
Stroke-Induced Central Pain: Overview of the Mechanisms, Management, and Emerging Targets of Central Post-Stroke Pain

 ,
,
Abstract
1. Introduction
2. Central Post-Stroke Pain (CPSP)
3. Epidemiology and Risk Factors
4. Clinical Features and Diagnosis
5. Mechanisms and Pathophysiology of CPSP
5.1. Route of Pain Transmission
5.2. Mechanisms Involved in Central Pain and CPSP
5.3. Pathophysiology of CPSP
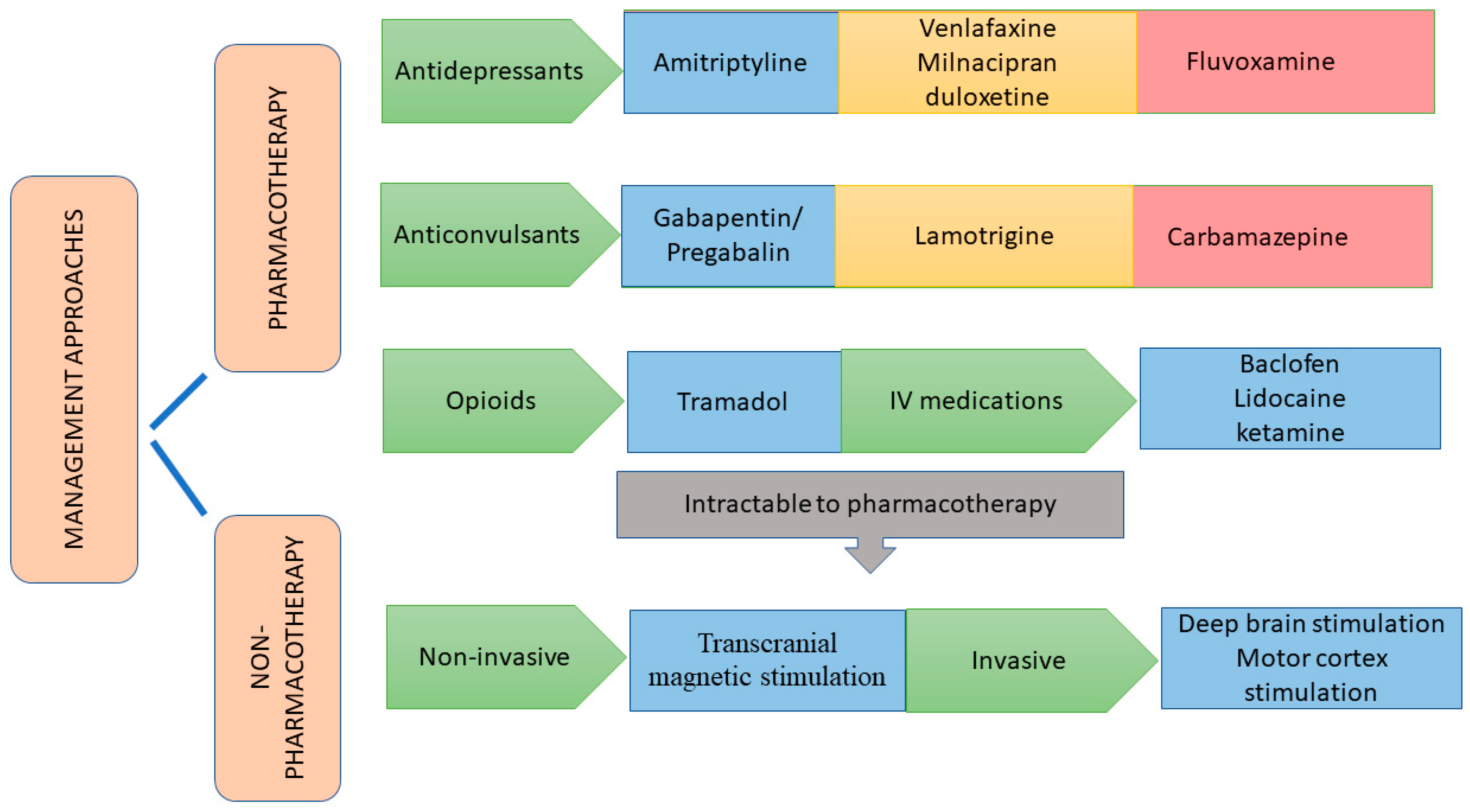
6. Management of CPSP
6.1. Pharmacotherapeutic Approaches
6.1.1. Antidepressants
6.1.2. Anticonvulsants
6.1.3. Opioid Analgesics
6.1.4. Miscellaneous Drugs
6.2. Non-Pharmacotherapeutic Approaches
7. Emerging Targets
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Watson, J.C.; Sandroni, P. Central Neuropathic pain syndromes. Mayo Clin. Proc. 2016, 91, 372–385. [Google Scholar] [CrossRef]
- Hassaballa, D.; Harveya, R.L. Central pain syndromes. NeuroRehabilitation 2020, 47, 285–297. [Google Scholar] [CrossRef]
- Huang-Lionnet, J.H.; Brummett, C.; Raja, S.N. Central Pain States. In Essentials of Pain Medicine; Elsevier: Amsterdam, The Netherlands, 2018; pp. 251–260. [Google Scholar]
- Harrison, R.A.; Field, T.S. Post stroke pain: Identification, assessment, and therapy. Cerebrovasc. Dis. 2015, 39, 190–201. [Google Scholar] [CrossRef]
- Haroutounian, S.; Ford, A.L.; Frey, K.; Nikolajsen, L.; Finnerup, N.B.; Neiner, A.; Kharasch, E.D.; Karlsson, P.; Bottros, M.M. How central is central poststroke pain? The role of afferent input in poststroke neuropathic pain: A prospective, open-label pilot study. Pain 2018, 159, 1317–1324. [Google Scholar] [CrossRef]
- Widyadharma, I.; Eka, P.; Tertia, C.; Wijayanti, S.; Barus, J.F.A. Central post stroke pain: What are the new insights? Rom. J. Neurol. 2021, 20, 28–34. [Google Scholar] [CrossRef]
- Mhangara, C.T.; Naidoo, V.; Ntsiea, M.V. The prevalence and management of central post-stroke pain at a hospital in Zimbabwe. Malawi Med. J. 2020, 32, 132–138. [Google Scholar]
- Akyuz, G.; Kuru, P. Systematic Review of Central Post Stroke Pain: What Is Happening in the Central Nervous System? Am. J. Phys. Med. Rehabil. 2016, 95, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart disease and stroke statistics—2019 update: A report from the American Heart Association. Circulation 2019, 139, e56. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, L.M.; da Silva, V.A.; de Lima Rodrigues, A.L.; Mendes Fernandes, D.T.R.; de Oliveira, R.A.A.; Galhardoni, R.; Yeng, L.T.; Junior, J.R.; Conforto, A.B.; Lucato, L.T.; et al. Dissecting central post-stroke pain: A controlled symptom-psychophysical characterization. Brain Commun. 2022, 4, fcac090. [Google Scholar] [CrossRef]
- Seifert, C.L.; Mallar Chakravarty, M.; Sprenger, T. The complexities of pain after stroke—A review with a focus on central post-stroke pain. Panminerva Med. 2013, 55, 1–10. [Google Scholar] [PubMed]
- Bashir, A.H.; Abdullahi, A.; Abba, M.A.; Mukhtar, N.B. Central Poststroke Pain: Its Profile among Stroke Survivors in Kano, Nigeria. Behav. Neurol. 2017, 2017, 9318597. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Seo, W. A Comprehensive Review of Central Post-Stroke Pain. Pain Manag. Nurs. 2015, 16, 804–818. [Google Scholar] [CrossRef] [PubMed]
- Raffaeli, W.; Minella, C.E.; Magnani, F.; Sarti, D. Population-based study of central post-stroke pain in Rimini district, Italy. J. Pain Res. 2013, 6, 705–711. [Google Scholar] [PubMed]
- Betancur, D.F.A.; Tarragó, M.D.G.L.; Torres, I.L.D.S.; Fregni, F.; Caumo, W. Central Post-Stroke Pain: An Integrative Review of Somatotopic Damage, Clinical Symptoms, and Neurophysiological Measures. Front. Neurol. 2021, 12, 678198. [Google Scholar] [CrossRef]
- Kumar, A.; Bhoi, S.K.; Kalita, J.; Misra, U.K. Central Poststroke Pain Can Occur with Normal Sensation. Clin. J. Pain 2016, 32, 955–960. [Google Scholar] [CrossRef]
- Harno, H.; Haapaniemi, E.; Putaala, J.; Haanpää, M.; Mäkelä, J.P.; Kalso, E.; Tatlisumak, T. Central poststroke pain in young ischemic stroke survivors in the Helsinki Young Stroke Registry. Neurology 2014, 83, 1147–1154. [Google Scholar] [CrossRef]
- Vukojevic, Z.; Dominovic Kovacevic, A.; Peric, S.; Grgic, S.; Bjelica, B.; Basta, I.; Lavrnic, D. Frequency and features of the central poststroke pain. J. Neurol. Sci. 2018, 391, 100–103. [Google Scholar] [CrossRef]
- Osama, A.; Abo Hagar, A.; Elkholy, S.; Negm, M.; Abd El-Razek, R.; Orabi, M. Central post-stroke pain: Predictors and relationship with magnetic resonance imaging and somatosensory evoked potentials. Egypt. J. Neurol. Psychiatr. Neurosurg. 2018, 54, 40. [Google Scholar] [CrossRef]
- Klit, H.; Finnerup, N.B.; Jensen, T.S. Central post-stroke pain: Clinical characteristics, pathophysiology, and management. Lancet Neurol. 2009, 8, 857–868. [Google Scholar] [CrossRef]
- Kumar, B.; Kalita, J.; Kumar, G.; Misra, U.K. Central poststroke pain: A review of pathophysiology and treatment. Anesth. Analg. 2009, 108, 1645–1657. [Google Scholar] [CrossRef]
- Corbetta, D.; Sarasso, E.; Agosta, F.; Filippi, M.; Gatti, R. Mirror therapy for an adult with central post-stroke pain: A case report. Arch. Physiother. 2018, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Ri, S. The Management of Poststroke Thalamic Pain: Update in Clinical Practice. Diagnostics 2022, 12, 1439. [Google Scholar] [CrossRef] [PubMed]
- Choi-Kwon, S.; Choi, S.H.; Suh, M.; Choi, S.; Cho, K.H.; Nah, H.W.; Song, H.; Kim, J.S. Musculoskeletal and central pain at 1 year post-stroke: Associated factors and impact on quality of life. Acta Neurol. Scand. 2017, 135, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Yam, M.F.; Loh, Y.C.; Tan, C.S.; Khadijah Adam, S.; Abdul Manan, N.; Basir, R. General Pathways of Pain Sensation and the Major Neurotransmitters Involved in Pain Regulation. Int. J. Mol. Sci. 2018, 19, 2164. [Google Scholar] [CrossRef]
- Treister, A.K.; Hatch, M.N.; Cramer, S.C.; Chang, E.Y. Demystifying Poststroke Pain: From Etiology to Treatment. Phys. Med. Rehabil. 2017, 9, 63–75. [Google Scholar] [CrossRef]
- Vartiainen, N.; Perchet, C.; Magnin, M.; Creac’h, C.; Convers, P.; Nighoghossian, N.; Mauguière, F.; Peyron, R.; Garcia-Larrea, L. Thalamic pain: Anatomical and physiological indices of prediction. Brain 2016, 139, 708–722. [Google Scholar] [CrossRef]
- Head, H.; Holmes, G. Sensory disturbances from cerebral lesions. Brain 1911, 34, 102–254. [Google Scholar] [CrossRef]
- Morishita, T.; Inoue, T. Brain Stimulation Therapy for Central Post-Stroke Pain from a Perspective of Interhemispheric Neural Network Remodeling. Front. Hum. Neurosci. 2016, 10, 166. [Google Scholar] [CrossRef]
- Nijs, J.; Lahousse, A.; Kapreli, E.; Bilika, P.; Saraçoğlu, İ.; Malfliet, A.; Coppieters, I.; De Baets, L.; Leysen, L.; Roose, E.; et al. Nociplastic Pain Criteria or Recognition of Central Sensitization? Pain Phenotyping in the Past, Present and Future. J. Clin. Med. 2021, 10, 3203. [Google Scholar] [CrossRef]
- Lu, J.; Guo, X.; Yan, M.; Yuan, X.; Chen, S.; Wang, Y.; Zhu, J.; Huang, S.; Shen, H.; Li, H.; et al. P2X4R Contributes to Central Disinhibition Via TNF-α/TNFR1/GABAaR Pathway in Post-stroke Pain Rats. J. Pain 2021, 22, 968–980. [Google Scholar] [CrossRef]
- Krause, T.; Asseyer, S.; Taskin, B.; Flöel, A.; Witte, A.V.; Mueller, K.; Fiebach, J.B.; Villringer, K.; Villringer, A.; Jungehulsing, G.J. The Cortical Signature of Central Poststroke Pain: Gray Matter Decreases in Somatosensory, Insular, and Prefrontal Cortices. Cereb. Cortex 2016, 26, 80–88. [Google Scholar] [CrossRef]
- Li, X.; Feng, Y.; Gao, F. Maladaptive reorganization in pain-related brain network contributing to the central post-stroke pain. Neuropsychiatry 2019, 9, 2186–2197. [Google Scholar]
- Gritsch, S.; Bali, K.K.; Kuner, R.; Vardeh, D. Functional characterization of a mouse model for central post-stroke pain. Mol. Pain 2016, 12, 1744806916629049. [Google Scholar] [CrossRef]
- He, C.; Liu, R.; Fan, Z.; Li, Y.; Yang, M.; Hou, W.; Lu, Z.; Fang, Z.; Su, B. Microglia in the Pathophysiology of Hemorrhagic Stroke and the Relationship Between Microglia and Pain After Stroke: A Narrative Review. Pain Ther. 2021, 10, 927–939. [Google Scholar] [CrossRef]
- Di Virgilio, F. P2X receptors and inflammation. Curr. Med. Chem. 2015, 22, 866–877. [Google Scholar] [CrossRef]
- Wan, L.; Li, Z.; Liu, T.; Chen, X.; Xu, Q.; Yao, W.; Zhang, C.; Zhang, Y. Epoxyeicosatrienoic acids: Emerging therapeutic agents for central post-stroke pain. Pharmacol. Res. 2020, 159, 104923. [Google Scholar] [CrossRef] [PubMed]
- Kuan, Y.H.; Shih, H.C.; Tang, S.C.; Jeng, J.S.; Shyu, B.C. Targeting P2X7 receptor for the treatment of central post-stroke pain in a rodent model. Neurobiol. Dis. 2015, 78, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Shih, H.C.; Kuan, Y.H.; Shyu, B.C. Targeting brain-derived neurotrophic factor in the medial thalamus for the treatment of central poststroke pain in a rodent model. Pain 2017, 158, 1302–1313. [Google Scholar] [CrossRef]
- Yang, F.; Luo, W.J.; Sun, W.; Wang, Y.; Wang, J.L.; Yang, F.; Li, C.L.; Wei, N.; Wang, X.L.; Guan, S.M.; et al. SDF1-CXCR4 Signaling Maintains Central Post-Stroke Pain through Mediation of Glial-Neuronal Interactions. Front. Mol. Neurosci. 2017, 10, 226. [Google Scholar] [CrossRef]
- Huang, T.; Fu, G.; Gao, J.; Zhang, Y.; Cai, W.; Wu, S.; Jia, S.; Xia, S.; Bachmann, T.; Bekker, A.; et al. Fgr contributes to hemorrhage-induced thalamic pain by activating NF-κB/ERK1/2 pathways. JCI Insight 2020, 5, e139987. [Google Scholar] [CrossRef] [PubMed]
- Li, H.L.; Lin, M.; Tan, X.P.; Wang, J.L. Role of Sensory Pathway Injury in Central Post-Stroke Pain: A Narrative Review of Its Pathogenetic Mechanism. J. Pain Res. 2023, 16, 1333–1343. [Google Scholar] [CrossRef]
- Choi, H.R.; Aktas, A.; Bottros, M.M. Pharmacotherapy to Manage Central Post-Stroke Pain. CNS Drugs 2021, 35, 151–160. [Google Scholar] [CrossRef]
- Flaster, M.; Meresh, E.; Rao, M.; Biller, J. Central poststroke pain: Current diagnosis and treatment. Top. Stroke Rehabil. 2013, 20, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Subedi, M.; Bajaj, S.; Kumar, M.S.; Mayur, Y.C. An overview of tramadol and its usage in pain management and future perspective. Biomed. Pharmacother. 2019, 111, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S. Pharmacological management of central post-stroke pain: A practical guide. CNS Drugs 2014, 28, 787–797. [Google Scholar] [CrossRef]
- Ramger, B.C.; Bader, K.A.; Davies, S.P.; Stewart, D.A.; Ledbetter, L.S.; Simon, C.B.; Feld, J.A. Effects of Non-Invasive Brain Stimulation on Clinical Pain Intensity and Experimental Pain Sensitivity Among Individuals with Central Post-Stroke Pain: A Systematic Review. J. Pain Res. 2019, 12, 3319–3329. [Google Scholar] [CrossRef] [PubMed]
- Lempka, S.F.; Malone, D.A., Jr.; Hu, B.; Baker, K.B.; Wyant, A.; Ozinga, J.G., 4th; Plow, E.B.; Pandya, M.; Kubu, C.S.; Ford, P.J.; et al. Randomized clinical trial of deep brain stimulation for poststroke pain. Ann. Neurol. 2017, 81, 653–663. [Google Scholar] [CrossRef]
- Boccard, S.G.J.; Prangnell, S.J.; Pycroft, L.; Cheeran, B.; Moir, L.; Pereira, E.A.C.; Fitzgerald, J.J.; Green, A.L.; Aziz, T.Z. Long-Term Results of Deep Brain Stimulation of the Anterior Cingulate Cortex for Neuropathic Pain. World Neurosurg. 2017, 106, 625–637. [Google Scholar] [CrossRef]
- Chen, X.; Li, Z.; Zhang, B.; Hu, R.; Li, J.; Feng, M.; Yao, W.; Zhang, C.; Wan, L.; Zhang, Y. Alleviation of Mechanical Allodynia by 14,15-Epoxyeicosatrienoic Acid in a Central Poststroke Pain Model: Possible Role of Allopregnanolone and δ-Subunit-Containing Gamma-Aminobutyric Acid A Receptors. J. Pain 2019, 20, 577–591. [Google Scholar] [CrossRef]
- Wagner, K.; Yang, J.; Inceoglu, B.; Hammock, B.D. Soluble epoxide hydrolase inhibition is antinociceptive in a mouse model of diabetic neuropathy. J. Pain 2014, 15, 907–914. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, L.J.; Wang, J.; Li, D.; Ren, W.J.; Peng, J.; Wei, X.; Xu, T.; Xin, W.J.; Pang, R.P.; et al. TNF-α Differentially Regulates Synaptic Plasticity in the Hippocampus and Spinal Cord by Microglia-Dependent Mechanisms after Peripheral Nerve Injury. J. Neurosci. 2017, 37, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Coppi, E.; Dettori, I.; Cherchi, F.; Bulli, I.; Venturini, M.; Pedata, F.; Pugliese, A.M. New Insight into the Role of Adenosine in Demyelination, Stroke and Neuropathic Pain. Front. Pharmacol. 2021, 11, 625662. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Author | Diagnosis Criteria |
| Ri S, 2022 [23] |
|
| Osama A et al., 2018 [19] |
|
| Choi-kwan et al., 2016 [24] |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohanan, A.T.; Nithya, S.; Nomier, Y.; Hassan, D.A.; Jali, A.M.; Qadri, M.; Machanchery, S. Stroke-Induced Central Pain: Overview of the Mechanisms, Management, and Emerging Targets of Central Post-Stroke Pain. Pharmaceuticals 2023, 16, 1103. https://doi.org/10.3390/ph16081103
Mohanan AT, Nithya S, Nomier Y, Hassan DA, Jali AM, Qadri M, Machanchery S. Stroke-Induced Central Pain: Overview of the Mechanisms, Management, and Emerging Targets of Central Post-Stroke Pain. Pharmaceuticals. 2023; 16(8):1103. https://doi.org/10.3390/ph16081103
Chicago/Turabian StyleMohanan, Anugeetha Thacheril, Sermugapandian Nithya, Yousra Nomier, Dalin A. Hassan, Abdulmajeed M. Jali, Marwa Qadri, and Shamna Machanchery. 2023. "Stroke-Induced Central Pain: Overview of the Mechanisms, Management, and Emerging Targets of Central Post-Stroke Pain" Pharmaceuticals 16, no. 8: 1103. https://doi.org/10.3390/ph16081103
APA StyleMohanan, A. T., Nithya, S., Nomier, Y., Hassan, D. A., Jali, A. M., Qadri, M., & Machanchery, S. (2023). Stroke-Induced Central Pain: Overview of the Mechanisms, Management, and Emerging Targets of Central Post-Stroke Pain. Pharmaceuticals, 16(8), 1103. https://doi.org/10.3390/ph16081103