
Discovery of New Uracil and Thiouracil Derivatives as Potential HDAC Inhibitors

 , , , , , ,
, , , , , ,  and
and
Abstract
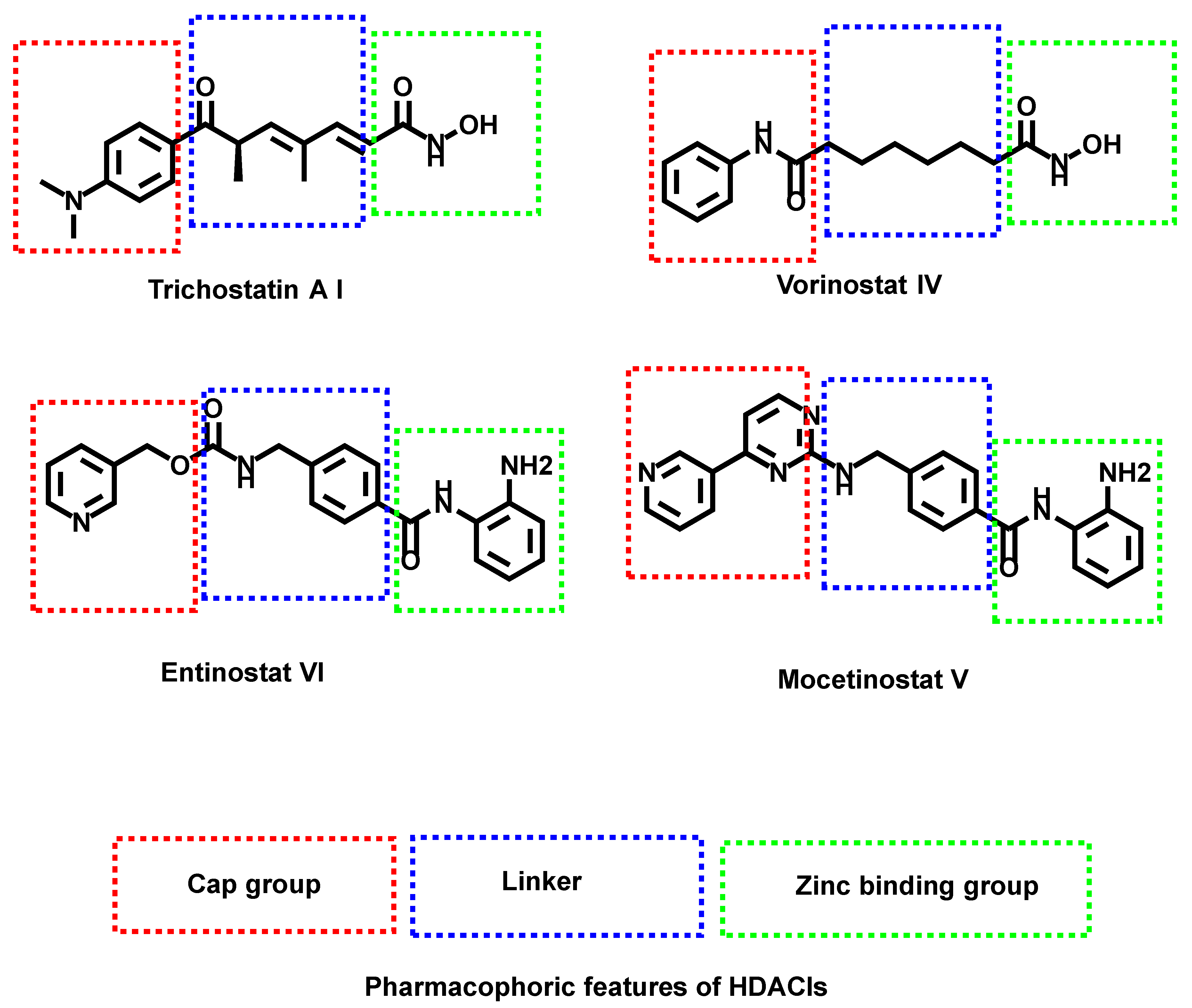
1. Introduction
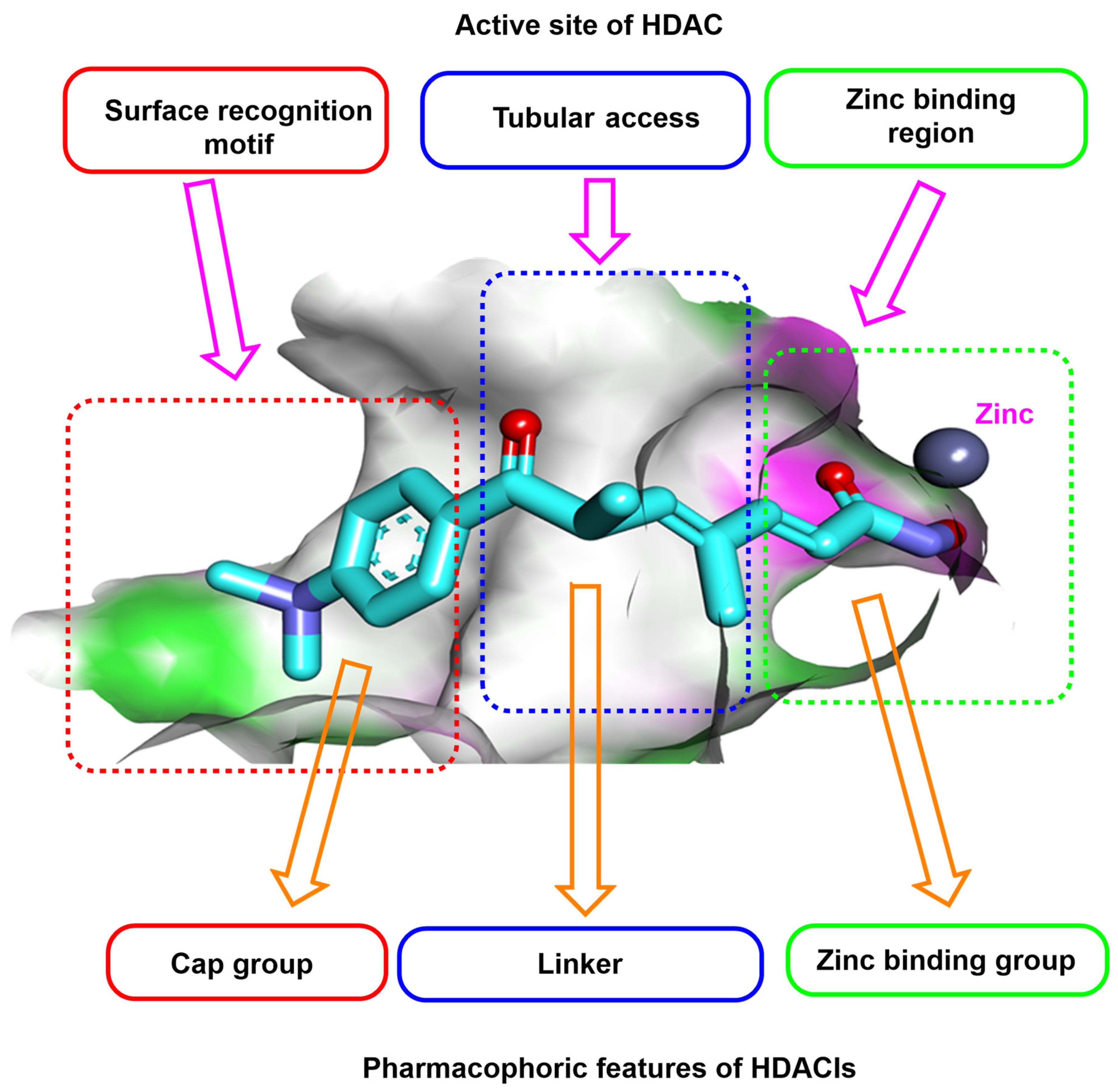
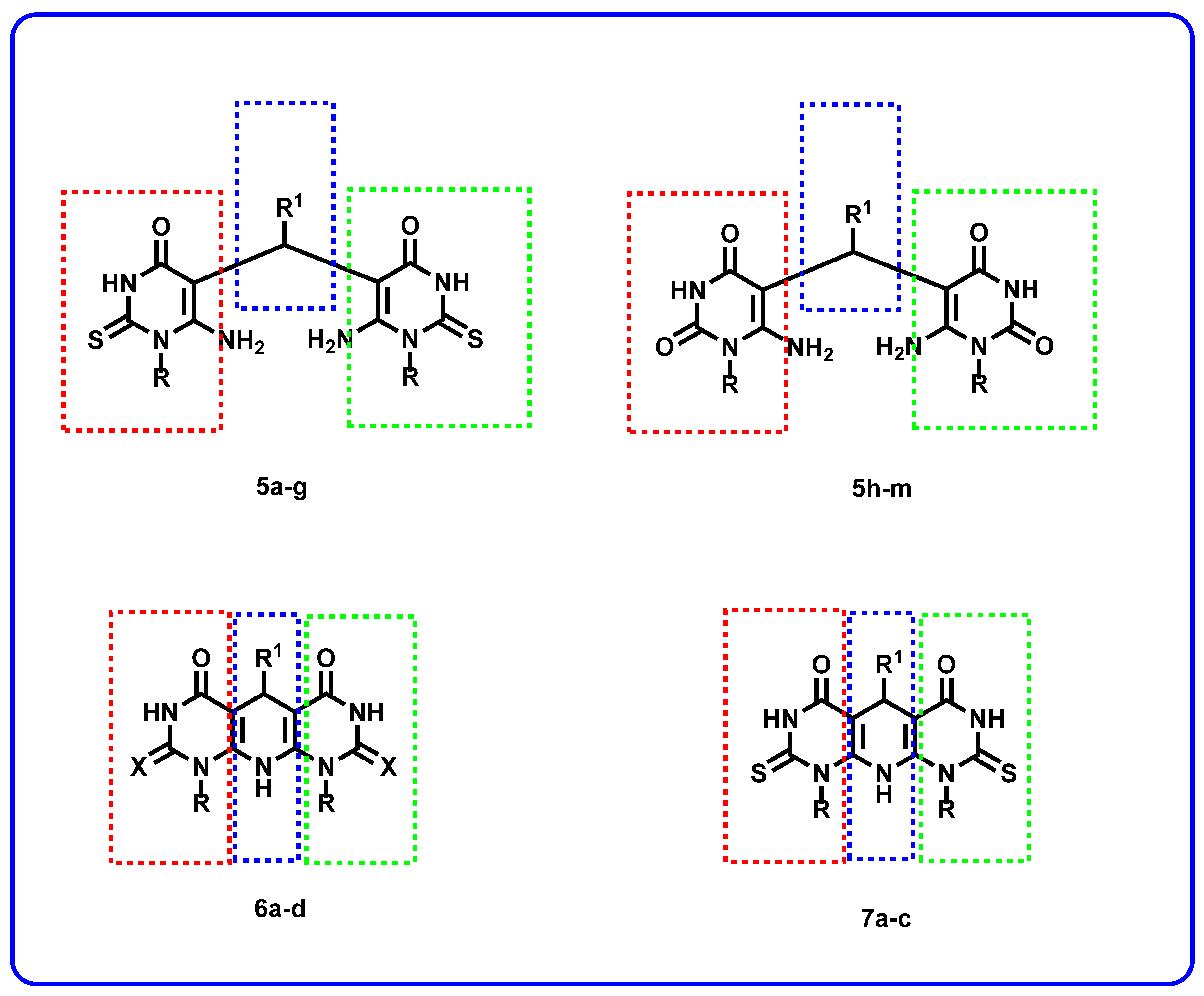
Rationale of Molecular Design
2. Results and Discussion
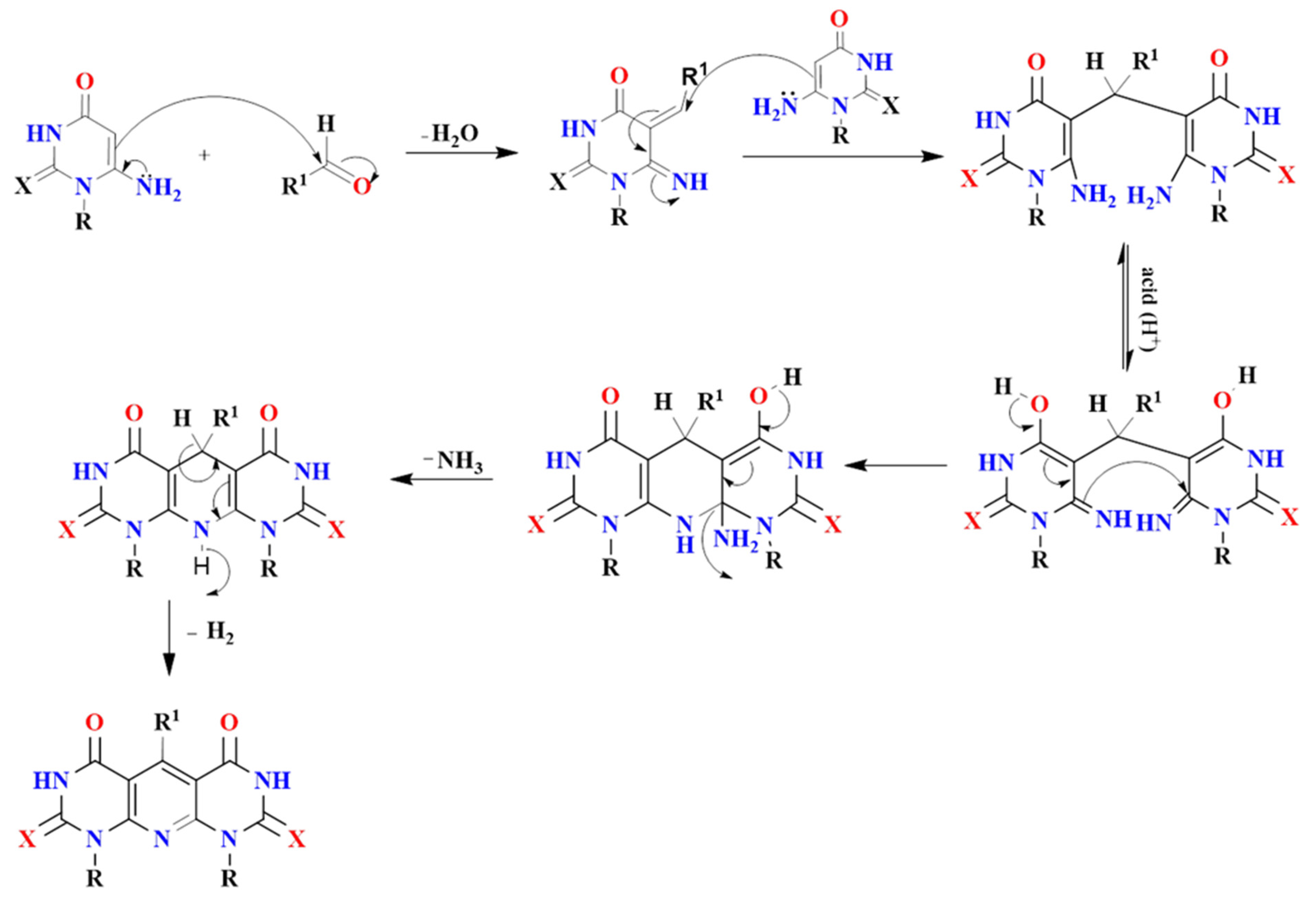
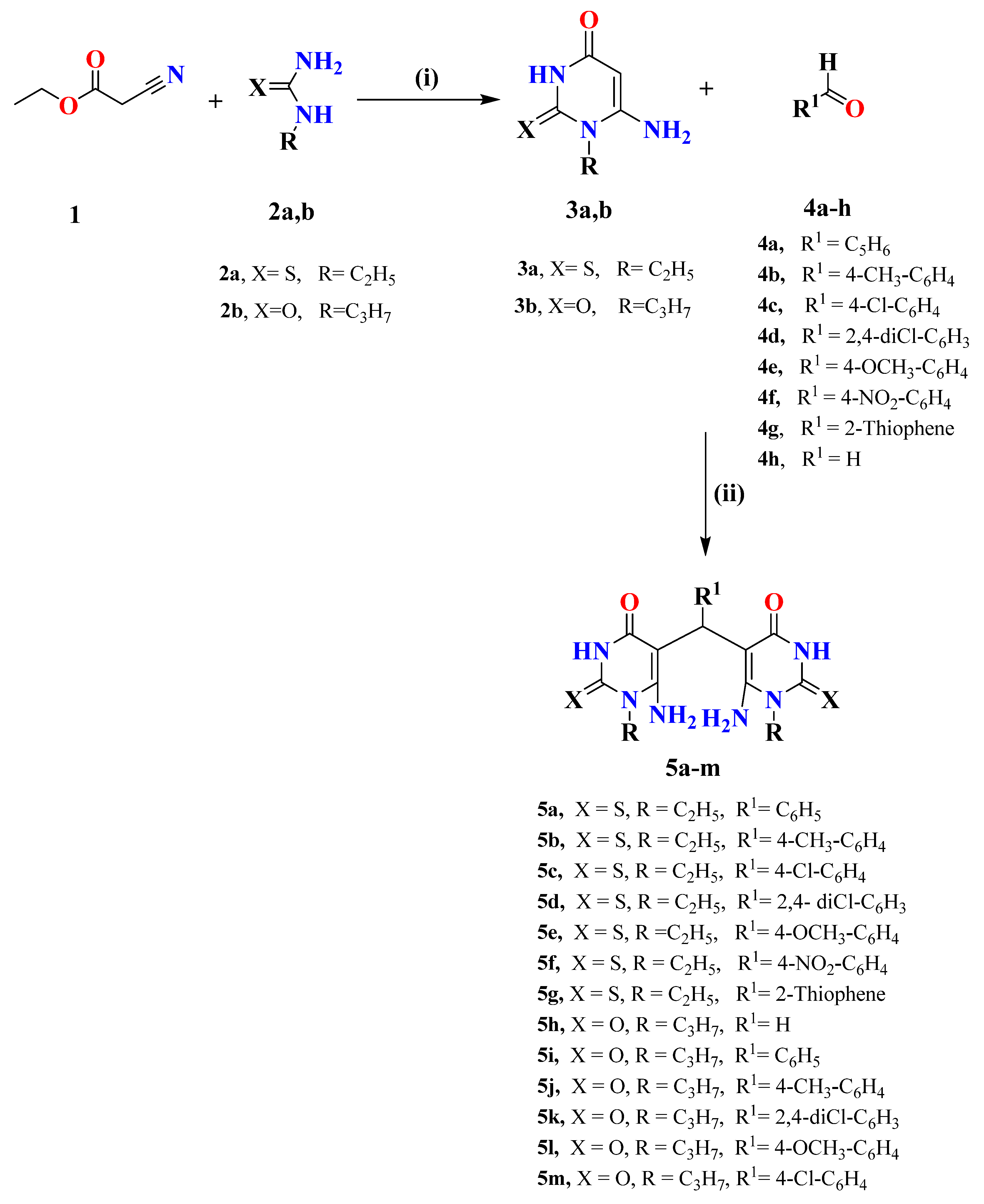
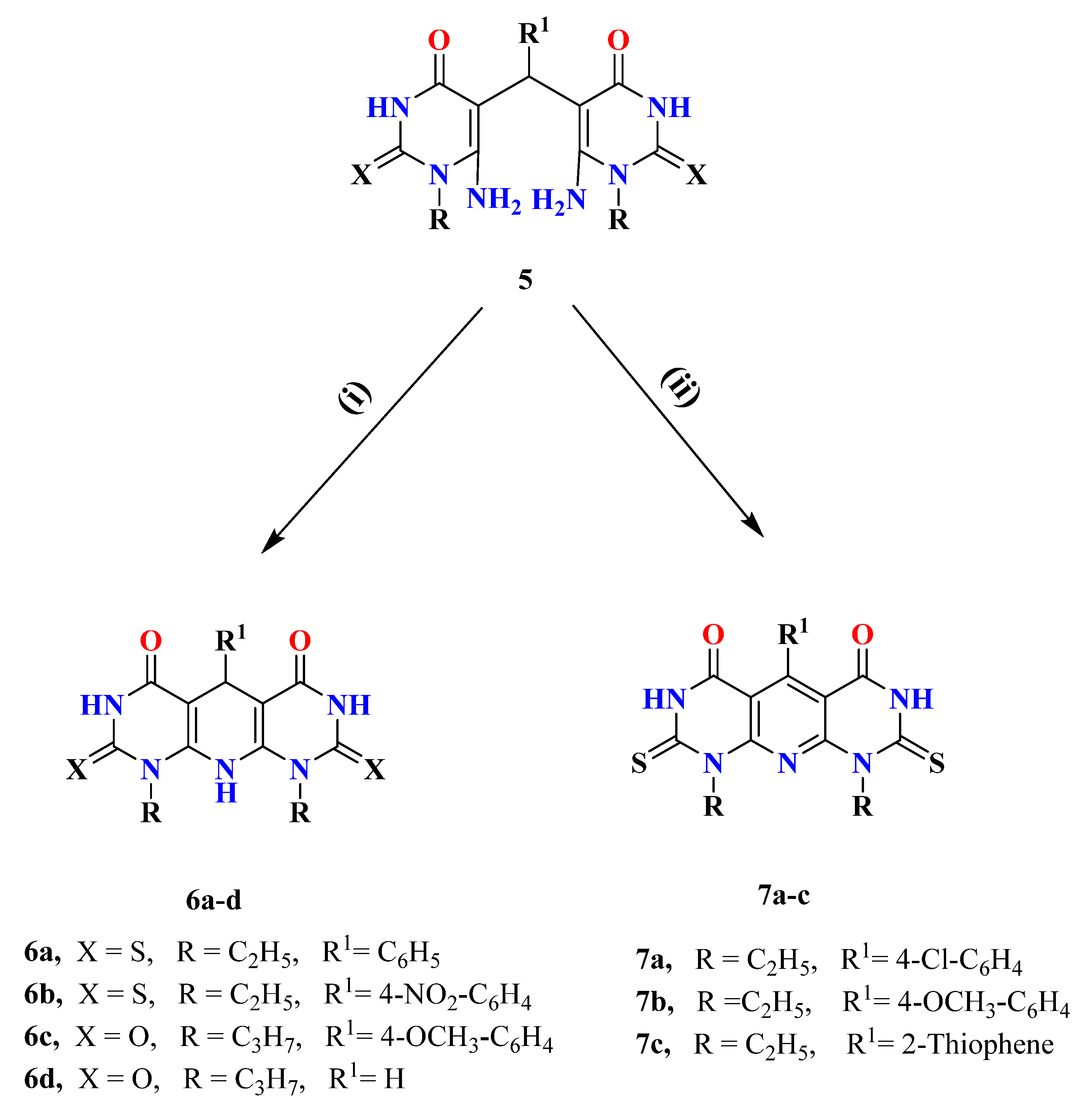
2.1. Chemistry
2.2. Biological Testing
2.2.1. In Vitro Cytotoxic Activities
2.2.2. Structure-Activity Relationship
2.2.3. HDAC1 and HDAC4 Inhibitory Assay
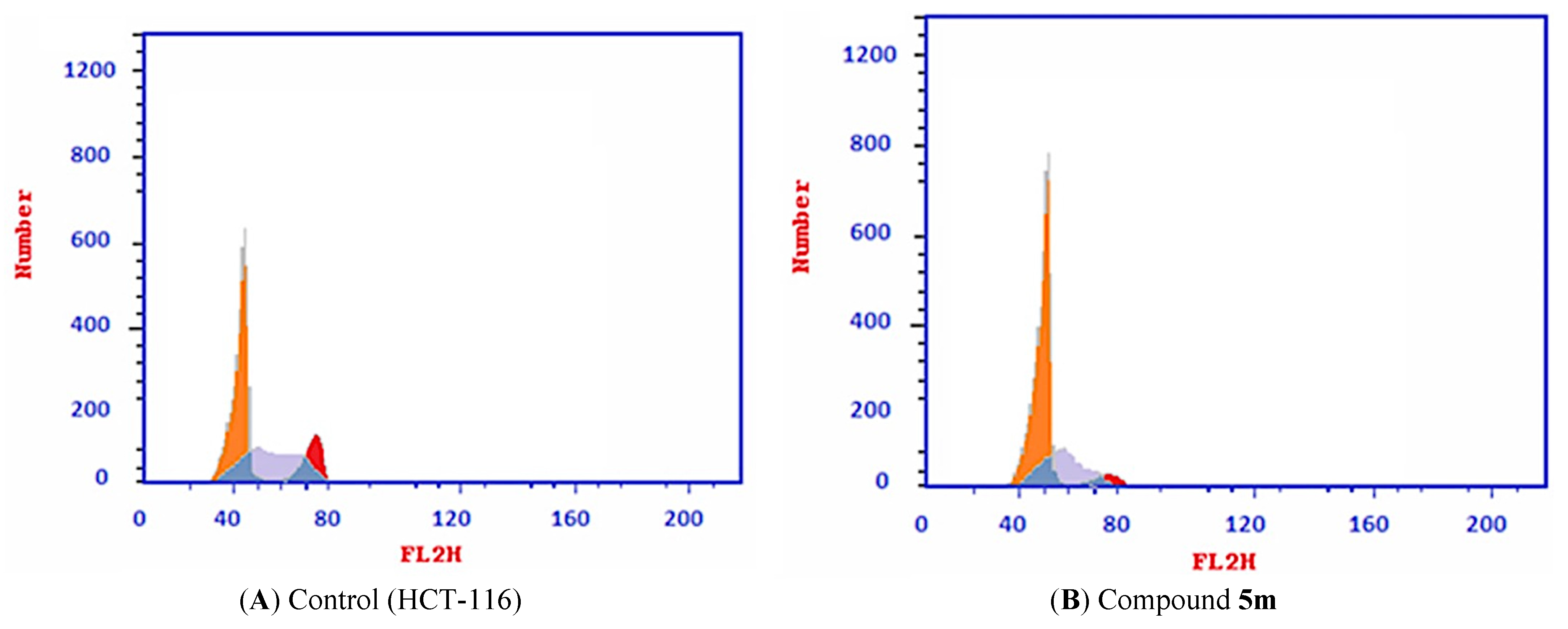
2.2.4. Cell Cycle Analysis
2.2.5. Apoptosis Analysis
2.2.6. Caspase-3 and Caspase-8 Determination
2.2.7. Cytotoxicity against Normal Cell Line
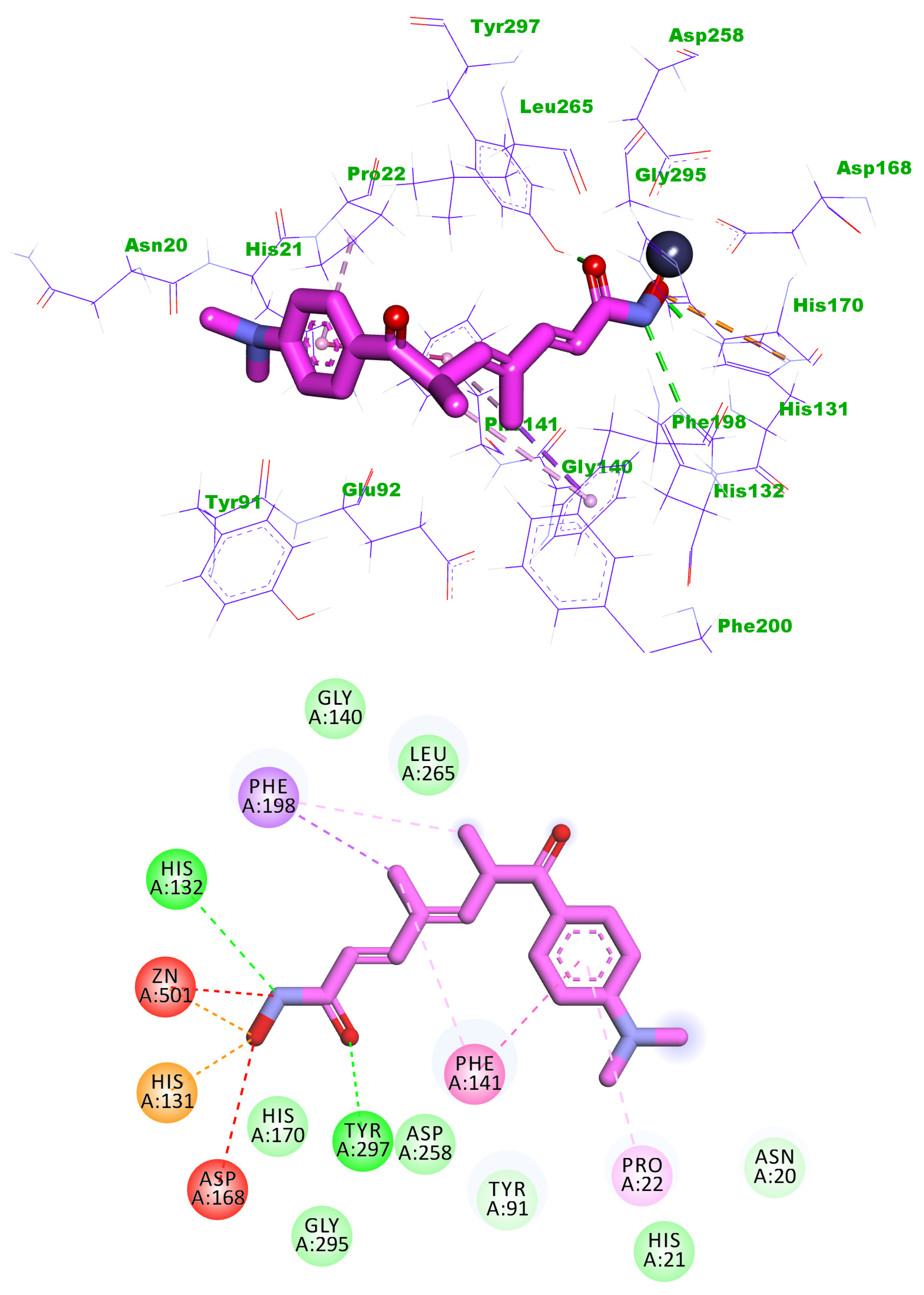
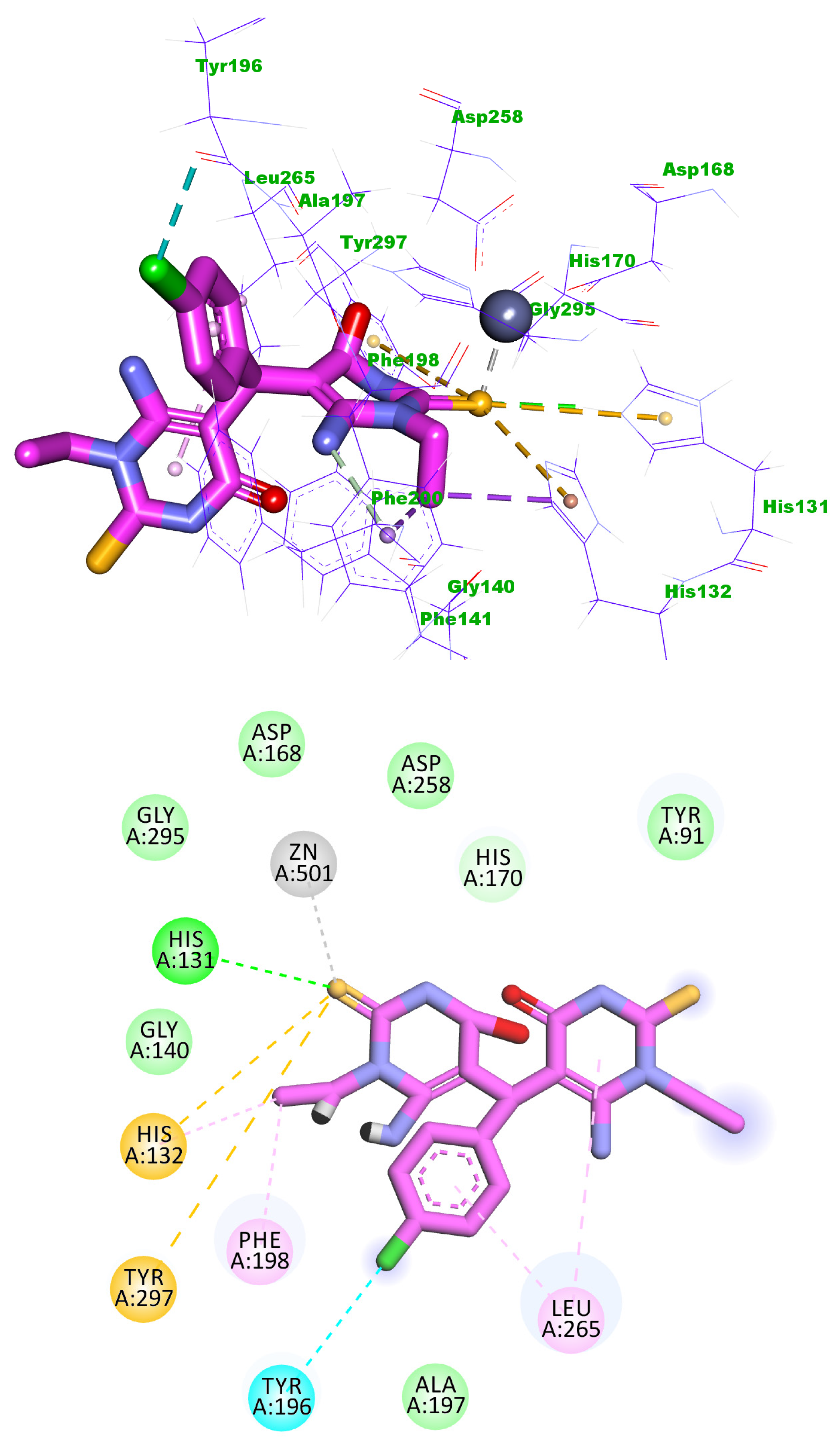
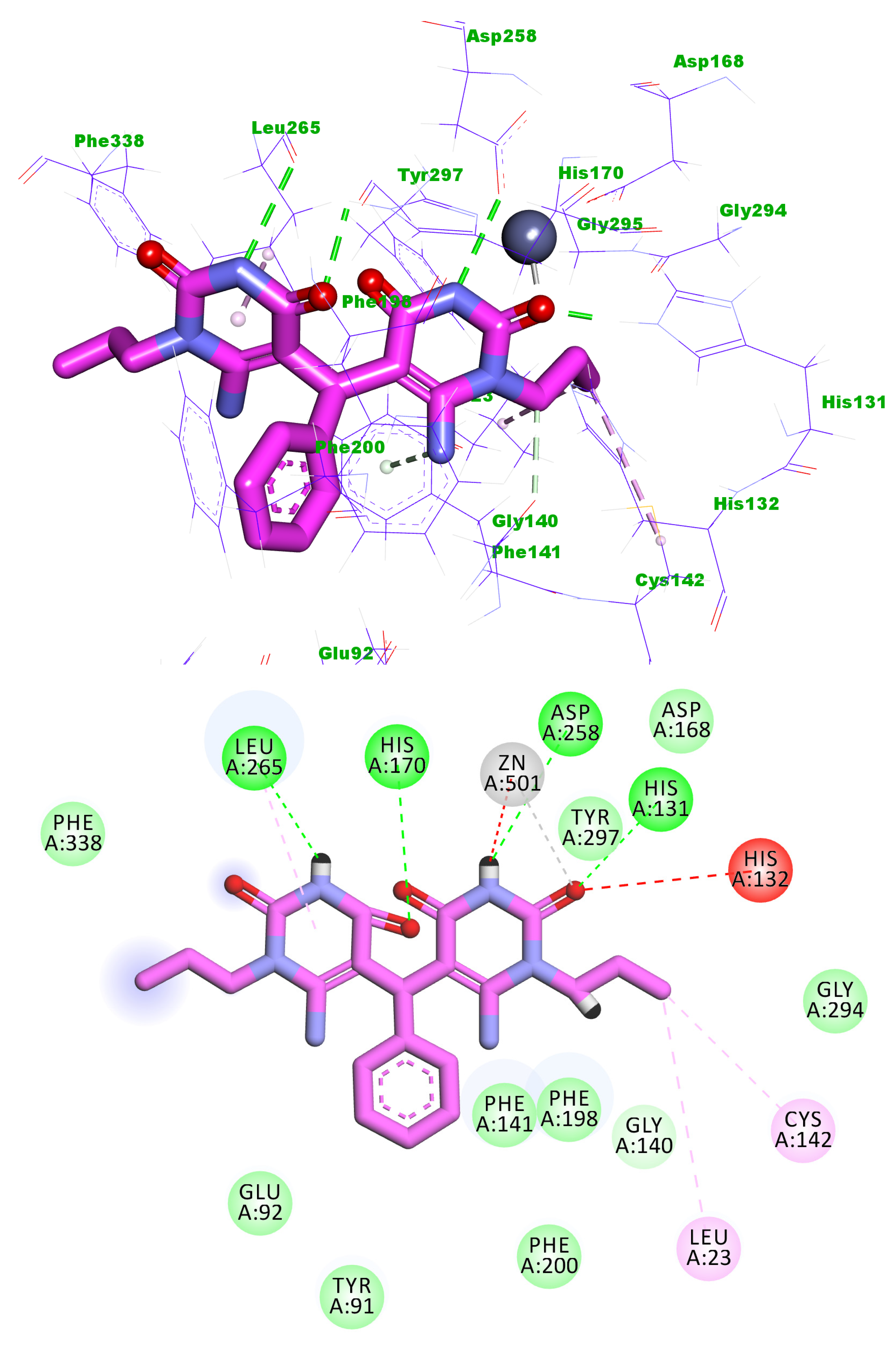
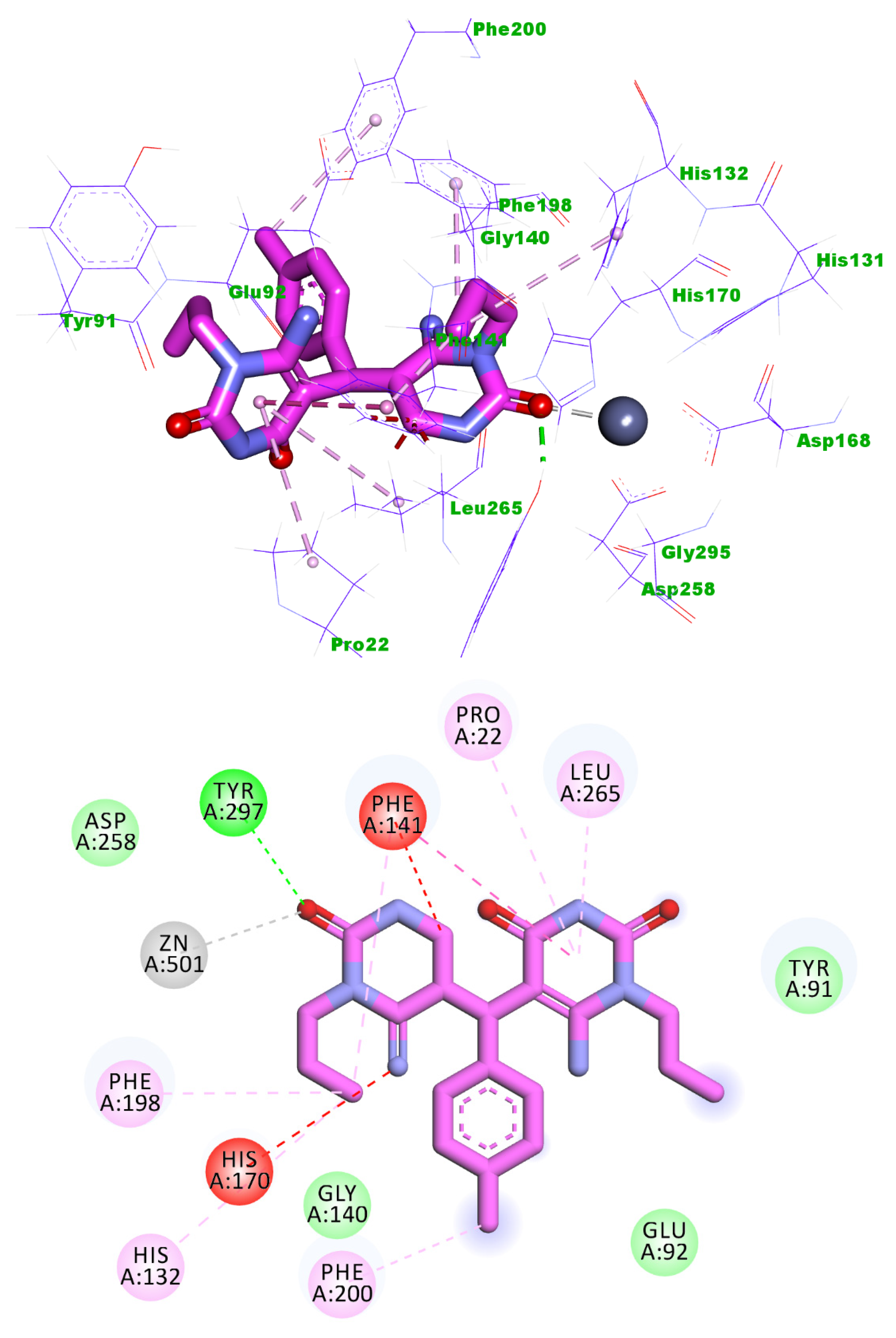
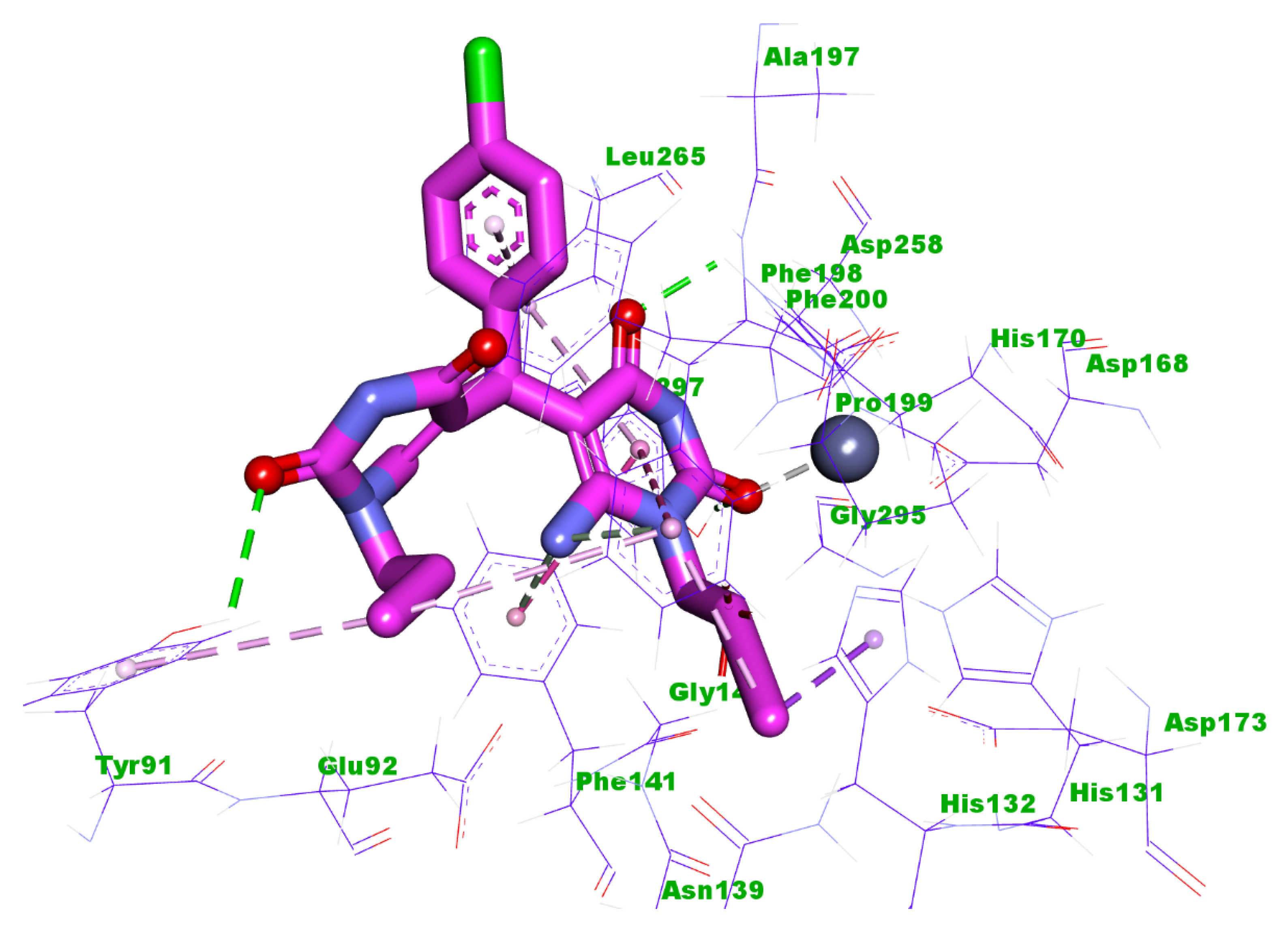
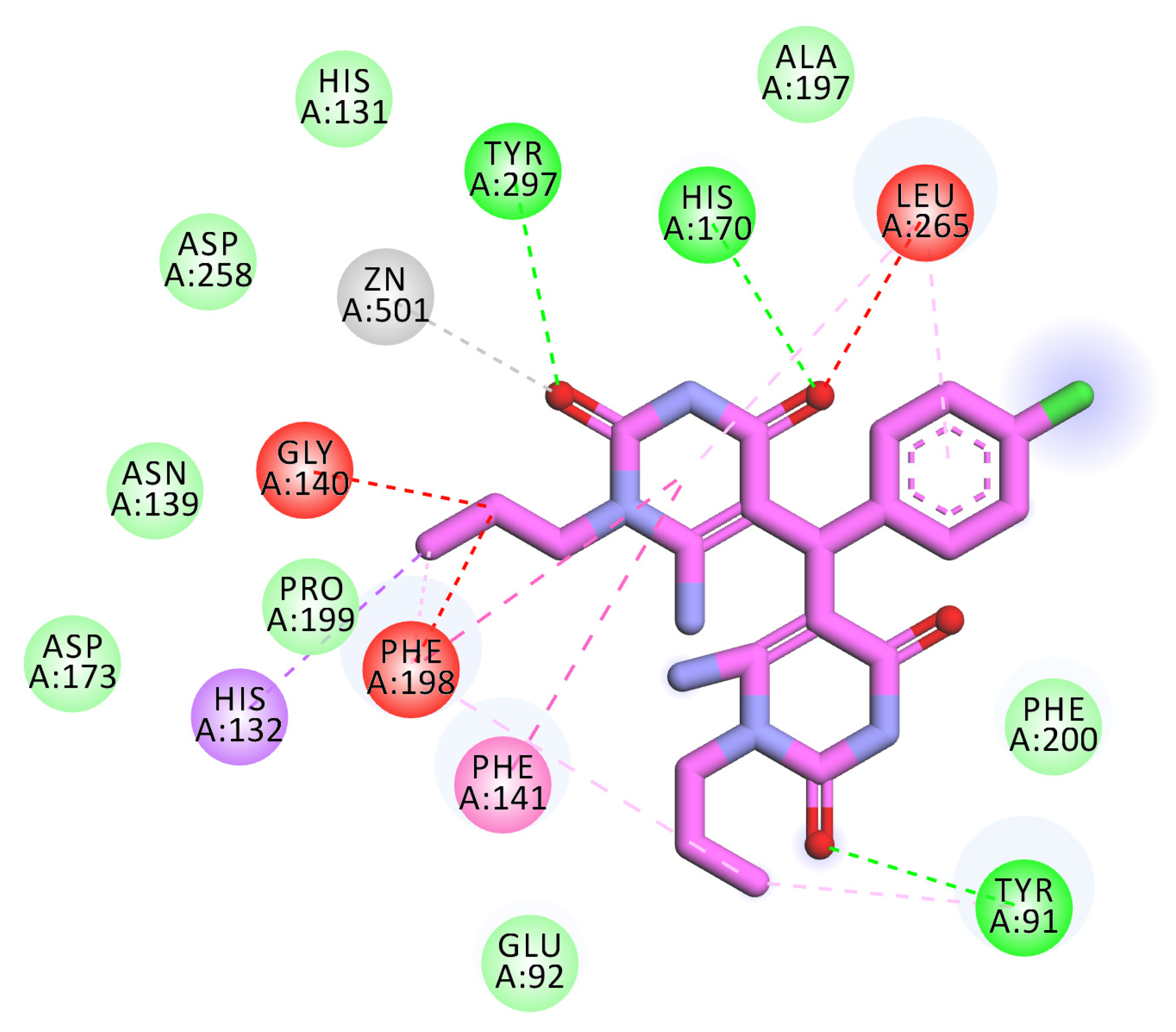
2.3. Docking Studies
3. Conclusions
4. Experimental
4.1. Chemistry
4.1.1. General
4.1.2. 6-Amino-1-alkyl-2-oxo/thioxo-2,3-dihydropyrimidinones (3a,b)
4.1.3. 5,5′-(Arylmethylene)bis(6-amino-1-alkyl-2-oxo/thioxo-2,3-dihydropyrimidinones) (5a–m)
5,5′-(Phenylmethylene)bis(6-amino-1-ethyl-2-thioxo-2,3-dihydropyrimidin-4(1H)-one) (5a)
5,5′-(p-Tolylmethylene)bis(6-amino-1-ethyl-2-thioxo-2,3-dihydropyrimidin-4(1H)-one) (5b)
5,5′-((4-Chlorophenyl)methylene)bis(6-amino-1-ethyl-2-thioxo-2,3-dihydropyrimidin- 4(1H)-one) (5c)
5,5′-((2,4-Dichlorophenyl)methylene)bis(6-amino-1-ethyl-2-thioxo-2,3-dihydro pyrimidin-4(1H)-one) (5d)
5,5′-((4-Methoxyphenyl)methylene)bis(6-amino-1-ethyl-2-thioxo-2,3-dihydropyrimidin -4(1H)-one) (5e)
5,5′-((4-Nitrophenyl)methylene)bis(6-amino-1-ethyl-2-thioxo-2,3-dihydropyrimidin-4(1H)-one) (5f)
5,5′-(Thiophen-2-ylmethylene)bis(6-amino-1-ethyl-2-thioxo-2,3-dihydropyrimidin-4(1H)-one) (5g)
5,5′-Methylenebis(6-amino-1-propylpyrimidine-2,4(1H,3H)-dione) (5h)
5,5′-(Phenylmethylene)bis(6-amino-1-propylpyrimidine-2,4(1H,3H)-dione) (5i)
5,5′-(p-Tolylmethylene)bis(6-amino-1-propylpyrimidine-2,4(1H,3H)-dione) (5j)
5,5′-((2,4-Dichlorophenyl)methylene)bis(6-amino-1-propylpyrimidine-2,4(1H,3H)-dione) (5k)
5,5′-((4-Methoxyphenyl)methylene)bis(6-amino-1-propylpyrimidine-2,4(1H,3H)-dione) (5l)
5,5′-((4-Chlorophenyl)methylene)bis(6-amino-1-propylpyrimidine-2,4(1H,3H)-dione) (5m)
4.1.4. 1,9-Dialkyl-2,3,5,8,9,10-hexahydropyrido[2,3-d:6,5-d′]dipyrimidinones (6a–d)
1,9-Diethyl-5-phenyl-2,8-dithioxo-2,3,5,8,9,10-hexahydropyrido[2,3-d:6,5-d′]dipyrimidine-4,6(1H,7H)-dione (6a)
1,9-Diethyl-5-(4-nitrophenyl)-2,8-dithioxo-2,3,5,8,9,10-hexahydropyrido[2,3-d:6,5-d′]dipyrimidine-4,6(1H,7H)-dione (6b)
5-(4-Methoxyphenyl)-1,9-dipropyl-5,10-dihydropyrido[2,3-d:6,5-d′]dipyrimidine-2,4,6,8(1H,3H,7H,9H)-tetraone (6c)
1,9-Dipropyl-5,10-dihydropyrido[2,3-d:6,5-d′]dipyrimidine-2,4,6,8 (1H,3H,7H,9H)-tetraone (6d)
4.1.5. 5Aryl-1,9-Diethyl-2,8-dithioxo-2,3,8,9-tetrahydropyrido[2,3-d:6,5-d′]dipyrimidine-4,6(1H,7H)-diones (7a–c)
5-(4-Chlorophenyl)-1,9-diethyl-2,8-dithioxo-2,3,8,9-tetrahydropyrido[2,3-d:6,5-d′]dipyrimidine-4,6(1H,7H)-dione (7a)
1,9-Diethyl-5-(4-methoxyphenyl)-2,8-dithioxo-2,3,8,9-tetrahydropyrido[2,3-d:6,5-d′]dipyrimidine-4,6(1H,7H)-dione (7b)
1,9-Diethyl-5-(thiophen-2-yl)-2,8-dithioxo-2,3,8,9-tetrahydropyrido[2,3-d:6,5-d′]dipyrimidine-4,6(1H,7H)-dione (7c)
4.2. Biological Testing
4.2.1. In Vitro Cytotoxic Activity
4.2.2. In vitro HDAC Assay
4.2.3. Flow Cytometry Analysis for Cell Cycle
4.2.4. Flow Cytometry Analysis for Apoptosis
4.3. Docking Studies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- WHO. Cancer. Available online: https://www.who.int/health-topics/cancer#tab=tab_1 (accessed on 1 October 2020).
- Badran, M.M.; Abouzid, K.A.M.; Hussein, M.H.M. Synthesis of certain substituted quinoxalines as antimicrobial agents (Part II). Arch. Pharmacal Res. 2003, 26, 107–113. [Google Scholar] [CrossRef]
- World Health Organisation. Cancer—Key Facts. 2018. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 1 March 2023).
- Chabner, B.A.; Roberts, T.G., Jr. Timeline: Chemotherapy and the war on cancer. Nat. Rev. Cancer 2005, 5, 65–72. [Google Scholar] [CrossRef]
- Krauss, G.; Schönbrunner, N.; Cooper, J. Biochemistry of Signal Transduction and Regulation; Wiley Online Library: Weinheim, NJ, USA, 2003; Volume 3. [Google Scholar]
- Nguyen, K.-S.H.; Kobayashi, S.; Costa, D.B. Acquired Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Non–Small-Cell Lung Cancers Dependent on the Epidermal Growth Factor Receptor Pathway. Clin. Lung Cancer 2009, 10, 281–289. [Google Scholar] [CrossRef]
- Waldmann, T.; Schneider, R. Targeting histone modifications—Epigenetics in cancer. Curr. Opin. Cell Biol. 2013, 25, 184–189. [Google Scholar] [CrossRef]
- Yang, X.-J.; Seto, E. Lysine Acetylation: Codified Crosstalk with Other Posttranslational Modifications. Mol. Cell 2008, 31, 449–461. [Google Scholar] [CrossRef]
- Yang, X.J.; Grégoire, S. Metabolism, cytoskeleton and cellular signalling in the grip of protein Nϵ-and O-acetylation. EMBO Rep. 2007, 8, 556–562. [Google Scholar] [CrossRef]
- Sun, X.-J.; Man, N.; Tan, Y.; Nimer, S.D.; Wang, L. The Role of Histone Acetyltransferases in Normal and Malignant Hematopoiesis. Front. Oncol. 2015, 5, 108. [Google Scholar] [CrossRef]
- Glauben, R.; Sonnenberg, E.; Zeitz, M.; Siegmund, B. HDAC inhibitors in models of inflammation-related tumorigenesis. Cancer Lett. 2009, 280, 154–159. [Google Scholar] [CrossRef]
- Hanessian, S.; Auzzas, L.; Giannini, G.; Marzi, M.; Cabri, W.; Barbarino, M.; Vesci, L.; Pisano, C. ω-Alkoxy analogues of SAHA (vorinostat) as inhibitors of HDAC: A study of chain-length and stereochemical dependence. Bioorganic Med. Chem. Lett. 2007, 17, 6261–6265. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, H.-F.; Lu, S.; Zheng, Y.-X.; Wu, Z.; Tang, W.-F.; Zhou, X.; Lu, T. Investigation on the isoform selectivity of histone deacetylase inhibitors using chemical feature based pharmacophore and docking approaches. Eur. J. Med. Chem. 2010, 45, 1777–1791. [Google Scholar] [CrossRef]
- Marks, P.A.; Rifkind, R.A.; Richon, V.M.; Breslow, R.; Miller, T.; Kelly, W.K. Histone deacetylases and cancer: Causes and therapies. Nat. Rev. Cancer 2001, 1, 194–202. [Google Scholar] [CrossRef]
- Ekou, L.; Ekou, T.; Opalinski, I.; Gesson, J.P. Histone Deacetylase Inhibitors: Synthesis of Tetrapeptide Analogue SAHA/TPX. E J. Chem. 2011, 8, S79–S84. [Google Scholar] [CrossRef]
- Grozinger, C.M.; Schreiber, S.L. Deacetylase Enzymes: Biological Functions and the Use of Small-Molecule Inhibitors. Chem. Biol. 2002, 9, 3–16. [Google Scholar] [CrossRef]
- Miller, T.A.; Witter, D.J.; Belvedere, S. Histone deacetylase inhibitors. J. Med. Chem. 2003, 46, 5097–5116. [Google Scholar] [CrossRef]
- Yoshida, M.; Horinouchi, S.; Beppu, T. Trichostatin A and trapoxin: Novel chemical probes for the role of histone acetylation in chromatin structure and function. Bioessays 1995, 17, 423–430. [Google Scholar] [CrossRef]
- Methot, J.L.; Chakravarty, P.K.; Chenard, M.; Close, J.; Cruz, J.C.; Dahlberg, W.K.; Fleming, J.; Hamblett, C.L.; Hamill, J.E.; Harrington, P. Exploration of the internal cavity of histone deacetylase (HDAC) with selective HDAC1/HDAC2 inhibitors (SHI-1: 2). Bioorganic Med. Chem. Lett. 2008, 18, 973–978. [Google Scholar] [CrossRef]
- Schäfer, S.; Saunders, L.; Eliseeva, E.; Velena, A.; Jung, M.; Schwienhorst, A.; Strasser, A.; Dickmanns, A.; Ficner, R.; Schlimme, S.; et al. Phenylalanine-containing hydroxamic acids as selective inhibitors of class IIb histone deacetylases (HDACs). Bioorganic Med. Chem. 2008, 16, 2011–2033. [Google Scholar] [CrossRef]
- Islam, N.M.; Kato, T.; Nishino, N.; Kim, H.-J.; Ito, A.; Yoshida, M. Bicyclic peptides as potent inhibitors of histone deacetylases: Optimization of alkyl loop length. Bioorganic Med. Chem. Lett. 2010, 20, 997–999. [Google Scholar] [CrossRef]
- Meinke, P.; Liberator, P. Histone Deacetylase: A Target for Antiproliferative and Antiprotozoal Agents. Curr. Med. Chem. 2001, 8, 211–235. [Google Scholar] [CrossRef]
- Yoshida, M.; Matsuyama, A.; Komatsu, Y.; Nishino, N. From discovery to the coming generation of histone deacetylase inhibitors. Curr. Med. Chem. 2003, 10, 2351–2358. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, P. Inside HDAC with HDAC inhibitors. Eur. J. Med. Chem. 2010, 45, 2095–2116. [Google Scholar] [CrossRef]
- Awad, S.M.; Zohny, Y.M.; Ali, S.A.; Mahgoub, S.; Said, A.M. Design, Synthesis, Molecular Modeling, and Biological Evaluation of Novel Thiouracil Derivatives as Potential Antithyroid Agents. Molecules 2018, 23, 2913. [Google Scholar] [CrossRef]
- El-Naggar, A.M.; Abou-El-Regal, M.M.; El-Metwally, S.A.; Sherbiny, F.F.; Eissa, I.H. Synthesis, characterization and molecular docking studies of thiouracil derivatives as potent thymidylate synthase inhibitors and potential anticancer agents. Mol. Divers. 2017, 21, 967–983. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Y.; Xie, N.; Xu, M.; Qian, P.; Zhao, Y.; Li, S. Design, synthesis and antiproliferative activities of novel benzamides derivatives as HDAC inhibitors. Eur. J. Med. Chem. 2015, 100, 270–276. [Google Scholar] [CrossRef]
- El-Kalyoubi, S.A.; Taher, E.S.; Ibrahim, T.S.; El-Behairy, M.F.; Al-Mahmoudy, A.M.M. Uracil as a Zn-Binding Bioisostere of the Allergic Benzenesulfonamide in the Design of Quinoline–Uracil Hybrids as Anticancer Carbonic Anhydrase Inhibitors. Pharmaceuticals 2022, 15, 494. [Google Scholar] [CrossRef]
- El-Kalyoubi, S.; Agili, F.; Adel, I.; Tantawy, M.A. Novel uracil derivatives depicted potential anticancer agents: In Vitro, molecular docking, and ADME study. Arab. J. Chem. 2022, 15, 103669. [Google Scholar] [CrossRef]
- El-Kalyoubi, S.A.; Ragab, A.; Abu Ali, O.A.; Ammar, Y.A.; Seadawy, M.G.; Ahmed, A.; Fayed, E.A. One-pot synthesis and molecular modeling studies of new bioactive spiro-oxindoles based on uracil derivatives as SARS-CoV-2 inhibitors targeting rna polymerase and spike glycoprotein. Pharmaceuticals 2022, 15, 376. [Google Scholar] [CrossRef]
- El-Kalyoubi, S.; Agili, F.; Zordok, W.A.; El-Sayed, A.S.A. Synthesis, In Silico Prediction and In Vitro Evaluation of Antimicrobial Activity, DFT Calculation and Theoretical Investigation of Novel Xanthines and Uracil Containing Imidazolone Derivatives. Int. J. Mol. Sci. 2021, 22, 10979. [Google Scholar] [CrossRef]
- El-Kalyoubi, S.; Agili, F. Synthesis, In Silico Prediction and In Vitro Evaluation of Antitumor Activities of Novel Pyrido [2, 3-d] pyrimidine, Xanthine and Lumazine Derivatives. Molecules 2020, 25, 5205. [Google Scholar] [CrossRef]
- El-Kalyoubi, S.; Agili, F.; Youssif, S. Novel 2-Thioxanthine and Dipyrimidopyridine Derivatives: Synthesis and Antimicrobial Activity. Molecules 2015, 20, 19263–19276. [Google Scholar] [CrossRef]
- Han, H.; Li, C.; Li, M.; Yang, L.; Zhao, S.; Wang, Z.; Liu, H.; Liu, D. Design, Synthesis, and Biological Evaluation of 8-Mercapto-3,7-Dihydro-1H-Purine-2,6-Diones as Potent Inhibitors of SIRT1, SIRT2, SIRT3, and SIRT5. Molecules 2020, 25, 2755. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Bandyopadhyay, D.; Okan, N.A.; Bales, E.; Nascimento, L.; Cole, P.A.; Medrano, E.E. Down-regulation of p300/CBP histone acetyltransferase activates a senescence checkpoint in human melanocytes. Cancer Res. 2002, 62, 6231–6239. [Google Scholar]
- Bandyopadhyay, D.; Mishra, A.; Medrano, E.E. Overexpression of Histone Deacetylase 1 Confers Resistance to Sodium Butyrate–Mediated Apoptosis in Melanoma Cells through a p53-Mediated Pathway. Cancer Res. 2004, 64, 7706–7710. [Google Scholar] [CrossRef]
- Wang, Z.; Qin, G.; Zhao, T.C. HDAC4: Mechanism of regulation and biological functions. Epigenomics 2014, 6, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Mai, A.; Perrone, A.; Nebbioso, A.; Rotili, D.; Valente, S.; Tardugno, M.; Massa, S.; De Bellis, F.; Altucci, L. Novel uracil-based 2-aminoanilide and 2-aminoanilide-like derivatives: Histone deacetylase inhibition and in-cell activities. Bioorganic Med. Chem. Lett. 2008, 18, 2530–2535. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, D.; Wang, X.; Wang, Y.; Ren, F.; Chang, D.; Chang, Z.; Jia, B. Caspase 3 is Activated through Caspase 8 instead of Caspase 9 during H2O2-induced Apoptosis in HeLa Cells. Cell. Physiol. Biochem. 2011, 27, 539–546. [Google Scholar] [CrossRef]
- Denizot, F.; Lang, R. Rapid colorimetric assay for cell growth and survival: Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods 1986, 89, 271–277. [Google Scholar] [CrossRef]
- Thabrew, M.I.; Hughes, R.D.; Mcfarlane, I.G. Screening of Hepatoprotective Plant Components using a HepG2 Cell Cytotoxicity Assay. J. Pharm. Pharmacol. 1997, 49, 1132–1135. [Google Scholar] [CrossRef]
- Al-Rashood, S.T.; Hamed, A.R.; Hassan, G.S.; Alkahtani, H.M.; Almehizia, A.A.; Alharbi, A.; Al-Sanea, M.M.; Eldehna, W.M. Antitumor properties of certain spirooxindoles towards hepatocellular carcinoma endowed with antioxidant activity. J. Enzym. Inhib. Med. Chem. 2020, 35, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Analysis of Cell Viability by the MTT Assay. Cold Spring Harb. Protoc. 2018, 2018, pdb-prot095505. [Google Scholar] [CrossRef]
- Fournel, M.; Bonfils, C.; Hou, Y.; Yan, P.T.; Trachy-Bourget, M.-C.; Kalita, A.; Liu, J.; Lu, A.-H.; Zhou, N.Z.; Robert, M.-F. MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol. Cancer Ther. 2008, 7, 759–768. [Google Scholar] [CrossRef] [PubMed]
- Heltweg, B.; Trapp, J.; Jung, M. In vitro assays for the determination of histone deacetylase activity. Methods 2005, 36, 332–337. [Google Scholar] [CrossRef]
- Eldehna, W.M.; Hassan, G.S.; Al-Rashood, S.T.; Al-Warhi, T.; Altyar, A.E.; Alkahtani, H.M.; Almehizia, A.A.; Abdel-Aziz, H.A. Synthesis and in vitro anticancer activity of certain novel 1-(2-methyl-6-arylpyridin-3-yl)-3-phenylureas as apoptosis-inducing agents. J. Enzym. Inhib. Med. Chem. 2019, 34, 322–332. [Google Scholar] [CrossRef]
- Yousef, R.G.; Sakr, H.M.; Eissa, I.H.; Mehany, A.B.; Metwaly, A.M.; Elhendawy, M.A.; Radwan, M.M.; ElSohly, M.A.; Abulkhair, H.S.; El-Adl, K. New quinoxaline-2 (1 H)-ones as potential VEGFR-2 inhibitors: Design, synthesis, molecular docking, ADMET profile and anti-proliferative evaluations. New J. Chem. 2021, 45, 16949–16964. [Google Scholar] [CrossRef]
- Pozarowski, P.; Darzynkiewicz, Z. Analysis of Cell Cycle by Flow Cytometry. In Checkpoint Controls and Cancer: Volume 2: Activation and Regulation Protocols; Springer: Berlin, Germany, 2004; pp. 301–311. [Google Scholar]
- Lo, K.K.-W.; Lee, T.K.-M.; Lau, J.S.-Y.; Poon, W.-L.; Cheng, S.-H. Luminescent Biological Probes Derived from Ruthenium(II) Estradiol Polypyridine Complexes. Inorg. Chem. 2007, 47, 200–208. [Google Scholar] [CrossRef]
- Elkady, H.; Elwan, A.; El-Mahdy, H.A.; Doghish, A.S.; Ismail, A.; Taghour, M.S.; Elkaeed, E.B.; Eissa, I.H.; Dahab, M.A.; Mahdy, H.A.; et al. New benzoxazole derivatives as potential VEGFR-2 inhibitors and apoptosis inducers: Design, synthesis, anti-proliferative evaluation, flowcytometric analysis, and in silico studies. J. Enzym. Inhib. Med. Chem. 2022, 37, 403–416. [Google Scholar] [CrossRef]
- Eray, M.; Mättö, M.; Kaartinen, M.; Andersson, L.C.; Pelkonen, J. Flow cytometric analysis of apoptotic subpopulations with a combination of Annexin V-FITC, propidium iodide, and SYTO 17. Cytom. J. Int. Soc. Anal. Cytol. 2001, 43, 134–142. [Google Scholar] [CrossRef]
- Balah, A.; Ezzat, O.; Akool, E.-S. Vitamin E inhibits cyclosporin A-induced CTGF and TIMP-1 expression by repressing ROS-mediated activation of TGF-β/Smad signaling pathway in rat liver. Int. Immunopharmacol. 2018, 65, 493–502. [Google Scholar] [CrossRef]
- Aborehab, N.M.; Elnagar, M.R.; Waly, N.E. Gallic acid potentiates the apoptotic effect of paclitaxel and carboplatin via overexpression of Bax and P53 on the MCF-7 human breast cancer cell line. J. Biochem. Mol. Toxicol. 2020, 35, e22638. [Google Scholar] [CrossRef] [PubMed]
- Elnagar, M.R.; Walls, A.B.; Helal, G.K.; Hamada, F.M.; Thomsen, M.S.; Jensen, A.A. Functional characterization of α7 nicotinic acetylcholine and NMDA receptor signaling in SH-SY5Y neuroblastoma cells in an ERK phosphorylation assay. Eur. J. Pharmacol. 2018, 826, 106–113. [Google Scholar] [CrossRef]
- Guo, Y.; Tong, Y.; Zhu, H.; Xiao, Y.; Guo, H.; Shang, L.; Zheng, W.; Ma, S.; Liu, X.; Bai, Y. Quercetin suppresses pancreatic ductal adenocarcinoma progression via inhibition of SHH and TGF-β/Smad signaling pathways. Cell Biol. Toxicol. 2021, 37, 479–496. [Google Scholar] [CrossRef]
- Jiao, C.; Chen, W.; Tan, X.; Liang, H.; Li, J.; Yun, H.; He, C.; Chen, J.; Ma, X.; Xie, Y.; et al. Ganoderma lucidum spore oil induces apoptosis of breast cancer cells in vitro and in vivo by activating caspase-3 and caspase-9. J. Ethnopharmacol. 2020, 247, 112256. [Google Scholar] [CrossRef]
- Ma, C.; Taghour, M.S.; Belal, A.; Mehany, A.B.M.; Mostafa, N.; Nabeeh, A.; Eissa, I.H.; Al-Karmalawy, A.A. Design and Synthesis of New Quinoxaline Derivatives as Potential Histone Deacetylase Inhibitors Targeting Hepatocellular Carcinoma: In Silico, In Vitro, and SAR Studies. Front. Chem. 2021, 9, 725135. [Google Scholar] [CrossRef]
- Elkady, M.A.; Doghish, A.S.; Elshafei, A.; Elshafey, M.M. MicroRNA-567 inhibits cell proliferation and induces cell apoptosis in A549 NSCLC cells by regulating cyclin-dependent kinase 8. Saudi J. Biol. Sci. 2021, 28, 2581–2590. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | Cytotoxicity IC50 (µM) a | |||
|---|---|---|---|---|
| MCF-7 | HepG-2 | HCT-116 | WI-38 | |
| 5a | 11 ± 1.6 | 39 ± 2.5 | 88 ± 2.4 | - |
| 5b | 55 ± 2.8 | 51 ± 4.0 | 21 ± 2.4 | - |
| 5c | 77 ± 2.3 | 89 ± 2.4 | 97 ± 1.2 | - |
| 5d | 62 ± 2.1 | 40 ± 2.7 | 281 ± 2.1 | - |
| 5e | 60 ± 0.49 | 42 ± 2.0 | 90 ± 3.0 | - |
| 5f | 9.3 ± 3.4 | 49 ± 0.17 | 71 ± 2.1 | - |
| 5g | 21 ± 2.2 | 33 ± 3.8 | 68 ± 1.8 | - |
| 5h | 88 ± 2.5 | 52 ± 3.2 | 91 ± 1.9 | - |
| 5i | 261 ± 2.5 | 4 ± 1.0 | 64 ± 2.1 | - |
| 5j | 71 ± 2.0 | 38 ± 1.8 | 400 ± 2.4 | - |
| 5k | 242 ± 2.0 | 5 ± 2.0 | 80 ± 2.6 | - |
| 5l | 381 ± 1.5 | 31 ± 2.0 | 83 ± 1.9 | - |
| 5m | 52 ± 3.5 | 3.3 ± 0.56 | 78 ± 2.0 | 65.67 ± 1.7 |
| 6a | 333 ± 2.9 | 220 ± 3.0 | 291 ± 3.5 | - |
| 6b | 577 ± 20 | 184 ± 3.7 | 299 ± 2.0 | - |
| 6c | 435 ± 4.3 | 114 ± 2.2 | 466 ± 4.6 | - |
| 6d | 413 ± 1.3 | 168 ± 1.5 | 361 ± 2.0 | - |
| 7a | 472 ± 3.2 | 189 ± 4.8 | 361 ± 1.9 | - |
| 7b | 871 ± 3.2 | 173 ± 4.5 | 301 ± 3.4 | - |
| 7c | 450 ± 2.2 | 221 ± 2.5 | 243 ± 2.2 | - |
| Sorafenib | 141 ± 3.0 | 17 ± 2.3.0 | 177 ± 0.9 | - |
| Staurosporine | - | - | - | 51.48 ± 2.2 |
| Comp. | HDAC1 IC50 (µg/mL) | HDAC4 IC50 (µg/mL) |
|---|---|---|
| 5a | 1.34 | 5.27 |
| 5b | 1.01 | 7.99 |
| 5f | 1.90 | 5.82 |
| 5i | 0.15 | 8.311 |
| 5k | 0.23 | 6.45 |
| 5m | 0.05 | 2.83 |
| Trichostatin A | 0.03 | 3.35 |
| Sample | %G0–G1 | %S | %G2/M |
|---|---|---|---|
| 5m/HCT116 | 55.31 | 34.88 | 9.81 |
| Cont. HCT116 | 43.82 | 41.16 | 15.02 |
| Sample | DNA Content (%) a | Necrosis | ||
|---|---|---|---|---|
| Total | Early | Late | ||
| Treated HCT116 with 5m | 37.59 | 22.36 | 13.14 | 2.09 |
| Untreated HCT116 | 2.17 | 0.43 | 0.18 | 1.56 |
| Sample ID and Treatment | RT-PCR (Fold Change) | |
|---|---|---|
| Caspase-3 | Caspase-8 | |
| Treated HCT116 with 5m | 4.891 | 2.655 |
| Treated HCT116 with Staurosporine | 6.8073 | 3.4529 |
| Untreated HCT116 | 1 | 1 |
| Comp. | Binding Energy | Comp. | Binding Energy |
|---|---|---|---|
| 5a | −17.21 | 5l | −15.43 |
| 5b | −14.13 | 5m | −14.99 |
| 5c | −16.16 | 6a | −12.57 |
| 5d | −14.72 | 6b | −13.81 |
| 5e | −16.76 | 6c | −15.75 |
| 5f | −17.16 | 6d | −17.51 |
| 5g | −17.48 | 7a | −12.99 |
| 5h | −17.49 | 7b | −15.06 |
| 5i | −17.35 | 7c | −14.13 |
| 5j | −20.48 | Trichostatin A | −19.11 |
| 5k | −15.36 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elbatrawy, O.R.; Hagras, M.; El Deeb, M.A.; Agili, F.; Hegazy, M.; El-Husseiny, A.A.; Mokhtar, M.M.; Elkhawaga, S.Y.; Eissa, I.H.; El-Kalyoubi, S. Discovery of New Uracil and Thiouracil Derivatives as Potential HDAC Inhibitors. Pharmaceuticals 2023, 16, 966. https://doi.org/10.3390/ph16070966
Elbatrawy OR, Hagras M, El Deeb MA, Agili F, Hegazy M, El-Husseiny AA, Mokhtar MM, Elkhawaga SY, Eissa IH, El-Kalyoubi S. Discovery of New Uracil and Thiouracil Derivatives as Potential HDAC Inhibitors. Pharmaceuticals. 2023; 16(7):966. https://doi.org/10.3390/ph16070966
Chicago/Turabian StyleElbatrawy, Omnia R., Mohamed Hagras, Moshira A. El Deeb, Fatimah Agili, Maghawry Hegazy, Ahmed A. El-Husseiny, Mahmoud Mohamed Mokhtar, Samy Y. Elkhawaga, Ibrahim H. Eissa, and Samar El-Kalyoubi. 2023. "Discovery of New Uracil and Thiouracil Derivatives as Potential HDAC Inhibitors" Pharmaceuticals 16, no. 7: 966. https://doi.org/10.3390/ph16070966
APA StyleElbatrawy, O. R., Hagras, M., El Deeb, M. A., Agili, F., Hegazy, M., El-Husseiny, A. A., Mokhtar, M. M., Elkhawaga, S. Y., Eissa, I. H., & El-Kalyoubi, S. (2023). Discovery of New Uracil and Thiouracil Derivatives as Potential HDAC Inhibitors. Pharmaceuticals, 16(7), 966. https://doi.org/10.3390/ph16070966