Med. Chem. 2019, 180, 486–508. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, C.; Lonneman, M.; Wu, C. Polypharmacological drug actions of recently FDA approved antibiotics. Eur. J. Med. Chem. 2021, 209, 112931. [Google Scholar] [CrossRef] [PubMed]
- Heravi, M.M.; Zadsirjan, V. Prescribed drugs containing nitrogen heterocycles: An overview. RSC Adv. 2020, 10, 44247–44311. [Google Scholar] [CrossRef]
- Inoue, M.; Sumii, Y.; Shibata, N. Contribution of Organofluorine Compounds to Pharmaceuticals. ACS Omega 2020, 5, 10633–10640. [Google Scholar] [CrossRef]
- Upadhyay, C.; Chaudhary, M.; De Oliveira, R.N.; Borbas, A.; Kempaiah, P.S.; Rathi, B. Fluorinated scaffolds for antimalarial drug discovery. Expert Opin. Drug Discov. 2020, 15, 705–718. [Google Scholar] [CrossRef]
- O’Hagan, D. Fluorine in health care: Organofluorine containing blockbuster drugs. J. Fluor. Chem. 2010, 131, 1071–1081. [Google Scholar] [CrossRef]
- Fried, J.; Sabo, E.F. 9α-Fluoro Derivatives of Cortisone and Hydrocortisone. J. Am. Chem. Soc. 1954, 76, 1455–1456. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Meanwell, N.A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.M.; Shu, Y.Z.; Zhuo, X.; Meanwell, N.A. Metabolic and Pharmaceutical Aspects of Fluorinated Compounds. J. Med. Chem. 2020, 63, 6315–6386. [Google Scholar] [CrossRef] [PubMed]
- Morgenthaler, M.; Schweizer, E.; Hoffmann-Röder, A.; Benini, F.; Martin, R.E.; Jaeschke, G.; Wagner, B.; Fischer, H.; Bendels, S.; Zimmerli, D.; et al. Predicting and Tuning Physicochemical Properties in Lead Optimization: Amine Basicities. ChemMedChem 2007, 2, 1100–1115. [Google Scholar] [CrossRef] [PubMed]
- Rowley, M.; Hallett, D.J.; Goodacre, S.; Moyes, C.; Crawforth, J.; Sparey, T.J.; Patel, S.; Marwood, R.; Patel, S.; Thomas, S.; et al. 3-(4-Fluoropiperidin-3-yl)-2-phenylindoles as High Affinity, Selective, and Orally Bioavailable h5-HT2A Receptor Antagonists. J. Med. Chem. 2001, 44, 1603–1614. [Google Scholar] [CrossRef]
- Giese, M.; Albrecht, M.; Rissanen, K. Anion–π interactions with fluoroarenes. Chem. Rev. 2015, 115, 8867–8895. [Google Scholar] [CrossRef]
- Mei, H.; Han, J.; Fustero, S.; Medio-Simon, M.; Sedgwick, D.M.; Santi, C.; Ruzziconi, R.; Soloshonok, V.A. Fluorine-Containing Drugs Approved by the FDA in 2018. Chem. Eur. J. 2019, 25, 11797–11819. [Google Scholar] [CrossRef]
- Knight, J.C.; Edwards, P.G.; Paisey, S.J. Fluorinated contrast agents for magnetic resonance imaging; a review of recent developments. RSC Adv. 2011, 1, 1415–1425. [Google Scholar] [CrossRef]
- Sznaidman, M.L.; Haffner, C.D.; Maloney, P.R.; Fivush, A.; Chao, E.; Goreham, D.; Sierra, M.L.; LeGrumelec, C.; Xu, H.E.; Montana, V.G.; et al. Novel selective small molecule agonists for peroxisome proliferator-activated receptor δ (PPARδ)—Synthesis and biological activity. Bioorg. Med. Chem. Lett. 2003, 13, 1517–1521. [Google Scholar] [CrossRef]
- Raub, T.J.; Wishart, G.N.; Kulanthaivel, P.; Staton, B.A.; Ajamie, R.T.; Sawada, G.A.; Lawrence, M.J.; Shannon, H.E.; Sanchez-Martinez, C.; De Dios, A. Brain exposure of two selective dual CDK4 and CDK6 inhibitors and the antitumor activity of CDK4 and CDK6 inhibition in combination with temozolomide in an intracranial glioblastoma xenograft. Drug Metab. Dispos. 2015, 43, 1360–1371. [Google Scholar] [CrossRef]
- Hou, J.Z.; Ye, J.C.; Pu, J.J.; Liu, H.; Ding, W.; Zheng, H.; Liu, D. Novel agents and regimens for hematological malignancies: Recent updates from 2020 ASH annual meeting. J. Hematol. Oncol. 2021, 14, 66. [Google Scholar] [CrossRef]
- Rathkopf, D.E.; Antonarakis, E.S.; Shore, N.D.; Tutrone, R.F.; Alumkal, J.J.; Ryan, C.J.; Saleh, M. Safety and antitumor activity of apalutamide (ARN-509) in metastatic castration-resistant prostate cancer with and without prior abiraterone acetate and prednisone. Clin. Cancer Res. 2017, 14, 3544–3551. [Google Scholar] [CrossRef]
- Xiao, H.; Dairaghi, D.J.; Powers, J.P.; Ertl, L.S.; Baumgart, T.; Wang, Y.; Seitz, L.C. C5a receptor (CD88) blockade protects against MPO-ANCA GN. Am. J. Soc. Nephrol. 2014, 2, 225. [Google Scholar] [CrossRef]
- Jorgensen, S.C.; Mercuro, N.J.; Davis, S.L.; Rybak, M.J. Delafloxacin: Place in therapy and review of microbiologic, clinical and pharmacologic properties. Infect. Dis. Ther. 2018, 7, 197–217. [Google Scholar] [CrossRef] [PubMed]
- Biggioggero, M.; Becciolini, A.; Crotti, C.; Agape, E.; Favalli, E.G. Upadacitinib and filgotinib: The role of JAK1 selective inhibition in the treatment of rheumatoid arthritis. Drugs Context 2019, 8, 212595. [Google Scholar] [CrossRef] [PubMed]
- Salam, K.A.; Akimitsu, N. Hepatitis C virus NS3 inhibitors: Current and future perspectives. Biomed Res. Intl. 2013, 2013, 467869. [Google Scholar] [CrossRef]
- Xu, R.; Wang, K.; Rizzi, J.P.; Huang, H.; Grina, J.A.; Schlachter, S.T.; Wang, B. 3-[(1 S, 2 S, 3 R)-2, 3-Difluoro-1-hydroxy-7-methylsulfonylindan-4-yl] oxy-5-fluorobenzonitrile (PT2977), a hypoxia-inducible factor 2α (HIF-2α) inhibitor for the treatment of clear cell Renal cell carcinoma. J. Med. Chem. 2019, 62, 6876–6893. [Google Scholar] [CrossRef] [PubMed]
- Thakare, R.; Dasgupta, A.; Chopra, S. Eravacycline for the treatment of patients with bacterial infections. Drugs Today 2018, 4, 245–254. [Google Scholar] [CrossRef]
- Sanchez, R.I.; Fillgrove, K.L.; Yee, K.L.; Liang, Y.; Lu, B.; Tatavarti, A.; Liu, R.; Anderson, M.S.; Behm, M.O.; Fan, L.; et al. Characterisation of the absorption, distribution, metabolism, excretion and mass balance of doravirine, a non-nucleoside reverse transcriptase inhibitor in humans. Xenobiotica 2019, 4, 422–432. [Google Scholar] [CrossRef]
- Bendell, J.C.; Javle, M.; Bekaii-Saab, T.S.; Finn, R.S.; Wainberg, Z.A.; Laheru, D.A.; Weekes, C.D. A phase 1 dose-escalation and expansion study of binimetinib (MEK162), a potent and selective oral MEK1/2 inhibitor. Br. J. Cancer. 2017, 5, 575–583. [Google Scholar] [CrossRef]
- Shockcor, J.P.; Wurm, R.M.; Frick, L.W.; Sanderson, P.N.; Farrant, R.D.; Sweatman, B.C.; Lindon, J.C. Hplc-nmr identification of the human urinary metabolites of (—)-cis-5-fluoro-1-[2-(hydroxymethyl)-1, 3-oxathiolan-5-yl] cytosine, a nucleoside analogue active against human immunodeficiency virus (HIV). Xenobiotica 1996, 26, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, T.M.; Kim, D.W.; Kim, S.; Kim, M.; Ahn, Y.O.; Heo, D.S. Acquired resistance of MET-amplified non-small cell lung cancer cells to the MET inhibitor capmatinib. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2019, 51, 951–962. [Google Scholar] [CrossRef]
- Engelman, J.A.; Zejnullahu, K.; Gale, C.M.; Lifshits, E.; Gonzales, A.J.; Shimamura, T.; Zhao, F. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007, 24, 11924–11932. [Google Scholar] [CrossRef] [PubMed]
- Nnoaham, K.E.; Hummelshoj, L.; Webster, P.; Hooghe, T.D.; Nardone, F.D.C.; Nardone, C.D.C.; Jenkinson, C.; Stephen, H.K.; Zondervan, K.T. World Endometriosis Research Foundation Global Study. Impact of endometriosis on quality of life and work productivity: A multicenter study across ten countries. Fertil. Steril. 2011, 2, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Strunecká, A.; Patočka, J.; Connett, P. Fluorine in medicine. J. Appl. Biomed. 2004, 2, 141–150. [Google Scholar] [CrossRef]
- Stein, E.M.; Dinardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; Flinn, I.; Kantarjian, H.M.; Collins, R.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. Am. J. Hematol. 2017, 6, 722–731. [Google Scholar] [CrossRef]
- Sacchettini, J.C.; Rubin, E.J.; Freundlich, J.S. Drugs versus bugs: In pursuit of the persistent predator Mycobacterium tuberculosis. Nat. Rev. Microbiol. 2008, 1, 41–52. [Google Scholar] [CrossRef]
- Verbitskiy, E.V.; Rusinov, G.L.; Charushin, V.N.; Chupakhin, O.N. Development of new antituberculosis drugs among of 1, 3-and 1, 4-diazines. Highlights and perspectives. Russ. Chem. Bull. 2019, 68, 2172–2189. [Google Scholar] [CrossRef]
- Reuter, U.; Israel, H.; Neeb, L. The pharmacological profile and clinical prospects of the oral 5-HT1F receptor agonist lasmiditan in the acute treatment of migraine. Ther. Adv. Neurol. Disord. 2015, 8, 46–54. [Google Scholar] [CrossRef]
- Li, Z.; Jiang, K.; Zhu, X.; Lin, G.; Song, F.; Zhao, Y.; Piao, Y. Encorafenib (LGX818), a potent BRAF inhibitor, induces senescence accompanied by autophagy in BRAFV600E melanoma cells. Cancer Lett. 2016, 2, 332–344. [Google Scholar] [CrossRef] [PubMed]
- Pavel, M.; Gross, D.J.; Benavent, M.; Perros, P.; Srirajaskanthan, R.; Warner, R.R.; Kulke, M.H.; Anthony, L.B.; Kunz, P.L.; Hörsch, D.; et al. Telotristat ethyl in carcinoid syndrome: Safety and efficacy in the TELECAST phase 3 trial. Endocr. Relat. Cancer. 2018, 25, 309–322. [Google Scholar] [CrossRef]
- Melendez, D.P.; Razonable, R.R. Letermovir and inhibitors of the terminase complex: A promising new class of investigational antiviral drugs against human cytomegalovirus. Infect. Drug Resist. 2015, 8, 269. [Google Scholar] [CrossRef]
- Duggan, S. Osilodrostat: First approval. Drugs 2020, 5, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Merchant, S.L.; Culos, K.; Wyatt, H. Ivosidenib: IDH1 Inhibitor for the Treatment of Acute Myeloid Leukemia. J. Advan. Pract. Oncol. 2019, 5, 494. [Google Scholar] [CrossRef]
- Drilon, A.; Nagasubramanian, R.; Blake, J.F.; Ku, N.; Tuch, B.B.; Ebata, K.; Smith, S. A Next-Generation TRK Kinase Inhibitor Overcomes Acquired Resistance to Prior TRK Kinase Inhibition in Patients with TRK Fusion–Positive Solid Tumors Next-Generation TRK Inhibitor Overcomes Acquired Resistance. Cancer Discov. 2017, 9, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Vyas, P.; Hwang, B.J.; Brašić, J.R. An evaluation of lumateperone tosylate for the treatment of schizophrenia. Expert Opin. Pharmacother. 2020, 2, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, N.; Razavi, S.; Nikzad, E. Multiple sclerosis: Pathogenesis, symptoms, diagnoses and cell-based therapy. Cell J. 2017, 1, 1–10. [Google Scholar] [CrossRef]
- Giustini, N.; Bernthal, N.M.; Bukata, S.V.; Singh, A.S. Tenosynovial giant cell tumor: Case report of a patient effectively treated with pexidartinib (PLX3397) and review of the literature. Clin. Sarcoma Res. 2018, 8, 14. [Google Scholar] [CrossRef]
- Aygören-Pürsün, E.; Bygum, A.; Grivcheva-Panovska, V.; Magerl, M.; Graff, J.; Steiner, U.C.; Fain, O. Oral plasma kallikrein inhibitor for prophylaxis in hereditary angioedema. N. Engl. J. Med. 2018, 4, 352–362. [Google Scholar] [CrossRef]
- Coyne, J.W. The first oral fixed-dose combination of netupitant and palonosetron for the treatment of chemotherapy-induced nausea and vomiting. J. Adv. Pract. Oncol. 2016, 1, 66–70. [Google Scholar] [CrossRef]
- Bagaglio, S.; Uberti-Foppa, C.; Morsica, G. Resistance mechanisms in hepatitis C virus: Implications for direct-acting antiviral use. Drugs 2017, 10, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Gainor, J.F.; Rahal, R.; Brubaker, J.D.; Kim, J.L.; Maynard, M.; Hu, W. Precision Targeted Therapy with BLU-667 for RET-Driven CancersBLU-667 Inhibits RET Alterations in Cancer. Cancer Discov. 2018, 7, 836–849. [Google Scholar] [CrossRef] [PubMed]
- Miwa, K.; Hitaka, T.; Imada, T.; Sasaki, S.; Yoshimatsu, M.; Kusaka, M.; Kitazaki, T. Discovery of 1-{4-[1-(2, 6-difluorobenzyl)-5-[(dimethylamino) methyl]-3-(6-methoxypyridazin-3-yl)-2, 4-dioxo-1, 2, 3, 4-tetrahydrothieno [2, 3-d] pyrimidin-6-yl] phenyl}-3-methoxyurea (TAK-385) as a potent, orally active, non-peptide antagonist of the human gonadotropin-releasing hormone receptor. J. Med. Chem. 2011, 14, 4998–5012. [Google Scholar] [CrossRef]
- Zahodne, L.B.; Fernandez, H.H. Pathophysiology and treatment of psychosis in Parkinson’s disease. Drugs Aging 2008, 8, 665–682. [Google Scholar] [CrossRef]
- Mazzocca, A.; Napolitano, A.; Silletta, M.; Spalato-Ceruso, M.; Santini, D.; Tonini, G.; Vincenzi, B. New frontiers in the medical management of gastrointestinal stromal tumours. Ther. Adv. Med. Oncol. 2019, 11, 1758835919841946. [Google Scholar] [CrossRef]
- Dockery, L.E.; Gunderson, C.C.; Moore, K.N. Rucaparib: The past, present, and future of a newly approved PARP inhibitor for ovarian cancer. OncoTargets Ther. 2017, 10, 3029–3037. [Google Scholar] [CrossRef]
- Dombi, E.; Baldwin, A.; Marcus, L.J.; Fisher, M.J.; Weiss, B.; Kim, A.; Whitcomb, P. Activity of selumetinib in neurofibromatosis type 1–related plexiform neurofibromas. N. Engl. J. Med. 2016, 26, 2550–2560. [Google Scholar] [CrossRef]
- Gounder, M.M.; Zer, A.; Tap, W.D.; Salah, S.; Dickson, M.A.; Gupta, A.A.; Keohan, M.L.; Loong, H.H.; D’Angelo, S.P.; Baker, S.; et al. Phase IB study of selinexor, a first-in-class inhibitor of nuclear export, in patients with advanced refractory bone or soft tissue sarcoma. J. Clin. Oncol. 2016, 26, 3166–3174. [Google Scholar] [CrossRef]
- Fiala, O.; Pesek, M.; Finek, J.; Benesova, L.; Belsanova, B.; Minarik, M. The dominant role of G12C over other KRAS mutation types in the negative prediction of efficacy of epidermal growth factor receptor tyrosine kinase inhibitors in non–small cell lung cancer. Cancer Genet. 2013, 1, 26–31. [Google Scholar] [CrossRef]
- Grosenbach, D.W.; Jordan, R.; Hruby, D.E. Development of the small-molecule antiviral ST-246® as a smallpox therapeutic. Future Virol. 2011, 5, 653–671. [Google Scholar] [CrossRef]
- Bourlière, M.; Gordon, S.C.; Flamm, S.L.; Cooper, C.L.; Ramji, A.; Tong, M.; Ravendhran, N. Sofosbuvir, velpatasvir, and voxilaprevir for previously treated HCV infection. N. Eng. J. Med. 2017, 22, 2134–2146. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Curtin, N.J. The role of PARP in DNA repair and its therapeutic exploitation. Br. J. Cancer 2011, 8, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- Rajapakse, S.; Rodrigo, C.; Fernando, S.D. Tafenoquine for preventing relapse in people with Plasmodium vivax malaria. Cochrane Database Syst. Rev. 2015, 4, CD010458. [Google Scholar] [CrossRef]
- Jordheim, L.P.; Durantel, D.; Zoulim, F. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 2013, 12, 447–464. [Google Scholar] [CrossRef]
- Freedman, A.; Jacobsen, E. Follicular lymphoma: 2020 update on diagnosis and management. Am. J. Hematol. 2020, 95, 316–327. [Google Scholar] [CrossRef]
- Goldstein, J.; Silberstein, S.D.; Saper, J.R.; Elkind, A.H.; Smith, T.R.; Gallagher, R.M.; Battikha, J.P.; Hoffman, H.; Baggish, J. Acetaminophen, aspirin, and caffeine versus sumatriptan succinate in the early treatment of migraine: Results from the ASSET trial. Headache J. Head Face Pain 2005, 8, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Matthes, E.; Lehmann, C.; Scholz, D.; Rosenthal, H.A.; Langen, P. Phosphorylation, anti-HIV activity and cytotoxicity of 3′-fluorothymidine. Biochem. Biophys. Res. Commun. 1998, 153, 825–831. [Google Scholar] [CrossRef]
- Jakate, A.; Blumenfeld, A.M.; Boinpally, R.; Butler, M.; Borbridge, L.; Contreras-De Lama, J.; Lipton, R.B. Pharmacokinetics and safety of ubrogepant when coadministered with calcitonin gene-related peptide-targeted monoclonal antibody migraine preventives in participants with migraine: A randomized phase 1b drug–drug interaction study. Headache J. Head. Face Pain 2021, 61, 642–652. [Google Scholar] [CrossRef]
- Fleischmann, R.; Pangan, A.L.; Song, I.H.; Mysler, E.; Bessette, L.; Peterfy, C.; Genovese, M.C. Upadacitinib versus placebo or adalimumab in patients with rheumatoid arthritis and an inadequate response to methotrexate: Results of a phase III, double-blind, randomized controlled trial. Arthritis Rheumatol. 2019, 71, 1788–1800. [Google Scholar] [CrossRef]
- Marquez, V.E.; Tseng, C.K.; Mitsuya, H.; Aoki, S.; Kelley, J.A.; Ford, H., Jr.; Roth, J.S.; Broder, S.; Johns, D.G.; Driscoll, J.S. Acid-stable 2′-fluoro purine dideoxynucleosides as active agents against HIV. J. Med. Chem. 1990, 33, 978–985. [Google Scholar] [CrossRef]
- Das, P.; Delost, M.D.; Qureshi, M.H.; Smith, D.T.; Njardarson, J.T. A Survey of the Structures of US FDA Approved Combination Drugs. J. Med. Chem. 2019, 62, 4265–4311. [Google Scholar] [CrossRef] [PubMed]
- Isanbor, C.; David, O.H. Fluorine in medicinal chemistry: A review of anti-cancer agents. J. Fluor. Chem. 2006, 127, 303–319. [Google Scholar] [CrossRef]
- Chopra, D.; Tayur, N.; Guru, R. Role of organic fluorine in crystal engineering. CrystEngComm 2011, 13, 2175–2186. [Google Scholar] [CrossRef]
- Cozzi, P.; Mongelli, N.; Suarato, A. Recent anticancer cytotoxic agents. Curr. Med. Chem. 2004, 4, 93–121. [Google Scholar] [CrossRef] [PubMed]
- Finger, V.; Kufa, M.; Soukup, O.; Castagnolo, D.; Roh, J.; Korabecny, J. Pyrimidine derivatives with antitubercular activity. Eur. J. Med. Chem. 2023, 246, 114946. [Google Scholar] [CrossRef]
- Basha, J.; Goudgaon, N.M. A comprehensive review on pyrimidine analogs-versatile scaffold with medicinal and biological potential. J. Mol. Struct. 2021, 1246, 131168. [Google Scholar] [CrossRef]
- Zarenezhad, E.; Farjam, M.; Iraji, A. Synthesis and biological activity of pyrimidines-containing hybrids: Focusing on pharmacological application. J. Mol. Struct. 2021, 1230, 129833. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Barry, W.T.; Birrer, M.; Westin, S.N.; Caddo, K.A.; Shapiro, G.I.; Mayer, E.L. Olaparib and α-specific PI3K inhibitor alpelisib for patients with epithelial ovarian cancer: A dose-escalation and dose-expansion phase 1b trial. Lancet Oncol. 2019, 4, 570–580. [Google Scholar] [CrossRef]
- Collier, T.L.; Maresca, K.P.; Normandin, M.D.; Richardson, P.; McCarthy, T.J.; Liang, S.H.; Waterhouse, R.N.; Vasdev, N. Brain penetration of the ROS1/ALK inhibitor lorlatinib confirmed by PET. Mol. Imaging 2017, 16, 1536012117736669. [Google Scholar] [CrossRef]
- Abraham, G.M.; Morton, J.B.; Saravolatz, L.D. Baloxavir: A novel antiviral agent in the treatment of influenza. Clin. Infect. Dis. 2020, 7, 1790–1794. [Google Scholar] [CrossRef]
- Braselmann, S.; Taylor, V.; Zhao, H.; Wang, S.; Sylvain, C.; Baluom, M.; Qu, K. R406, an orally available spleen tyrosine kinase inhibitor blocks fc receptor signaling and reduces immune complex-mediated inflammation. J. Pharmacol. Exp. Ther. 2006, 3, 998–1008. [Google Scholar] [CrossRef] [PubMed]
- Chase, S.P.; Freimanis, A.K. Abdominal echography. Ohio State Med. J. 1971, 10, 901–906. [Google Scholar]
- Bowers, G.D.; Culp, A.; Reese, M.J.; Tabolt, G.; Moss, L.; Piscitelli, S.; Huynh, P. Disposition and metabolism of cabotegravir: A comparison of biotransformation and excretion between different species and routes of administration in humans. Xenobiotica 2016, 2, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Weidmann, H.; Heikaus, L.; Long, A.T.; Naudin, C.; Schlüter, H.; Renné, T. The plasma contact system, a protease cascade at the nexus of inflammation, coagulation and immunity. BBA Mol. Cell Res. 2017, 11, 2118–2127. [Google Scholar] [CrossRef]
- Taylor-Cousar, J.L.; Mall, M.A.; Ramsey, B.W.; McKone, E.F.; Tullis, E.; Marigowda, G.; McKee, C.M. Clinical development of triple-combination CFTR modulators for cystic fibrosis patients with one or two F508del alleles. ERJ Open Res. 2019, 5, 00082–02019. [Google Scholar] [CrossRef]
- Beuckmann, C.T.; Suzuki, M.; Uneo, T.; Nagaoka, K.; Arai, T.; Higashiyama, H. In vitro and in silico characterization of lemborexant (E2006), a novel dual orexin receptor antagonist. J. Pharmacol. Exp. Ther. 2017, 2, 287–295. [Google Scholar] [CrossRef]
- Follmann, M.; Ackerstaff, J.; Redlich, G.; Wunder, F.; Lang, D.; Kern, A.; Stasch, J.P. Discovery of the soluble guanylate cyclase stimulator vericiguat (BAY 1021189) for the treatment of chronic heart failure. J. Med. Chem. 2017, 12, 5146–5161. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Name | Structure | Mode of Action/Uses | References |
|---|---|---|---|
| Flortaucipir F-18 |  | Small lipophilic tracer containing 18F capable of crossing the blood–brain barrier and binding to aggregated tau proteins; used in PET imaging for the diagnosis of Alzheimer’s disease. | [43] |
| Fluciclovine F-18 |  | Transported into prostate cancer cells via ASCT2 and LAT1 transporters. | [44] |
| Fluorodopa F-18 |  | Decarboxylated by aromatic amino acid decarboxylase (AADC) in the striatum to fluorodopa F-18. Fluorodopa F-18 is then further metabolized by monoamine oxidase (MAO) to yield 18F 6-fluoro-3,4-dihydroxyphenylacetic acid. | [45] |
| Fluoroestradiol F-18 |  | Radioactive diagnostic agent for the detection of estrogen receptor-positive lesions. | [46] |
| Piflufolastat F-18 |  | Binds to PSMA and allows for the visualization of PSMA-positive lesions associated with prostate cancer. | [47] |
| Name | Mode of Action with Therapeutic Uses | References |
|---|---|---|
| Givosiran | 5-aminolevulinic acid synthase-directed small interfering RNA (siRNA) used in the prophylaxis of acute hepatic porphyria. | [51] |
| Inclisiran | PCSK9-targeting siRNA that lowers plasma LDL cholesterol levels. | [49] |
| Lumasiran | siRNA that silences hydroxyacid oxidase 1 for the treatment of primary hyperoxaluria type 1. | [50] |
| Vutrisiran | Transthyretin-directed siRNA used to treat polyneuropathy associated with hereditary transthyretin-mediated amyloidosis. | [48] |
| Name | Structure | Mode of Action | References |
|---|---|---|---|
| Abemaciclib |  | CDK4/cyclin D1 complex inhibitor | [70] |
| Alpelisib |  | Phosphatidylinositol 3-kinase (PI3K) inhibitor | [71] |
| Atogepant |  | CGRP receptor antagonist | [72] |
| Apalutamide |  | Androgen receptor inhibitor | [73] |
| Asciminib |  | ABL/BCR–ABL1 tyrosine kinase inhibitor | [74] |
| Avacopan |  | 5a receptor (C5aR) antagonist | [75] |
| Avapritinib |  | Selective tyrosine kinase inhibitor | [76] |
| Baloxavir marboxil |  | Endonuclease inhibitor | [77] |
| Belzutifan |  | Hypoxia-inducible factor 2 inhibitor | [78] |
| Berotralstat |  | Plasma kallikrein inhibitor | [79] |
| Bictegravir |  | Integrase inhibitor | [80] |
| Binimetinib |  | MEK inhibitor | [81] |
| Cabotegravir |  | HIV-1 integrase inhibitor | [82] |
| Capmatinib |  | Kinase inhibitor | [83] |
| Dacomitinib |  | EGFR protein inhibitor, including EGFR/HER1, HER2, and HER4 | [84] |
| Delafloxacin |  | Bacterial DNA gyrase and topoisomerase IV inhibitor | [85] |
| Doravirine |  | Non-nucleoside reverse transcriptase inhibitor | [86] |
| Elagolix |  | Gonadotropin-releasing hormone receptor antagonist (GnRH) | [87] |
| Elexacaftor |  | CFTR corrector | [88] |
| Emtricitabine |  | Nucleoside reverse transcriptase inhibitor (NRTI) | [89] |
| Enasidenib |  | Isocitrate dehydrogenase-2 inhibitor | [90] |
| Encorafenib |  | Kinase inhibitor | [91] |
| Eravacycline |  | Binds to the bacterial ribosomal 30S subunit | [92] |
| Fostamatinib |  | Tyrosine kinase inhibitor | [93] |
| Glecaprevir |  | NS3/4A and NS5A inhibitor | [94] |
| Ivosidenib |  | Isocitrate dehydrogenase-1 (IDH1) inhibitor | [95] |
| Larotrectinib |  | Tropomyosin receptor kinase (Trk) inhibitor | [96] |
| Lasmiditan |  | High-affinity serotonin (5-HT) 1F receptor agonist | [87] |
| Lemborexant |  | OX1R and OX2R antagonist | [97] |
| Letermovir |  | DNA terminase CMV complex inhibitor | [98] |
| Lorlatinib |  | Tyrosine kinase inhibitor | [99] |
| Lumateperone |  | Receptor antagonist of 5-HT2A receptor and antagonist of several dopamine receptors (D1, D2, and D4) | [100] |
| Melphalan flufenamide |  | Peptidase-enhanced cytotoxic (PEnC) | [101] |
| Netupitant |  | Selective neurokinin 1 (NK1) receptor antagonist | [100] |
| Osilodrostat |  | Cortisol synthesis inhibitor | [102] |
| Pexidartinib |  | Tyrosine kinase inhibitor | [103] |
| Pibrentasvir |  | NS3/4A and an NS5A inhibitor | [94] |
| Pralsetinib |  | Kinase inhibitor | [104] |
| Pretomanid |  | Inhibitor of cell wall biosynthesis via blockage of the oxidation of hydroxymycolate to ketomycolate | [72] |
| Pimavanserin |  | Serotonin 5-HT2A receptor antagonist | [105] |
| Relugolix |  | Nonpeptide GnRH receptor antagonist | [106] |
| Ripretinib |  | Protein kinase inhibitor | [107] |
| Rucaparib |  | Polymerase inhibitor | [108] |
| Selinexor |  | Selective nuclear transport (SINE) inhibitor | [109] |
| Selumetinib |  | Selective MEK 1 and MEK 2 inhibitor | [110] |
| Siponimod |  | Sphingosine 1-phosphate receptor (S1P) modulator | [111] |
| Sofosbuvir |  | Hepatitis C NS5B protein inhibitor | [112] |
| Sotorasib |  | KRAS G12C inhibitor | [113] |
| Tafenoquine |  | Antiparasitic agent | [114] |
| Talazoparib |  | PARP1/2 enzyme inhibitor | [115] |
| Tecovirimat |  | Orthopoxvirus VP37 inhibitor | [116] |
| Telotristat Ethyl |  | Tryptophan hydroxylase inhibitor | [117] |
| Tezacaftor |  | CFTR potentiator | [88] |
| Ubrogepant |  | CGRP antagonist | [114] |
| Umbralisib |  | Kinase inhibitor | [118] |
| Upadacitinib |  | Selective Janus kinase-1 (JAK-1) inhibitor | [119] |
| Vericiguat |  | Guanylate cyclase (sGC) direct stimulant | [120] |
| Voxilaprevir |  | Protease inhibitor | [121] |
| Name | Structure | Therapeutic Uses |
|---|---|---|
| Abemaciclib |  | Solid tumors, including glioblastoma, melanoma, and lymphoma |
| Alpelisib |  | Strong anticancer efficacy |
| Apalutamide |  | Non-metastatic castration-resistant prostate cancer |
| Avapritinib |  | Gastrointestinal cancers that are resistant to many drugs |
| Belzutifan |  | Antineoplastic for the treatment of some tumors connected to Von Hippel–Lindau (VHL) disease |
| Binimetinib |  | Chemotherapy drug with anti-tumor properties |
| Capmatinib |  | Non-small-cell lung cancer (NSCLC) |
| Dacomitinib |  | EGRF-mutated NSCLC |
| Encorafenib |  | Metastatic melanoma |
| Ivosidenib |  | Malignancies with a sensitive IDH1 mutation |
| Larotrectinib |  | Solid tumors |
| Lorlatinib |  | NSCLC |
| Pexidartinib |  | Tenosynovial giant cell tumor (TGCT) |
| Pralsetinib |  | NSCLC |
| Relugolix |  | Prostate cancer |
| Ripretinib |  | Gastrointestinal stromal tumors |
| Rucaparib |  | Adult patients with recurrent prostate and ovarian cancer |
| Selumetinib |  | NSCLC |
| Sotorasib |  | Mutant NSCLC harboring the KRAS G12C mutation |
| Talazoparib |  | Germline BRCA-mutant, HER2-negative, locally advanced, or metastatic breast cancer |
| Name | Structure | Therapeutic Uses |
|---|---|---|
| Baloxavir marboxil |  | Influenza |
| Fostamatinib |  | Acute respiratory distress syndrome (ARDS) in patients with severe COVID-19 |
| Glecaprevir |  | Chronic hepatitis C virus |
| Letermovir |  | CMV infection as well as the prevention of infection following bone marrow transplantation. |
| Osilodrostat |  | Cushing’s disease |
| Pibrentasvir |  | Chronic hepatitis C virus |
| Siponimod |  | Multiple sclerosis |
| Sofosbuvir |  | Hepatitis C in both adults and children older than 3 years |
| Tecovirimat |  | Human smallpox sickness in adults and children |
| Umbralisib |  | Non-Hodgkin lymphoma |
| Voxilaprevir |  | Hepatitis C infection |
| Name | Structure | Therapeutic Uses |
|---|---|---|
| Bictegravir |  | Treatment of HIV-1 infection |
| Cabotegravir |  | Treatment and prevention of HIV-1 infection |
| Doravirine |  | Treatment of HIV-1 infection in adult patients |
| Emtricitabine |  | Treatment of HIV infection in adults and prevention of HIV infection in high-risk adults and adolescents when coupled with tenofovir alafenamide |
| Name | Structure | Therapeutic Uses |
|---|---|---|
| Berotralstat |  | Prevention of attacks of hereditary angioedema (HAE) |
| Elexacaftor |  | Treatment of cystic fibrosis |
| Eravacycline |  | Antibiotic |
| Pretomanid |  | Treatment of pulmonary tuberculosis |
| Telotristat Ethyl |  | Treatment of diarrhea caused by carcinoid syndrome |
| Tezacaftor |  | Treatment of cystic fibrosis |
| Name | Structure | Therapeutic Uses |
|---|---|---|
| Asciminib |  | Chronic-phase chronic myeloid leukemia (Ph+ CML) |
| Atogepant |  | Episodic migraine headaches |
| Avacopan |  | Granulomatosis polyangiitis (GPA) or microscopic polyangiitis |
| Delafloxacin |  | Skin structural infections |
| Elagolix |  | Endometriosis-related discomfort |
| Enasidenib |  | Acute myeloid leukemia |
| Lasmiditan |  | Acute migraine headaches with or without aura |
| Lemborexant |  | Insomnia that interferes with falling asleep and/or staying asleep |
| Lumateperone |  | Positive and negative symptoms in schizophrenia patients |
| Melphalan flufenamide |  | Relapsed or resistant multiple myeloma |
| Netupitant |  | Nausea and vomiting brought on by chemotherapy in addition to other medications |
| Pimavanserin |  | Anti-psychotic drug |
| Selinexor |  | Multiple myeloma |
| Tafenoquine |  | Treatment and prevention of vivax malaria |
| Upadacitinib |  | Moderate to severe forms of rheumatoid arthritis, active psoriatic arthritis, ankylosing spondylitis, and severe atopic dermatitis |
| Ubrogepant |  | Acute migraines both with and without aura |
| Vericiguat |  | Systolic heart failure. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shabir, G.; Saeed, A.; Zahid, W.; Naseer, F.; Riaz, Z.; Khalil, N.; Muneeba; Albericio, F. Chemistry and Pharmacology of Fluorinated Drugs Approved by the FDA (2016–2022). Pharmaceuticals 2023, 16, 1162. https://doi.org/10.3390/ph16081162
Shabir G, Saeed A, Zahid W, Naseer F, Riaz Z, Khalil N, Muneeba, Albericio F. Chemistry and Pharmacology of Fluorinated Drugs Approved by the FDA (2016–2022). Pharmaceuticals. 2023; 16(8):1162. https://doi.org/10.3390/ph16081162
Chicago/Turabian StyleShabir, Ghulam, Aamer Saeed, Wajeeha Zahid, Fatima Naseer, Zainab Riaz, Nafeesa Khalil, Muneeba, and Fernando Albericio. 2023. "Chemistry and Pharmacology of Fluorinated Drugs Approved by the FDA (2016–2022)" Pharmaceuticals 16, no. 8: 1162. https://doi.org/10.3390/ph16081162
APA StyleShabir, G., Saeed, A., Zahid, W., Naseer, F., Riaz, Z., Khalil, N., Muneeba, & Albericio, F. (2023). Chemistry and Pharmacology of Fluorinated Drugs Approved by the FDA (2016–2022). Pharmaceuticals, 16(8), 1162. https://doi.org/10.3390/ph16081162