
Metabotropic Glutamate Receptor Subtype 5 Positron-Emission-Tomography Radioligands as a Tool for Central Nervous System Drug Development: Between Progress and Setbacks
 , , and
, , and
Abstract
1. Introduction
1.1. mGluR5 as a Target of Interest
1.2. Quantitative PET Imaging in Drug Development
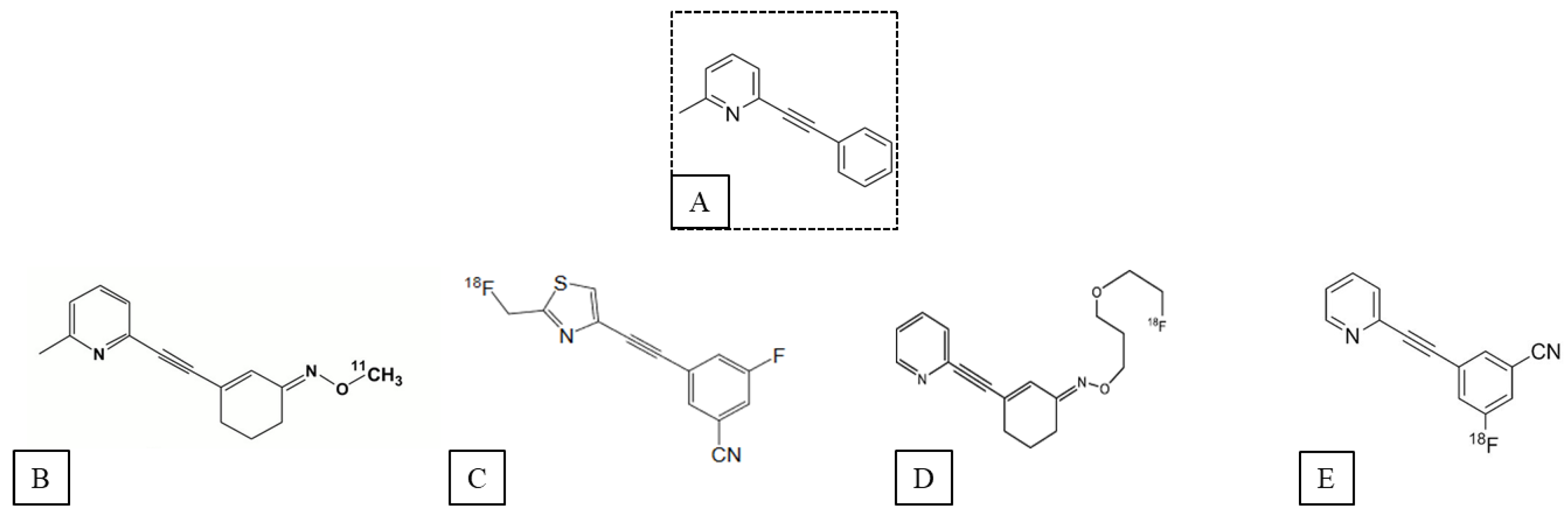
2. mGluR5 PET Ligands
2.1. Ligands of the Orthosteric Site of mGluR5
2.2. Ligands of the Allosteric Sites of mGluR5
2.2.1. “Cold” Ligands of the mGluR5 Allosteric Site
2.2.2. Allosteric Radioligands of mGluR5
3. mGluR5 PET Imaging and Its Impact in CNS Drug Development
- (i)
- Imaging the pathological hallmarks of disease;
- (ii)
- Receptor occupancies studies;
- (iii)
- Detection of a drug’s distribution and tissue kinetics;
- (iv)
- Monitoring treatment effect.
4. Discussion
- In the original competition model, where the endogenous ligand and the radioligand share the same binding site, the PET signal reflects the number of available receptors not occupied by the endogenous ligand. Most mGlur5 PET tracers are negative allosteric modulators (=non-competitive antagonist) that do not share the same binding site as endogenous glutamate. Thus, the theory that increasing synaptic concentrations of endogenous ligand reduces the number of receptors available for the radiotracer is challenged with an allosteric radiotracer. However, modeling studies have shown that minor changes in structure (which can occur during metabolism) can change the mode of pharmacology (e.g., from PAM to NAM or SAM or vice versa), the affinity to the allosteric site and even the selectivity to the receptor subtype. This phenomenon is called the “molecular switch” [4].
- Besides allosterism, the impact of the endogenous neuromodulator on radioligand binding can significantly limit the interpretation of various clinical PET studies and likely accounts for certain ongoing controversies within the field. Numerous preclinical and clinical studies have evaluated the influence of endogenous glutamate concentration on mGluR5 radioligand binding using a glutamatergic modulator such as N-acetylcystéine, ceftriaxone or ketamine [48,49,88,89,90,91,92,93,94]. Overall, it appears that [11C]ABP688 is more sensitive to changes in endogenous glutamate than the other mGluR5 radioligands. However, the precise mechanism behind this alteration in [11C]ABP688 binding remains unclear and cannot be attributed solely to straightforward direct competition.
- The mechanism responsible for this change in [11C]ABP688 binding is not clearly identified and cannot be explained by simple direct competition.
- The pharmacokinetic approach adapted to functional neuroimaging has also be discussed in a number of studies. Dynamic PET acquisitions consisting of a series of temporal images acquired over a certain time (frame) allow a precise measurement of the radiotracer kinetics. It depends on the number of receptors in the target organ, its affinity, non-specific binding, cerebral blood flow and the concentration of endogenous competitors. For the estimation of the detailed parameters (receptor density, KD, BP), the use of activity–time pharmacokinetic modeling of the tracer is required. The standard pharmacokinetic model for neuroreceptors is based on the three-compartment, two-tissue model. From the arterial blood as the first compartment, the free exchangeable radioligand in the plasma passes into the second compartment called the free compartment. The third compartment is the region of specific binding, the region of interest. The fourth compartment is a non-specific exchange compartment with the free compartment. In practice, for most radioligands, the non-specific binding compartment is in rapid equilibrium with the free compartment and the two compartments are treated as a single compartment. Yet, there is great heterogeneity in the pharmacokinetic models used and there is no consensus on the reference region to quantify the non-specific binding.
- Finally, the concept of receptor internalization by endogenous agonist stimulation is now well described for GPCR. But very little data are available on the ability of an allosteric radioligand to bind to its transmembrane allosteric binding site when mGluR5 is internalized.
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, Y.; Danbolt, N.C. Glutamate as a Neurotransmitter in the Healthy Brain. J. Neural Transm. Vienna Austria 1996 2014, 121, 799–817. [Google Scholar] [CrossRef]
- Jenner, P.; Caccia, C. The Role of Glutamate in the Healthy Brain and in the Pathophysiology of Parkinson’s Disease. Eur. Neurol. Rev. 2019, 14, 2–12. [Google Scholar]
- Mayer, M.L.; Armstrong, N. Structure and Function of Glutamate Receptor Ion Channels. Annu. Rev. Physiol. 2004, 66, 161–181. [Google Scholar] [CrossRef]
- Wood, M.R.; Hopkins, C.R.; Brogan, J.T.; Conn, P.J.; Lindsley, C.W. “Molecular Switches” on MGluR Allosteric Ligands that Modulate Modes of Pharmacology. Biochemistry 2011, 50, 2403–2410. [Google Scholar] [CrossRef]
- Pin, J.P.; Duvoisin, R. The Metabotropic Glutamate Receptors: Structure and Functions. Neuropharmacology 1995, 34, 1–26. [Google Scholar] [CrossRef]
- Shigemoto, R.; Nomura, S.; Ohishi, H.; Sugihara, H.; Nakanishi, S.; Mizuno, N. Immunohistochemical Localization of a Metabotropic Glutamate Receptor, MGluR5, in the Rat Brain. Neurosci. Lett. 1993, 163, 53–57. [Google Scholar] [CrossRef]
- Ferraguti, F.; Shigemoto, R. Metabotropic Glutamate Receptors. Cell Tissue Res. 2006, 326, 483–504. [Google Scholar] [CrossRef]
- Biber, K.; Laurie, D.J.; Berthele, A.; Sommer, B.; Tölle, T.R.; Gebicke-Härter, P.J.; van Calker, D.; Boddeke, H.W. Expression and Signaling of Group I Metabotropic Glutamate Receptors in Astrocytes and Microglia. J. Neurochem. 1999, 72, 1671–1680. [Google Scholar] [CrossRef]
- Matosin, N.; Newell, K.A. Metabotropic Glutamate Receptor 5 in the Pathology and Treatment of Schizophrenia. Neurosci. Biobehav. Rev. 2013, 37, 256–268. [Google Scholar] [CrossRef]
- Kumaresan, V.; Yuan, M.; Yee, J.; Famous, K.R.; Anderson, S.M.; Schmidt, H.D.; Pierce, R.C. Metabotropic Glutamate Receptor 5 (MGluR5) Antagonists Attenuate Cocaine Priming- and Cue-Induced Reinstatement of Cocaine Seeking. Behav. Brain Res. 2009, 202, 238–244. [Google Scholar] [CrossRef]
- Terbeck, S.; Akkus, F.; Chesterman, L.P.; Hasler, G. The Role of Metabotropic Glutamate Receptor 5 in the Pathogenesis of Mood Disorders and Addiction: Combining Preclinical Evidence with Human Positron Emission Tomography (PET) Studies. Front. Neurosci. 2015, 9, 86. [Google Scholar] [CrossRef]
- Dölen, G.; Bear, M.F. Role for Metabotropic Glutamate Receptor 5 (MGluR5) in the Pathogenesis of Fragile X Syndrome. J. Physiol. 2008, 586, 1503–1508. [Google Scholar] [CrossRef]
- Mehta, M.V.; Gandal, M.J.; Siegel, S.J. MGluR5-Antagonist Mediated Reversal of Elevated Stereotyped, Repetitive Behaviors in the VPA Model of Autism. PLoS ONE 2011, 6, e26077. [Google Scholar] [CrossRef]
- Pin, J.-P.; Galvez, T.; Prézeau, L. Evolution, Structure, and Activation Mechanism of Family 3/C G-Protein-Coupled Receptors. Pharmacol. Ther. 2003, 98, 325–354. [Google Scholar] [CrossRef]
- Monod, J.; Wyman, J.; Changeux, J.P. On the Nature of Allosteric Transitions: A Plausible Model. J. Mol. Biol. 1965, 12, 88–118. [Google Scholar] [CrossRef]
- Yin, S.; Niswender, C.M. Progress toward Advanced Understanding of Metabotropic Glutamate Receptors: Structure, Signaling and Therapeutic Indications. Cell. Signal. 2014, 26, 2284–2297. [Google Scholar] [CrossRef]
- Conn, P.J.; Christopoulos, A.; Lindsley, C.W. Allosteric Modulators of GPCRs: A Novel Approach for the Treatment of CNS Disorders. Nat. Rev. Drug Discov. 2009, 8, 41–54. [Google Scholar] [CrossRef]
- Christopoulos, A. Allosteric Binding Sites on Cell-Surface Receptors: Novel Targets for Drug Discovery. Nat. Rev. Drug Discov. 2002, 1, 198–210. [Google Scholar] [CrossRef]
- Levenga, J.; de Vrij, F.M.S.; Oostra, B.A.; Willemsen, R. Potential Therapeutic Interventions for Fragile X Syndrome. Trends Mol. Med. 2010, 16, 516–527. [Google Scholar] [CrossRef]
- Hagerman, R.; Jacquemont, S.; Berry-Kravis, E.; Des Portes, V.; Stanfield, A.; Koumaras, B.; Rosenkranz, G.; Murgia, A.; Wolf, C.; Apostol, G.; et al. Mavoglurant in Fragile X Syndrome: Results of Two Open-Label, Extension Trials in Adults and Adolescents. Sci. Rep. 2018, 8, 16970. [Google Scholar] [CrossRef]
- Lindemann, L.; Porter, R.H.; Scharf, S.H.; Kuennecke, B.; Bruns, A.; von Kienlin, M.; Harrison, A.C.; Paehler, A.; Funk, C.; Gloge, A.; et al. Pharmacology of Basimglurant (RO4917523, RG7090), a Unique Metabotropic Glutamate Receptor 5 Negative Allosteric Modulator in Clinical Development for Depression. J. Pharmacol. Exp. Ther. 2015, 353, 213–233. [Google Scholar] [CrossRef]
- Wellendorph, P.; Bräuner-Osborne, H. Molecular Basis for Amino Acid Sensing by Family C G-Protein-Coupled Receptors. Br. J. Pharmacol. 2009, 156, 869–884. [Google Scholar] [CrossRef]
- Flor, P.J.; Acher, F.C. Orthosteric versus Allosteric GPCR Activation: The Great Challenge of Group-III MGluRs. Biochem. Pharmacol. 2012, 84, 414–424. [Google Scholar] [CrossRef]
- Porter, R.H.; Roberts, P.J.; Jane, D.E.; Watkins, J.C. (S)-Homoquisqualate: A Potent Agonist at the Glutamate Metabotropic Receptor. Br. J. Pharmacol. 1992, 106, 509–510. [Google Scholar] [CrossRef]
- Doherty, A.J.; Palmer, M.J.; Henley, J.M.; Collingridge, G.L.; Jane, D.E. (RS)-2-Chloro-5-Hydroxyphenylglycine (CHPG) Activates MGlu5, but No MGlu1, Receptors Expressed in CHO Cells and Potentiates NMDA Responses in the Hippocampus. Neuropharmacology 1997, 36, 265–267. [Google Scholar] [CrossRef]
- Thomsen, C.; Boel, E.; Suzdak, P.D. Actions of Phenylglycine Analogs at Subtypes of the Metabotropic Glutamate Receptor Family. Eur. J. Pharmacol. 1994, 267, 77–84. [Google Scholar] [CrossRef]
- Brabet, I.; Mary, S.; Bockaert, J.; Pin, J.P. Phenylglycine Derivatives Discriminate between MGluR1- and MGluR5-Mediated Responses. Neuropharmacology 1995, 34, 895–903. [Google Scholar] [CrossRef]
- Sengmany, K.; Gregory, K.J. Metabotropic Glutamate Receptor Subtype 5: Molecular Pharmacology, Allosteric Modulation and Stimulus Bias. Br. J. Pharmacol. 2016, 173, 3001–3017. [Google Scholar] [CrossRef]
- Mølck, C.; Harpsøe, K.; Gloriam, D.E.; Mathiesen, J.M.; Nielsen, S.M.; Bräuner-Osborne, H. MGluR5: Exploration of Orthosteric and Allosteric Ligand Binding Pockets and Their Applications to Drug Discovery. Neurochem. Res. 2014, 39, 1862–1875. [Google Scholar] [CrossRef]
- Gregory, K.J.; Conn, P.J. Molecular Insights into Metabotropic Glutamate Receptor Allosteric Modulation. Mol. Pharmacol. 2015, 88, 188–202. [Google Scholar] [CrossRef]
- Doré, A.S.; Okrasa, K.; Patel, J.C.; Serrano-Vega, M.; Bennett, K.; Cooke, R.M.; Errey, J.C.; Jazayeri, A.; Khan, S.; Tehan, B.; et al. Structure of Class C GPCR Metabotropic Glutamate Receptor 5 Transmembrane Domain. Nature 2014, 511, 557–562. [Google Scholar] [CrossRef]
- Christopoulos, A.; Kenakin, T. G Protein-Coupled Receptor Allosterism and Complexing. Pharmacol. Rev. 2002, 54, 323–374. [Google Scholar] [CrossRef]
- Varney, M.A.; Cosford, N.D.; Jachec, C.; Rao, S.P.; Sacaan, A.; Lin, F.F.; Bleicher, L.; Santori, E.M.; Flor, P.J.; Allgeier, H.; et al. SIB-1757 and SIB-1893: Selective, Noncompetitive Antagonists of Metabotropic Glutamate Receptor Type 5. J. Pharmacol. Exp. Ther. 1999, 290, 170–181. [Google Scholar]
- Gasparini, F.; Lingenhöhl, K.; Stoehr, N.; Flor, P.J.; Heinrich, M.; Vranesic, I.; Biollaz, M.; Allgeier, H.; Heckendorn, R.; Urwyler, S.; et al. 2-Methyl-6-(Phenylethynyl)-Pyridine (MPEP), a Potent, Selective and Systemically Active MGlu5 Receptor Antagonist. Neuropharmacology 1999, 38, 1493–1503. [Google Scholar] [CrossRef]
- Cosford, N.D.P.; Roppe, J.; Tehrani, L.; Schweiger, E.J.; Seiders, T.J.; Chaudary, A.; Rao, S.; Varney, M.A. [3H]-Methoxymethyl-MTEP and [3H]-Methoxy-PEPy: Potent and Selective Radioligands for the Metabotropic Glutamate Subtype 5 (MGlu5) Receptor. Bioorg. Med. Chem. Lett. 2003, 13, 351–354. [Google Scholar] [CrossRef]
- Lea, P.M.; Faden, A.I. Metabotropic Glutamate Receptor Subtype 5 Antagonists MPEP and MTEP. CNS Drug Rev. 2006, 12, 149–166. [Google Scholar] [CrossRef]
- Gregory, K.J.; Nguyen, E.D.; Malosh, C.; Mendenhall, J.L.; Zic, J.Z.; Bates, B.S.; Noetzel, M.J.; Squire, E.F.; Turner, E.M.; Rook, J.M.; et al. Identification of Specific Ligand-Receptor Interactions That Govern Binding and Cooperativity of Diverse Modulators to a Common Metabotropic Glutamate Receptor 5 Allosteric Site. ACS Chem. Neurosci. 2014, 5, 282–295. [Google Scholar] [CrossRef]
- Hammond, A.S.; Rodriguez, A.L.; Townsend, S.D.; Niswender, C.M.; Gregory, K.J.; Lindsley, C.W.; Conn, P.J. Discovery of a Novel Chemical Class of MGlu(5) Allosteric Ligands with Distinct Modes of Pharmacology. ACS Chem. Neurosci. 2010, 1, 702–716. [Google Scholar] [CrossRef]
- Rook, J.M.; Tantawy, M.N.; Ansari, M.S.; Felts, A.S.; Stauffer, S.R.; Emmitte, K.A.; Kessler, R.M.; Niswender, C.M.; Daniels, J.S.; Jones, C.K.; et al. Relationship between in Vivo Receptor Occupancy and Efficacy of Metabotropic Glutamate Receptor Subtype 5 Allosteric Modulators with Different in Vitro Binding Profiles. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2015, 40, 755–765. [Google Scholar] [CrossRef]
- Ametamey, S.M.; Kessler, L.J.; Honer, M.; Wyss, M.T.; Buck, A.; Hintermann, S.; Auberson, Y.P.; Gasparini, F.; Schubiger, P.A. Radiosynthesis and Preclinical Evaluation of 11C-ABP688 as a Probe for Imaging the Metabotropic Glutamate Receptor Subtype 5. J. Nucl. Med. 2006, 47, 698–705. [Google Scholar]
- Wang, J.-Q.; Tueckmantel, W.; Zhu, A.; Pellegrino, D.; Brownell, A.-L. Synthesis and Preliminary Biological Evaluation of 3-[18F]Fluoro-5-(2-Pyridinylethynyl)Benzonitrile as a PET Radiotracer for Imaging Metabotropic Glutamate Receptor Subtype 5. Synapse 2007, 61, 951–961. [Google Scholar] [CrossRef]
- Siméon, F.G.; Brown, A.K.; Zoghbi, S.S.; Patterson, V.M.; Innis, R.B.; Pike, V.W. Synthesis and Simple 18F-Labeling of 3-Fluoro-5-(2-(2-(Fluoromethyl)Thiazol-4-Yl)Ethynyl)Benzonitrile as a High Affinity Radioligand for Imaging Monkey Brain Metabotropic Glutamate Subtype-5 Receptors with Positron Emission Tomography. J. Med. Chem. 2007, 50, 3256–3266. [Google Scholar] [CrossRef]
- Sephton, S.M.; Dennler, P.; Leutwiler, D.S.; Mu, L.; Wanger-Baumann, C.A.; Schibli, R.; Krämer, S.D.; Ametamey, S.M. Synthesis, Radiolabelling and in Vitro and in Vivo Evaluation of a Novel Fluorinated ABP688 Derivative for the PET Imaging of Metabotropic Glutamate Receptor Subtype 5. Am. J. Nucl. Med. Mol. Imaging 2012, 2, 14. [Google Scholar]
- Varnäs, K.; Juréus, A.; Finnema, S.J.; Johnström, P.; Raboisson, P.; Amini, N.; Takano, A.; Stepanov, V.; Halldin, C.; Farde, L. The Metabotropic Glutamate Receptor 5 Radioligand [11C]AZD9272 Identifies Unique Binding Sites in Primate Brain. Neuropharmacology 2018, 135, 455–463. [Google Scholar] [CrossRef]
- Kawamura, K.; Yamasaki, T.; Kumata, K.; Furutsuka, K.; Takei, M.; Wakizaka, H.; Fujinaga, M.; Kariya, K.; Yui, J.; Hatori, A.; et al. Binding Potential of (E)-[11C]ABP688 to Metabotropic Glutamate Receptor Subtype 5 Is Decreased by the Inclusion of Its 11C-Labelled Z-Isomer. Nucl. Med. Biol. 2014, 41, 17–23. [Google Scholar] [CrossRef]
- Akkus, F.; Treyer, V.; Ametamey, S.M.; Johayem, A.; Buck, A.; Hasler, G. Metabotropic Glutamate Receptor 5 Neuroimaging in Schizophrenia. Schizophr. Res. 2017, 183, 95–101. [Google Scholar] [CrossRef]
- Akkus, F.; Mihov, Y.; Treyer, V.; Ametamey, S.M.; Johayem, A.; Senn, S.; Rösner, S.; Buck, A.; Hasler, G. Metabotropic Glutamate Receptor 5 Binding in Male Patients with Alcohol Use Disorder. Transl. Psychiatry 2018, 8, 17. [Google Scholar] [CrossRef]
- DeLorenzo, C.; DellaGioia, N.; Bloch, M.; Sanacora, G.; Nabulsi, N.; Abdallah, C.; Yang, J.; Wen, R.; Mann, J.J.; Krystal, J.H.; et al. In Vivo Ketamine-Induced Changes in [11C]ABP688 Binding to Metabotropic Glutamate Receptor Subtype 5. Biol. Psychiatry 2015, 77, 266–275. [Google Scholar] [CrossRef]
- Wyckhuys, T.; Verhaeghe, J.; Wyffels, L.; Langlois, X.; Schmidt, M.; Stroobants, S.; Staelens, S. N-Acetylcysteine- and MK-801-Induced Changes in Glutamate Levels Do Not Affect In Vivo Binding of Metabotropic Glutamate 5 Receptor Radioligand 11C-ABP688 in Rat Brain. J. Nucl. Med. 2013, 54, 1954–1961. [Google Scholar] [CrossRef]
- Lucatelli, C.; Honer, M.; Salazar, J.-F.; Ross, T.L.; Schubiger, P.A.; Ametamey, S.M. Synthesis, Radiolabeling, in Vitro and in Vivo Evaluation of [18F]-FPECMO as a Positron Emission Tomography Radioligand for Imaging the Metabotropic Glutamate Receptor Subtype 5. Nucl. Med. Biol. 2009, 36, 613–622. [Google Scholar] [CrossRef]
- Sephton, S.M.; Dennler, P.; Leutwiler, D.S.; Mu, L.; Schibli, R.; Krämer, S.D.; Ametamey, S.M. Development of [(18)F]-PSS223 as a PET Tracer for Imaging of Metabotropic Glutamate Receptor Subtype 5 (MGluR5). Chimia 2012, 66, 201–204. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Warnock, G.; Sommerauer, M.; Mu, L.; Pla Gonzalez, G.; Geistlich, S.; Treyer, V.; Schibli, R.; Buck, A.; Krämer, S.D.; Ametamey, S.M. A First-in-Man PET Study of [18F]PSS232, a Fluorinated ABP688 Derivative for Imaging Metabotropic Glutamate Receptor Subtype 5. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1041–1051. [Google Scholar] [CrossRef]
- Wong, D.F.; Waterhouse, R.; Kuwabara, H.; Kim, J.; Brasic, J.R.; Chamroonrat, W.; Stabins, M.; Holt, D.P.; Dannals, R.F.; Hamill, T.G.; et al. 18F-FPEB, a PET Radiopharmaceutical for Quantifying Metabotropic Glutamate 5 Receptors: A First-in-Human Study of Radiochemical Safety, Biokinetics, and Radiation Dosimetry. J. Nucl. Med. 2013, 54, 388–396. [Google Scholar] [CrossRef]
- Park, E.; Sullivan, J.M.; Planeta, B.; Gallezot, J.-D.; Lim, K.; Lin, S.-F.; Ropchan, J.; McCarthy, T.J.; Ding, Y.-S.; Morris, E.D.; et al. Test-Retest Reproducibility of the Metabotropic Glutamate Receptor 5 Ligand [18F]FPEB with Bolus plus Constant Infusion in Humans. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 1530–1541. [Google Scholar] [CrossRef]
- Sullivan, J.M.; Lim, K.; Labaree, D.; Lin, S.-F.; McCarthy, T.J.; Seibyl, J.P.; Tamagnan, G.; Huang, Y.; Carson, R.E.; Ding, Y.-S.; et al. Kinetic Analysis of the Metabotropic Glutamate Subtype 5 Tracer [(18)F]FPEB in Bolus and Bolus-plus-Constant-Infusion Studies in Humans. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2013, 33, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Lohith, T.G.; Tsujikawa, T.; Siméon, F.G.; Veronese, M.; Zoghbi, S.S.; Lyoo, C.H.; Kimura, Y.; Morse, C.L.; Pike, V.W.; Fujita, M.; et al. Comparison of Two PET Radioligands, [11C]FPEB and [11C]SP203, for Quantification of Metabotropic Glutamate Receptor 5 in Human Brain. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2017, 37, 2458–2470. [Google Scholar] [CrossRef] [PubMed]
- Raboisson, P.; Breitholtz-Emanuelsson, A.; Dahllöf, H.; Edwards, L.; Heaton, W.L.; Isaac, M.; Jarvie, K.; Kers, A.; Minidis, A.B.E.; Nordmark, A.; et al. Discovery and Characterization of AZD9272 and AZD6538-Two Novel MGluR5 Negative Allosteric Modulators Selected for Clinical Development. Bioorg. Med. Chem. Lett. 2012, 22, 6974–6979. [Google Scholar] [CrossRef]
- Nag, S.; Varnas, K.; Arakawa, R.; Jahan, M.; Schou, M.; Farde, L.; Halldin, C. Synthesis, Biodistribution and Radiation Dosimetry of a Novel MGluR5 Radioligand: [18F]AZD9272. ACS Chem. Neurosci. 2020, 11, 1048–1057. [Google Scholar] [CrossRef]
- Deschwanden, A.; Karolewicz, B.; Feyissa, A.M.; Treyer, V.; Ametamey, S.M.; Johayem, A.; Burger, C.; Auberson, Y.P.; Sovago, J.; Stockmeier, C.A.; et al. Reduced Metabotropic Glutamate Receptor 5 Density in Major Depression Determined by [(11)C]ABP688 PET and Postmortem Study. Am. J. Psychiatry 2011, 168, 727–734. [Google Scholar] [CrossRef]
- Kågedal, M.; Cselényi, Z.; Nyberg, S.; Raboisson, P.; Ståhle, L.; Stenkrona, P.; Varnäs, K.; Halldin, C.; Hooker, A.C.; Karlsson, M.O. A Positron Emission Tomography Study in Healthy Volunteers to Estimate MGluR5 Receptor Occupancy of AZD2066—Estimating Occupancy in the Absence of a Reference Region. NeuroImage 2013, 82, 160–169. [Google Scholar] [CrossRef]
- Mathews, W.B.; Kuwabara, H.; Stansfield, K.; Valentine, H.; Alexander, M.; Kumar, A.; Hilton, J.; Dannals, R.F.; Wong, D.F.; Gasparini, F. Dose-Dependent, Saturable Occupancy of the Metabotropic Glutamate Subtype 5 Receptor by Fenobam as Measured with [11 C]ABP688 PET Imaging. Synapse 2014, 68, 565–573. [Google Scholar] [CrossRef] [PubMed]
- DeLorenzo, C.; Sovago, J.; Gardus, J.; Xu, J.; Yang, J.; Behrje, R.; Kumar, J.S.D.; Devanand, D.P.; Pelton, G.H.; Mathis, C.A.; et al. Characterization of Brain MGluR5 Binding in a Pilot Study of Late-Life Major Depressive Disorder Using Positron Emission Tomography and [11C]ABP688. Transl. Psychiatry 2015, 5, e693. [Google Scholar] [CrossRef] [PubMed]
- Leuzy, A.; Zimmer, E.R.; Dubois, J.; Pruessner, J.; Cooperman, C.; Soucy, J.-P.; Kostikov, A.; Schirmaccher, E.; Désautels, R.; Gauthier, S.; et al. In Vivo Characterization of Metabotropic Glutamate Receptor Type 5 Abnormalities in Behavioral Variant FTD. Brain Struct. Funct. 2016, 221, 1387–1402. [Google Scholar] [CrossRef]
- Bertoglio, D.; Kosten, L.; Verhaeghe, J.; Thomae, D.; Wyffels, L.; Stroobants, S.; Wityak, J.; Dominguez, C.; Mrzljak, L.; Staelens, S. Longitudinal Characterization of MGluR5 Using 11C-ABP688 PET Imaging in the Q175 Mouse Model of Huntington Disease. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2018, 59, 1722–1727. [Google Scholar] [CrossRef] [PubMed]
- Treyer, V.; Gietl, A.F.; Suliman, H.; Gruber, E.; Meyer, R.; Buchmann, A.; Johayem, A.; Unschuld, P.G.; Nitsch, R.M.; Buck, A.; et al. Reduced Uptake of [11C]-ABP688, a PET Tracer for Metabolic Glutamate Receptor 5 in Hippocampus and Amygdala in Alzheimer’s Dementia. Brain Behav. 2020, 10, e01632. [Google Scholar] [CrossRef]
- Streffer, J.; Treyer, V.; Buck, A.; Ametamey, S.M.; Blagoev, M.; Maguire, R.P.; Gautier, A.; Auberson, Y.P.; Schmidt, M.E.; Vranesic, I.-T.; et al. Regional Brain MGlu5 Receptor Occupancy Following Single Oral Doses of Mavoglurant as Measured by [11C]-ABP688 PET Imaging in Healthy Volunteers. NeuroImage 2021, 230, 117785. [Google Scholar] [CrossRef]
- Kim, J.-H.; Joo, Y.-H.; Son, Y.-D.; Kim, H.-K.; Kim, J.-H. Differences in MGluR5 Availability Depending on the Level of Social Avoidance in Drug-Naïve Young Patients with Major Depressive Disorder. Neuropsychiatr. Dis. Treat. 2022, 18, 2041–2053. [Google Scholar] [CrossRef] [PubMed]
- Müller Herde, A.; Schibli, R.; Weber, M.; Ametamey, S.M. Metabotropic Glutamate Receptor Subtype 5 Is Altered in LPS-Induced Murine Neuroinflammation Model and in the Brains of AD and ALS Patients. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 407–420. [Google Scholar] [CrossRef]
- Zasadny, K.; Chen, L.; Skaddan, M. Measurement of Receptor Occupancy of MGluR5 Negative Allosteric Modulator AFQ056 in Non Human Primate by 18FPEB PET. Mol. Imaging Biol. 2012, 14 (Suppl. S2), 998. [Google Scholar]
- Brownell, A.-L.; Kuruppu, D.; Kil, K.-E.; Jokivarsi, K.; Poutiainen, P.; Zhu, A.; Maxwell, M. PET Imaging Studies Show Enhanced Expression of MGluR5 and Inflammatory Response during Progressive Degeneration in ALS Mouse Model Expressing SOD1-G93A Gene. J. Neuroinflammation 2015, 12, 217. [Google Scholar] [CrossRef]
- Abdallah, C.G.; Hannestad, J.; Mason, G.F.; Holmes, S.E.; DellaGioia, N.; Sanacora, G.; Jiang, L.; Matuskey, D.; Satodiya, R.; Gasparini, F.; et al. Metabotropic Glutamate Receptor 5 and Glutamate Involvement in Major Depressive Disorder: A Multimodal Imaging Study. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2017, 2, 449–456. [Google Scholar] [CrossRef]
- Fatemi, S.H.; Wong, D.F.; Brašić, J.R.; Kuwabara, H.; Mathur, A.; Folsom, T.D.; Jacob, S.; Realmuto, G.M.; Pardo, J.V.; Lee, S. Metabotropic Glutamate Receptor 5 Tracer [18F]-FPEB Displays Increased Binding Potential in Postcentral Gyrus and Cerebellum of Male Individuals with Autism: A Pilot PET Study. Cerebellum Ataxias 2018, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Leurquin-Sterk, G.; Ceccarini, J.; Crunelle, C.L.; de Laat, B.; Verbeek, J.; Deman, S.; Neels, H.; Bormans, G.; Peuskens, H.; Van Laere, K. Lower Limbic Metabotropic Glutamate Receptor 5 Availability in Alcohol Dependence. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2018, 59, 682–690. [Google Scholar] [CrossRef]
- Lee, M.; Lee, H.-J.; Park, I.S.; Park, J.-A.; Kwon, Y.J.; Ryu, Y.H.; Kim, C.H.; Kang, J.H.; Hyun, I.Y.; Lee, K.C.; et al. Aβ Pathology Downregulates Brain MGluR5 Density in a Mouse Model of Alzheimer. Neuropharmacology 2018, 133, 512–517. [Google Scholar] [CrossRef] [PubMed]
- de Laat, B.; Weerasekera, A.; Leurquin-Sterk, G.; Bormans, G.; Himmelreich, U.; Casteels, C.; Van Laere, K. Glutamatergic Biomarkers for Cocaine Addiction: A Longitudinal Study Using MR Spectroscopy and MGluR5 PET in Self-Administering Rats. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2018, 59, 952–959. [Google Scholar] [CrossRef]
- Kang, Y.; Henchcliffe, C.; Verma, A.; Vallabhajosula, S.; He, B.; Kothari, P.J.; Pryor, K.O.; Mozley, P.D. 18F-FPEB PET/CT Shows MGluR5 Upregulation in Parkinson’s Disease. J. Neuroimaging Off. J. Am. Soc. Neuroimaging 2019, 29, 97–103. [Google Scholar] [CrossRef]
- Cai, G.; Wang, M.; Wang, S.; Liu, Y.; Zhao, Y.; Zhu, Y.; Zhao, S.; Zhang, M.; Guo, B.; Yao, H.; et al. Brain MGluR5 in Shank3B-/- Mice Studied With in Vivo [18F]FPEB PET Imaging and Ex Vivo Immunoblotting. Front. Psychiatry 2019, 10, 38. [Google Scholar] [CrossRef]
- Mecca, A.P.; McDonald, J.W.; Michalak, H.R.; Godek, T.A.; Harris, J.E.; Pugh, E.A.; Kemp, E.C.; Chen, M.-K.; Salardini, A.; Nabulsi, N.B.; et al. PET Imaging of MGluR5 in Alzheimer’s Disease. Alzheimers Res. Ther. 2020, 12, 15. [Google Scholar] [CrossRef] [PubMed]
- Mota, F.; Sementa, T.; Taddei, C.; Moses, N.; Bordoloi, J.; Hader, S.; Eykyn, T.; Cash, D.; Turkheimer, F.; Veronese, M.; et al. Investigating the Effects of Ebselen, a Potential New Lithium Mimetic, on Glutamate Transmission. Synapse 2020, 74, e22151. [Google Scholar] [CrossRef] [PubMed]
- Brašić, J.R.; Nandi, A.; Russell, D.S.; Jennings, D.; Barret, O.; Mathur, A.; Slifer, K.; Sedlak, T.; Martin, S.D.; Brinson, Z.; et al. Reduced Expression of Cerebral Metabotropic Glutamate Receptor Subtype 5 in Men with Fragile X Syndrome. Brain Sci. 2020, 10, 899. [Google Scholar] [CrossRef]
- Brašić, J.R.; Nandi, A.; Russell, D.S.; Jennings, D.; Barret, O.; Martin, S.D.; Slifer, K.; Sedlak, T.; Seibyl, J.P.; Wong, D.F.; et al. Cerebral Expression of Metabotropic Glutamate Receptor Subtype 5 in Idiopathic Autism Spectrum Disorder and Fragile X Syndrome: A Pilot Study. Int. J. Mol. Sci. 2021, 22, 2863. [Google Scholar] [CrossRef] [PubMed]
- Mody, M.; Petibon, Y.; Han, P.; Kuruppu, D.; Ma, C.; Yokell, D.; Neelamegam, R.; Normandin, M.D.; Fakhri, G.E.; Brownell, A.-L. In Vivo Imaging of MGlu5 Receptor Expression in Humans with Fragile X Syndrome towards Development of a Potential Biomarker. Sci. Rep. 2021, 11, 15897. [Google Scholar] [CrossRef]
- Esterlis, I.; DeBonee, S.; Cool, R.; Holmes, S.; Baldassari, S.R.; Maruff, P.; Pietrzak, R.H.; Davis, M.T. Differential Role of MGluR5 in Cognitive Processes in Posttraumatic Stress Disorder and Major Depression. Chronic Stress Thousand Oaks Calif 2022, 6, 24705470221105804. [Google Scholar] [CrossRef] [PubMed]
- Holmes, S.E.; Asch, R.H.; Davis, M.T.; DellaGioia, N.; Pashankar, N.; Gallezot, J.-D.; Nabulsi, N.; Matuskey, D.; Sanacora, G.; Carson, R.E.; et al. Differences in Quantification of the Metabotropic Glutamate Receptor 5 Across Bipolar Disorder and Major Depressive Disorder. Biol. Psychiatry 2023, 93, 1099–1107. [Google Scholar] [CrossRef]
- Galineau, L.; Arlicot, N.; Dupont, A.-C.; Briend, F.; Houy-Durand, E.; Tauber, C.; Gomot, M.; Gissot, V.; Barantin, L.; Lefevre, A.; et al. Glutamatergic Synapse in Autism: A Complex Story for a Complex Disorder. Mol. Psychiatry 2023, 28, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Van de Bittner, G.C.; Ricq, E.L.; Hooker, J.M. A Philosophy for CNS Radiotracer Design. Acc. Chem. Res. 2014, 47, 3127–3134. [Google Scholar] [CrossRef]
- Berry-Kravis, E.; Des Portes, V.; Hagerman, R.; Jacquemont, S.; Charles, P.; Visootsak, J.; Brinkman, M.; Rerat, K.; Koumaras, B.; Zhu, L.; et al. Mavoglurant in Fragile X Syndrome: Results of Two Randomized, Double-Blind, Placebo-Controlled Trials. Sci. Transl. Med. 2016, 8, 321ra5. [Google Scholar] [CrossRef] [PubMed]
- Sandiego, C.M.; Nabulsi, N.; Lin, S.-F.; Labaree, D.; Najafzadeh, S.; Huang, Y.; Cosgrove, K.; Carson, R.E. Studies of the Metabotropic Glutamate Receptor 5 Radioligand [11C]ABP688 with N-Acetylcysteine Challenge in Rhesus Monkeys: ABP688 and NAC in Monkeys. Synapse 2013, 67, 489–501. [Google Scholar] [CrossRef]
- Miyake, N.; Skinbjerg, M.; Easwaramoorthy, B.; Kumar, D.; Girgis, R.R.; Xu, X.; Slifstein, M.; Abi-Dargham, A. Imaging Changes in Glutamate Transmission In Vivo with the Metabotropic Glutamate Receptor 5 Tracer [11C] ABP688 and N-Acetylcysteine Challenge. Biol. Psychiatry 2011, 69, 822–824. [Google Scholar] [CrossRef] [PubMed]
- O’Gorman Tuura, R.; Warnock, G.; Ametamey, S.; Treyer, V.; Noeske, R.; Buck, A.; Sommerauer, M. Imaging Glutamate Redistribution after Acute N-Acetylcysteine Administration: A Simultaneous PET/MR Study. NeuroImage 2019, 184, 826–833. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, E.R.; Parent, M.J.; Leuzy, A.; Aliaga, A.; Aliaga, A.; Moquin, L.; Schirrmacher, E.S.; Soucy, J.-P.; Skelin, I.; Gratton, A.; et al. Imaging in Vivo Glutamate Fluctuations with [(11)C]ABP688: A GLT-1 Challenge with Ceftriaxone. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2015, 35, 1169–1174. [Google Scholar] [CrossRef] [PubMed]
- Kosten, L.; Verhaeghe, J.; Wyffels, L.; Stroobants, S.; Staelens, S. Acute Ketamine Infusion in Rat Does Not Affect In Vivo [11C]ABP688 Binding to Metabotropic Glutamate Receptor Subtype 5. Mol. Imaging 2018, 17, 1536012118788636. [Google Scholar] [CrossRef]
- Esterlis, I.; DellaGioia, N.; Pietrzak, R.H.; Matuskey, D.; Nabulsi, N.; Abdallah, C.G.; Yang, J.; Pittenger, C.; Sanacora, G.; Krystal, J.H.; et al. Ketamine-Induced Reduction in MGluR5 Availability Is Associated with an Antidepressant Response: An [11C]ABP688 and PET Imaging Study in Depression. Mol. Psychiatry 2018, 23, 824–832. [Google Scholar] [CrossRef] [PubMed]
- Dupont, A.-C.; Serrière, S.; Barantin, L.; Vercouillie, J.; Tauber, C.; Gissot, V.; Bodard, S.; Chicheri, G.; Chalon, S.; Bonnet-Brilhault, P.F.; et al. Study of Influence of the Glutamatergic Concentration of [18F]FPEB Binding to Metabotropic Glutamate Receptor Subtype 5 with N-Acetylcysteine Challenge in Rats and SRM/PET Study in Human Healthy Volunteers. Transl. Psychiatry 2021, 11, 66. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Radioligand | PET Application in Drug Development | Drug/Disease | Population | Main Findings | Reference |
|---|---|---|---|---|---|
| [11C]ABP688 | Hallmark of disease | Major depressive disorder | Clinical: 11 un-medicated individuals with MDD and 11 matched healthy comparison subjects | Lower levels of regional mGluR5 binding in the prefrontal cortex, the cingulate cortex, the insula, the thalamus and the hippocampus in the depression group relative to the comparison group. | (2011) [59] |
| RO study | AZD2066 | Clinical: 6 healthy volunteers after different doses of AZD2066 | AZD2066 displaced [11C]ABP688 from mGluR5 binding sites in the human brain. The estimated Ki was around 1200 nM, suggesting that approximately 50% occupancy was achieved at Cmax with the highest dose (13.5 mg). | (2013) [60] | |
| RO study | Fenobam | Preclinical: 4 baboons’ PET at baseline condition vs. after intravenous treatment with fenobam at different dose levels (0.3–1.33 mg/kg) | In vivo binding of [11C]ABP688 was blocked by pre-treatment with fenobam in a dose-dependent, saturable manner, approaching close to full occupancy (>90%) at a dose of 1.33 mg/kg. | (2014) [61] | |
| Hallmark of disease | Major depressive disorder | Clinical: 20 elderly (mean age: 63.0 ± 6.3) subjects with MDD and 22 healthy volunteers in the same age range | No significant difference in [11C]ABP688 binding was observed between elderly subjects with MDD and healthy volunteers. | (2015) [62] | |
| Hallmark of disease | Behavioral variant frontotemporal dementia | Clinical: 5 bvFTD patients and 10 healthy volunteers | BvFTD patients showed widespread decrements in [11C]ABP688 BPND throughout frontal, temporal and subcortical areas. | (2016) [63] | |
| Hallmark of disease | Schizophrenia | Clinical: 15 individuals with schizophrenia and 15 healthy controls. | Distribution volume ratio in the 15 individuals with schizophrenia did not differ from that of the 15 controls. | (2017) [46] | |
| Hallmark of disease | Huntington’s disease | Preclinical: 18 heterozygous mice (Q175 Mouse Model of Huntington’s Disease) and 18 wild-type (WT) at 3 different time points (6, 9 and 13 months old) | Reduction in [11C]ABP688 binding in the striatum and cortex of heterozygous mice, compared with WT mice, as well as a temporal decline. | (2018) [64] | |
| Hallmark of disease | Alzheimer’s disease | Clinical: 9 subjects with AD and 10 cognitively healthy controls | Reduction in mGluR5 binding in the hippocampus and amygdala in AD group. | (2020) [65] | |
| Drug distribution and RO study | Mavoglurant | Clinical: 6 subjects divided into 2 cohorts at different doses (25,100, 200, 400 mg) of mavoglurant and different periods. | Mavoglurant passes the blood–brain barrier and induces a dose/exposure-dependent displacement of [11C]ABP688 bound to mGlu5 receptors in humans in vivo. A single oral dose of 400 mg induced an estimated displacement of 63% at a scan time of 3–4 h post dose, inferring a receptor occupancy estimate of nearly 85%. | (2021) [66] | |
| Hallmark of disease | Major depressive disorder | Clinical: 20 non-smoking MDD patients and 18 matched non-smoking healthy controls. | Significant differences in frontal mGluR5 availability depending on the level of social avoidance in drug-naïve non-smoking MDD patients. | (2022) [67] | |
| [18F]PSS232 | Hallmark of disease | Neuroinflammation model | Preclinical: 4 LPS-induced animal models of neuroinflammation and 4 control mice | LPS-induced neuroinflammation increased mGluR5 levels in mouse brain. | (2019) [68] |
| [18F]FPEB | RO study | Mavoglurant | Preclinical: 2 male cynomolgus monkeys PET at baseline condition vs. after intravenous treatment with mavoglurant at 2 different doses (0.074 and 0.34 mg/kg) | Measured RO for mavoglurant was 73% at the 0.34 mg/kg dose and 51% at 0.074 mg/kg. The current data would predict that ≥80% RO is required for efficacy. | (2012) [69] |
| RO study | -VU0409106 (NAM) -VU0092273 (ago-PAM) -VU0360172 (PAM) | Preclinical: 8, 5 and 7 rats, respectively, after IP injection of increasing doses of treatment (3–100 mg/kg) | VU0409106: ED50 = 7.5 mg/kg VU0092273: ED50 = 17.8 mg/kg VU0360172 does not significantly displace [18F]FPEB binding to mGlu5 in vivo, demonstrating that RO does not predict in vivo efficacy for this mGlu5 PAM. | (2015) [39] | |
| Hallmark of disease | Amyotrophic lateral sclerosis | Preclinical: 4 ALS mice expressing SOD1-G93A gene and 4 control base mice (C57/BL6) | In the whole brain, the binding potential increased by 49 ± 9% from base mice to ALS-type mice and further enhanced by 23 ± 4% during disease progression. | (2015) [70] | |
| Hallmark of disease | Major depressive disorder | Clinical: 30 MDD and 35 HC | No significant between-group differences were observed in mGluR5 VT or DVR | (2017) [71] | |
| Hallmark of disease | Autism spectrum disorder | Clinical: 6 ASD patients and 3 control subjects | Significantly higher [18F]FPEB binding potential in the postcentral gyrus and cerebellum of individuals with autism | (2018) [72] | |
| Hallmark of disease | Alcohol Dependence | Clinical: 16 recently abstinent alcohol-dependent subjects and 32 age-matched controls | mGluR5 availability was lower mainly in limbic regions of alcohol-dependent subjects than in controls, ranging from 14% in the posterior cingulate cortex to 36% in the caudate nucleus. | (2018) [73] | |
| Hallmark of disease | Alzheimer disease | Preclinical: 4 10-month-old male 5xFAD transgenic mice models and 4 10-month-old wild type (WT) mice were used as control | mGluR5 in the hippocampus and the striatum was significantly lower in 5xFAD mice compared to control animals. | (2018) [74] | |
| Hallmark of disease | Cocaine addiction | Preclinical: 42 rats before and after sucrose or intravenous cocaine self-administration, during withdrawal and during relapse. | Only cocaine self-administration induced a decrease in [18F]FPEB binding | (2018) [75] | |
| Hallmark of disease | Parkinson’s disease | Clinical: 9 patients with PD and 8 healthy volunteers (HV) | [18F]FPEB BPND values were slightly more than 20% higher in PD than HVs in several mesocortical regions, including the bilateral putamen, hippocampus and amygdala. | (2018) [76] | |
| Hallmark of disease | Autism spectrum disorder | Preclinical: 6 Shank3B−/− mice and 6 control mice | Shank3B−/− mice showed significantly increased BPND compared to the control mice in the hippocampus, thalamus, striatum and amygdala | (2019) [77] | |
| Hallmark of disease | Alzheimer disease | Clinical: 16 individuals with amnestic mild cognitive impairment (MCI) due to AD or mild AD dementia who were positive for brain amyloid were compared to 15 cognitively normal (CN) participants who were negative for brain amyloid. | Significant reduction (43%) in mGluR5 binding in the hippocampus of AD compared to participants. | (2020) [78] | |
| Monitoring treatment effect | Ebselen | Preclinical: Dawley rats were randomized to receive either ebselen (5 mg/kg, n = 4) or vehicle (n = 4). | Acute administration of ebselen potentially decreases synaptic glutamate levels, as measured by an increased brain uptake of [18F]FPEB | (2020) [79] | |
| Hallmark of disease | Fragile X Syndrome | Clinical: 9 men with FXS and 8 with typical development (TD) | mGluR5 expression was significantly reduced in cortical and subcortical regions of men with FXS in contrast to age-matched men with TD. | (2020) [80] | |
| Hallmark of disease | Autism spectrum disorder and Fragile X Syndrome | Clinical: 10 men with FXS, 7 with ASD and 19 with typical development (TD) | In contrast to participants with TD, mGluR5 expression was significantly increased in the cortical regions of participants with IASD and significantly reduced in all regions of men with FXS. | (2021) [81] | |
| Hallmark of disease | Fragile X Syndrome | Clinical: 8 males with FXS and 8 age- and gender-matched controls | Patients with FXS showed lower [18F]FPEB binding potential, reflecting reduced mGluR5 availability, than the healthy controls throughout the brain, with significant group differences in insula, anterior cingulate, parahippocampal, inferior temporal and olfactory cortices. | (2021) [82] | |
| Hallmark of disease | Post-traumatic stress disorder and major depressive disorder | Clinical: 28 PTSD, 21 MDD and 28 healthy adults were matched for age, gender and smoking status. | Significant relationship between frontolimbic mGluR5 availability and performance on tests of attention in individuals with MDD and PTSD | (2022) [83] | |
| Hallmark of disease | Bipolar disorder and major depressive disorder | Clinical: Individuals with BD (n = 17 depressed; n = 10 euthymic), MDD (n = 17) and healthy control (HC) individuals (n = 18) | mGluR5 was lower in BD versus MDD and HC groups, with no difference between MDD and HC groups. | (2022) [84] | |
| Hallmark of disease | Autism spectrum disorder | Clinical: 12 adult males with ASD and 14 healthy adult males | mGluR5 binding was significantly increased in the brain of ASD vs. controls groups | (2023) [85] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dupont, A.-C.; Arlicot, N.; Vercouillie, J.; Serrière, S.; Maia, S.; Bonnet-Brilhault, F.; Santiago-Ribeiro, M.-J. Metabotropic Glutamate Receptor Subtype 5 Positron-Emission-Tomography Radioligands as a Tool for Central Nervous System Drug Development: Between Progress and Setbacks. Pharmaceuticals 2023, 16, 1127. https://doi.org/10.3390/ph16081127
Dupont A-C, Arlicot N, Vercouillie J, Serrière S, Maia S, Bonnet-Brilhault F, Santiago-Ribeiro M-J. Metabotropic Glutamate Receptor Subtype 5 Positron-Emission-Tomography Radioligands as a Tool for Central Nervous System Drug Development: Between Progress and Setbacks. Pharmaceuticals. 2023; 16(8):1127. https://doi.org/10.3390/ph16081127
Chicago/Turabian StyleDupont, Anne-Claire, Nicolas Arlicot, Johnny Vercouillie, Sophie Serrière, Serge Maia, Frédérique Bonnet-Brilhault, and Maria-Joao Santiago-Ribeiro. 2023. "Metabotropic Glutamate Receptor Subtype 5 Positron-Emission-Tomography Radioligands as a Tool for Central Nervous System Drug Development: Between Progress and Setbacks" Pharmaceuticals 16, no. 8: 1127. https://doi.org/10.3390/ph16081127
APA StyleDupont, A.-C., Arlicot, N., Vercouillie, J., Serrière, S., Maia, S., Bonnet-Brilhault, F., & Santiago-Ribeiro, M.-J. (2023). Metabotropic Glutamate Receptor Subtype 5 Positron-Emission-Tomography Radioligands as a Tool for Central Nervous System Drug Development: Between Progress and Setbacks. Pharmaceuticals, 16(8), 1127. https://doi.org/10.3390/ph16081127