
A VEGFB-Based Peptidomimetic Inhibits VEGFR2-Mediated PI3K/Akt/mTOR and PLCγ/ERK Signaling and Elicits Apoptotic, Antiangiogenic, and Antitumor Activities

 , ,
, , 
 , ,
, ,  and
and
Abstract
1. Introduction
2. Results
2.1. Peptidomimetic Design
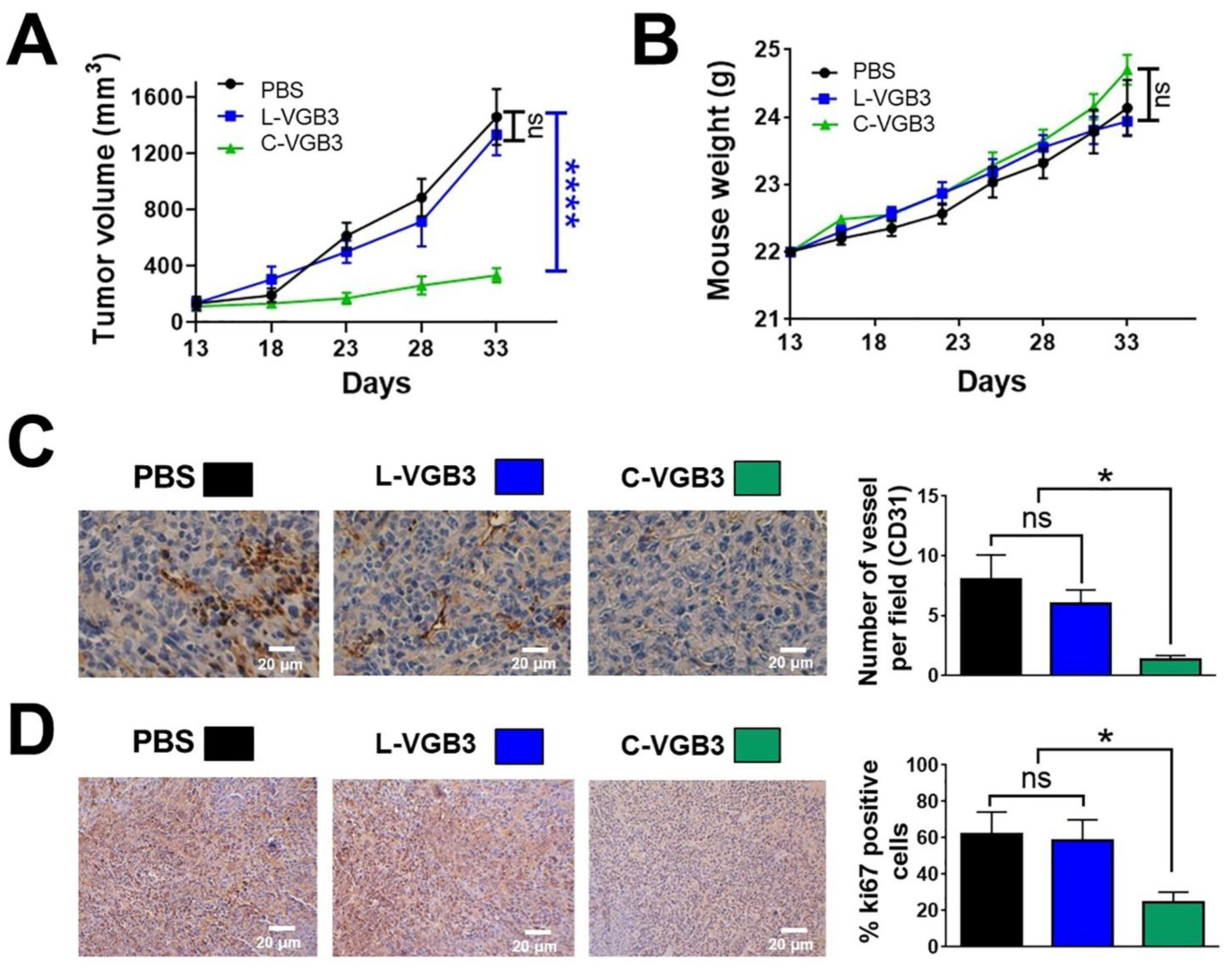
2.2. Loop Formation Is Essential for the Antitumor Activity of VGB3
2.3. Cyclic but Not Linear VGB3 Binds VEGFR2
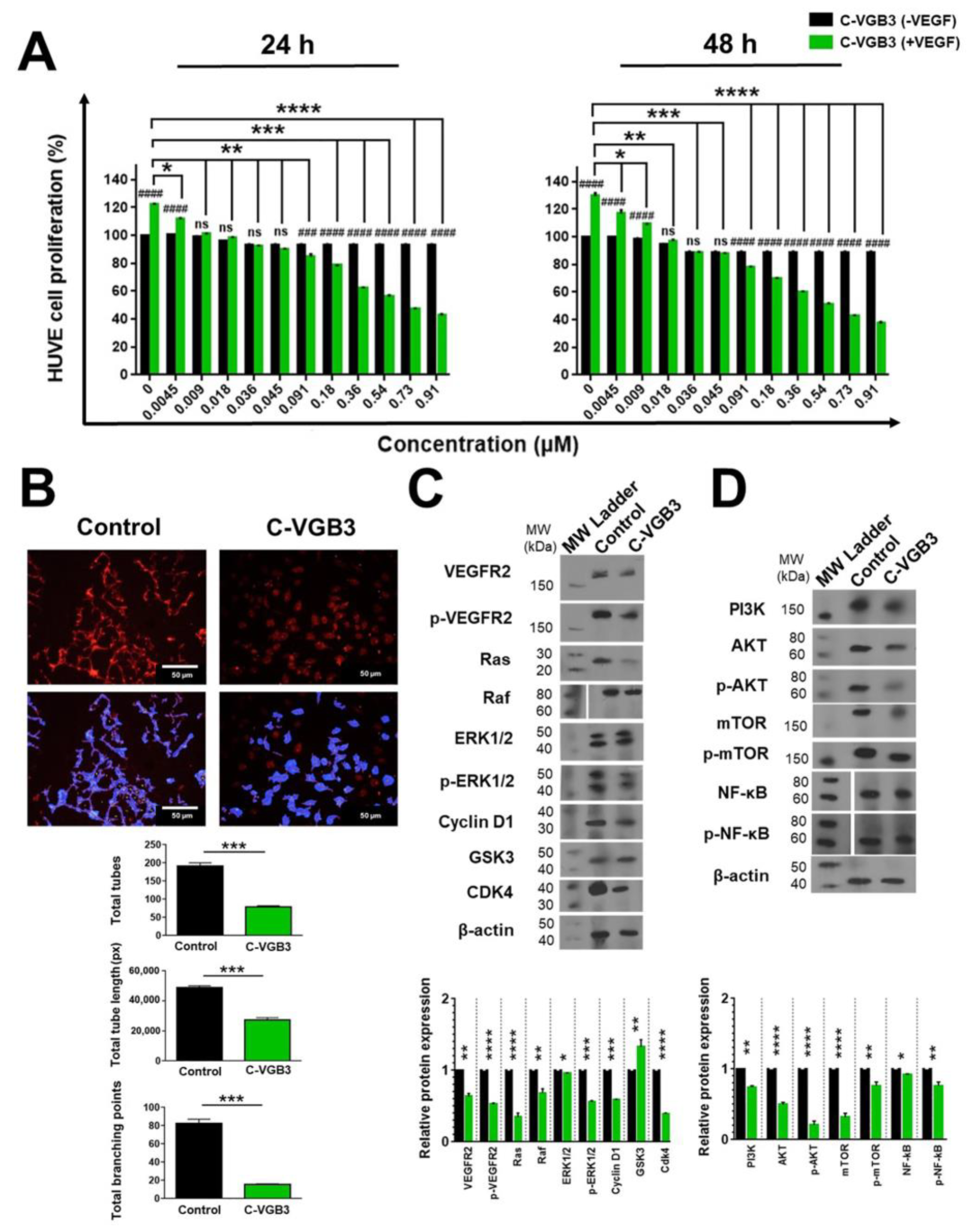
2.4. C-VGB3 Inhibited EC Proliferation and Tube Formation through Abrogation of VEGFR2-Mediated Signaling Pathways
2.5. C-VGB3 Inhibited Proliferation and VEGFR2 Signaling Pathways in 4T1 MCT Cells
2.6. C-VGB3 Induces Endothelial and Tumor Cell Apoptosis
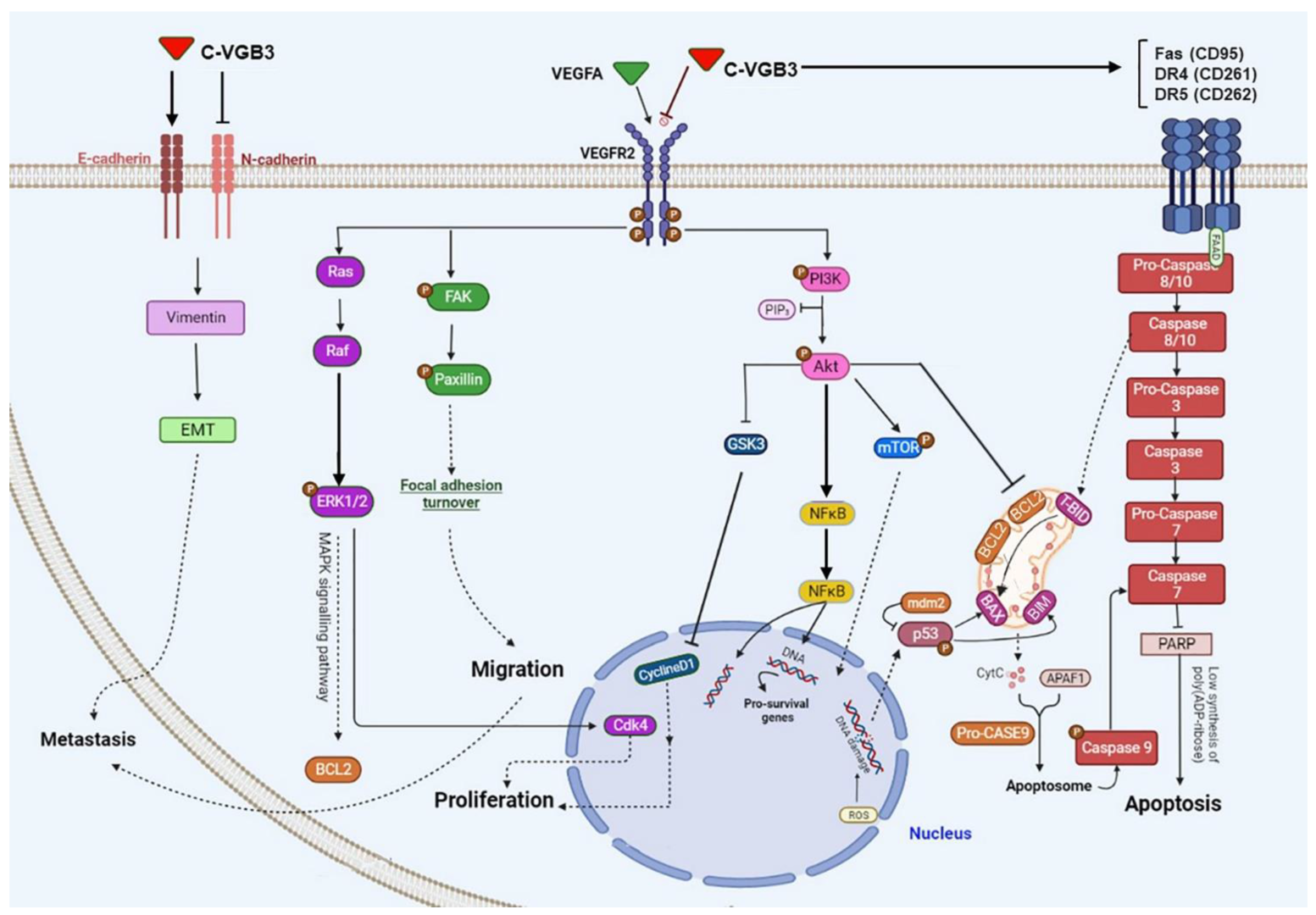
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Evaluation of Antitumor Activity
4.3. Immunohistochemistry Analysis
4.4. VEGFR2 Binding Assay
4.5. Molecular Docking
4.6. MTT Assay
4.7. Tubulogenesis Assay
4.8. Annexin V/PI Flow Cytometry
4.9. Western Blot Analysis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef]
- Takahashi, H.; Shibuya, M. The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clin. Sci. 2005, 109, 227–241. [Google Scholar] [CrossRef]
- Goel, H.L.; Mercurio, A.M. VEGF targets the tumour cell. Nat. Rev. Cancer 2013, 13, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef]
- Sadremomtaz, A.; Mansouri, K.; Alemzadeh, G.; Safa, M.; Esmaeili Rastaghi, A.R.; Asghari, S.M. Dual blockade of VEGFR1 and VEGFR2 by a novel peptide abrogates VEGF-driven angiogenesis, tumor growth, and metastasis through PI3K/AKT and MAPK/ERK1/2 pathway. Biochim. Biophys. Acta Gen Subj. 2018, 1862, 2688–2700. [Google Scholar] [CrossRef] [PubMed]
- Farzaneh Behelgardi, M.; Zahri, S.; Mashayekhi, F.; Mansouri, K.; Asghari, S.M. A peptide mimicking the binding sites of VEGF-A and VEGF-B inhibits VEGFR-1/-2 driven angiogenesis, tumor growth and metastasis. Sci. Rep. 2018, 8, 17924. [Google Scholar] [CrossRef] [PubMed]
- Assareh, E.; Mehrnejad, F.; Mansouri, K.; Esmaeili Rastaghi, A.R.; Naderi-Manesh, H.; Asghari, S.M. A cyclic peptide reproducing the α1 helix of VEGF-B binds to VEGFR-1 and VEGFR-2 and inhibits angiogenesis and tumor growth. Biochem. J. 2019, 476, 645–663. [Google Scholar] [CrossRef]
- Farzaneh Behelgardi, M.; Zahri, S.; Gholami Shahvir, Z.; Mashayekhi, F.; Mirzanejad, L.; Asghari, S.M. Targeting signaling pathways of VEGFR1 and VEGFR2 as a potential target in the treatment of breast cancer. Mol. Biol. Rep. 2020, 47, 2061–2071. [Google Scholar] [CrossRef] [PubMed]
- Sadremomtaz, A.; Ali, A.M.; Jouyandeh, F.; Balalaie, S.; Navari, R.; Broussy, S.; Mansouri, K.; Groves, M.R.; Asghari, S.M. Molecular docking, synthesis and biological evaluation of Vascular Endothelial Growth Factor (VEGF) B based peptide as antiangiogenic agent targeting the second domain of the Vascular Endothelial Growth Factor Receptor 1 (VEGFR1D2) for anticancer application. Signal Transduct. Target. Ther. 2020, 5, 76. [Google Scholar] [CrossRef] [PubMed]
- Muller, Y.A.; Li, B.; Christinger, H.W.; Wells, J.A.; Cunningham, B.C.; De Vos, A.M. Vascular endothelial growth factor: Crystal structure and functional mapping of the kinase domain receptor binding site. Proc. Natl. Acad. Sci. USA 1997, 94, 7192–7197. [Google Scholar] [CrossRef] [PubMed]
- Sadremomtaz, A.; Kobarfard, F.; Mansouri, K.; Mirzanejad, L.; Asghari, S.M. Suppression of migratory and metastatic pathways via blocking VEGFR1 and VEGFR2. J. Recept. Signal Transduct. 2018, 38, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bove, A.M.; Simone, G.; Ma, B. Molecular bases of VEGFR-2-mediated physiological function and pathological role. Front. Cell Dev. Biol. 2020, 8, 599281. [Google Scholar] [CrossRef] [PubMed]
- Guangqi, E.; Cao, Y.; Bhattacharya, S.; Dutta, S.; Wang, E.; Mukhopadhyay, D. Endogenous vascular endothelial growth factor-A (VEGF-A) maintains endothelial cell homeostasis by regulating VEGF receptor-2 transcription. J. Biol. Chem. 2012, 287, 3029–3041. [Google Scholar] [CrossRef]
- Yuan, J.; Dong, X.; Yap, J.; Hu, J. The MAPK and AMPK signalings: Interplay and implication in targeted cancer therapy. J. Hematol. Oncol. 2020, 13, 113. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M. VEGFR and type-V RTK activation and signaling. Cold Spring Harb. Perspect. Biol. 2013, 5, a009092. [Google Scholar] [CrossRef] [PubMed]
- Herber, B.; Truss, M.; Beato, M.; Müller, R. Inducible regulatory elements in the human cyclin D1 promoter. Oncogene 1994, 9, 1295–1304. [Google Scholar] [PubMed]
- Karar, J.; Maity, A. PI3K/AKT/mTOR pathway in angiogenesis. Front. Mol. Neurosci. 2011, 4, 51. [Google Scholar] [CrossRef]
- Tabruyn, S.P.; Griffioen, A.W. NF-κB: A new player in angiostatic therapy. Angiogenesis 2008, 11, 101–106. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.-g. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef]
- Walther, W.; Kobelt, D.; Bauer, L.; Aumann, J.; Stein, U. Chemosensitization by diverging modulation by short-term and long-term TNF-α action on ABCB1 expression and NF-κB signaling in colon cancer. Int. J. Oncol. 2015, 47, 2276–2285. [Google Scholar] [CrossRef]
- Lian, L.; Li, X.-L.; Xu, M.-D.; Li, X.-M.; Wu, M.-Y.; Zhang, Y.; Tao, M.; Li, W.; Shen, X.-M.; Zhou, C. VEGFR2 promotes tumorigenesis and metastasis in a pro-angiogenic-independent way in gastric cancer. BMC Cancer 2019, 19, 183. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zhou, W. The emerging regulation of VEGFR-2 in triple-negative breast cancer. Front. Endocrinol. 2015, 6, 159. [Google Scholar] [CrossRef]
- Xia, Y.; Shen, S.; Verma, I.M. NF-κB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Mittal, V. Epithelial mesenchymal transition in tumor metastasis. Annu. Rev. Pathol. 2018, 13, 395–412. [Google Scholar] [CrossRef]
- Zanjanchi, P.; Asghari, S.M.; Mohabatkar, H.; Shourian, M.; Shafiee Ardestani, M. Conjugation of VEGFR1/R2-targeting peptide with gold nanoparticles to enhance antiangiogenic and antitumoral activity. J. Nanobiotechnology 2022, 20, 7. [Google Scholar] [CrossRef] [PubMed]
- Abraham, A.G.; O’Neill, E. PI3K/Akt-mediated regulation of p53 in cancer. Biochem. Soc. Trans. 2014, 42, 798–803. [Google Scholar] [CrossRef]
- Hemann, M.; Lowe, S. The p53-BCL-2 connection. Cell Death Differ. 2006, 13, 1256. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef] [PubMed]
- Billen, L.; Shamas-Din, A.; Andrews, D. Bid: A Bax-like BH3 protein. Oncogene 2008, 27, S93–S104. [Google Scholar] [CrossRef]
- Zhu, S.; Evans, S.; Yan, B.; Povsic, T.J.; Tapson, V.; Goldschmidt-Clermont, P.J.; Dong, C. Transcriptional Regulation of Bim by FOXO3a and Akt Mediates Scleroderma Serum–Induced Apoptosis in Endothelial Progenitor Cells. Circulation 2008, 118, 2156–2165. [Google Scholar] [CrossRef] [PubMed]
- Takahashi; Yamaguchi, S.; Chida, K.; Shibuya, M. A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-γ and DNA synthesis in vascular endothelial cells. EMBO J. 2001, 20, 2768–2778. [Google Scholar] [CrossRef]
- Iyer, S.; Darley, P.I.; Acharya, K.R. Structural insights into the binding of vascular endothelial growth factor-B by VEGFR-1D2: Recognition and specificity. J. Biol. Chem. 2010, 285, 23779–23789. [Google Scholar] [CrossRef]
- Allahmoradi, H.; Asghari, S.M.; Ahmadi, A.; Assareh, E.; Nazari, M. Anti-tumor and anti-metastatic activity of the FGF2 118-126 fragment dependent on the loop structure. Biochem. J. 2022, 479, 1285–1302. [Google Scholar] [CrossRef] [PubMed]
- Chamani, R.; Asghari, S.M.; Alizadeh, A.M.; Eskandari, S.; Mansouri, K.; Khodarahmi, R.; Taghdir, M.; Heidari, Z.; Gorji, A.; Aliakbar, A. Engineering of a disulfide loop instead of a Zn binding loop restores the anti-proliferative, anti-angiogenic and anti-tumor activities of the N-terminal fragment of endostatin: Mechanistic and therapeutic insights. Vascul. Pharmacol. 2015, 72, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Tjin Tham Sjin, R.M.; Satchi-Fainaro, R.; Birsner, A.E.; Ramanujam, V.S.; Folkman, J.; Javaherian, K. A 27-amino-acid synthetic peptide corresponding to the NH2-terminal zinc-binding domain of endostatin is responsible for its antitumor activity. Cancer Res. 2005, 65, 3656–3663. [Google Scholar] [CrossRef] [PubMed]
- Baek, Y.-Y.; Lee, D.-K.; Kim, J.; Kim, J.-H.; Park, W.; Kim, T.; Han, S.; Jeoung, D.; You, J.C.; Lee, H. Arg-Leu-Tyr-Glu tetrapeptide inhibits tumor progression by suppressing angiogenesis and vascular permeability via VEGF receptor-2 antagonism. Oncotarget 2016, 8, 11763–11777. [Google Scholar] [CrossRef]
- D’Andrea, L.D.; Iaccarino, G.; Fattorusso, R.; Sorriento, D.; Carannante, C.; Capasso, D.; Trimarco, B.; Pedone, C. Targeting angiogenesis: Structural characterization and biological properties of a de novo engineered VEGF mimicking peptide. Proc. Natl. Acad. Sci. USA 2005, 102, 14215–14220. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Jezequel, S.; Löhr, M.; Shaikh, S.; Davis, D.; Soker, S.; Selwood, D.; Zachary, I. Peptides encoded by exon 6 of VEGF inhibit endothelial cell biological responses and angiogenesis induced by VEGF. Biochem. Biophys. Res. Commun. 2001, 283, 164–173. [Google Scholar] [CrossRef]
- Michaloski, J.S.; Redondo, A.R.; Magalhães, L.S.; Cambui, C.C.; Giordano, R.J. Discovery of pan-VEGF inhibitory peptides directed to the extracellular ligand-binding domains of the VEGF receptors. Sci. Adv. 2016, 2, e1600611. [Google Scholar] [CrossRef] [PubMed]
- Zilberberg, L.; Shinkaruk, S.; Lequin, O.; Rousseau, B.; Hagedorn, M.; Costa, F.; Caronzolo, D.; Balke, M.; Canron, X.; Convert, O. Structure and inhibitory effects on angiogenesis and tumor development of a new vascular endothelial growth inhibitor. J. Biol. Chem. 2003, 278, 35564–35573. [Google Scholar] [CrossRef]
- Farzaneh Behelgardi, M.; Gholami Shahvir, Z.; Asghari, S.M. Apoptosis induction in human lung and colon cancer cells via impeding VEGF signaling pathways. Mol. Biol. Rep. 2022, 49, 3637–3647. [Google Scholar] [CrossRef] [PubMed]
- Cudmore, M.J.; Hewett, P.W.; Ahmad, S.; Wang, K.-Q.; Cai, M.; Al-Ani, B.; Fujisawa, T.; Ma, B.; Sissaoui, S.; Ramma, W. The role of heterodimerization between VEGFR-1 and VEGFR-2 in the regulation of endothelial cell homeostasis. Nat. Commun. 2012, 3, 972. [Google Scholar] [CrossRef]
- Yu, D.-C.; Lee, J.-S.; Yoo, J.Y.; Shin, H.; Deng, H.; Wei, Y.; Yun, C.-O. Soluble vascular endothelial growth factor decoy receptor FP3 exerts potent antiangiogenic effects. Mol. Ther. 2012, 20, 938–947. [Google Scholar] [CrossRef]
- Koch, S.; Tugues, S.; Li, X.; Gualandi, L.; Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Biochem. J. 2011, 437, 169–183. [Google Scholar] [CrossRef]
- Lee, J.; Ku, T.; Yu, H.; Chong, K.; Ryu, S.-W.; Choi, K.; Choi, C. Blockade of VEGF-A suppresses tumor growth via inhibition of autocrine signaling through FAK and AKT. Cancer Lett. 2012, 318, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Mak, P.; Leav, I.; Pursell, B.; Bae, D.; Yang, X.; Taglienti, C.A.; Gouvin, L.M.; Sharma, V.M.; Mercurio, A.M. ERβ impedes prostate cancer EMT by destabilizing HIF-1α and inhibiting VEGF-mediated snail nuclear localization: Implications for Gleason grading. Cancer Cell 2010, 17, 319–332. [Google Scholar] [CrossRef]
- Wanami, L.S.; Chen, H.-Y.; Peiró, S.; de Herreros, A.G.; Bachelder, R.E. Vascular endothelial growth factor-A stimulates Snail expression in breast tumor cells: Implications for tumor progression. Exp. Cell Res. 2008, 314, 2448–2453. [Google Scholar] [CrossRef]
- Chavakis, E.; Dimmeler, S. Regulation of endothelial cell survival and apoptosis during angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.; Yu, Z.; Qiu, M.; Dai, Y.; Dong, Q.; Shen, J.; Dong, P.; Wang, X.; Liu, Y.; Quan, Z. Knockdown of VEGFR2 inhibits proliferation and induces apoptosis in hemangioma-derived endothelial cells. Eur. J. Histochem. 2014, 58, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, N.; Teshima-Kondo, S.; Masuda, K.; Nishida, K.; Kuwano, Y.; Dang, D.T.; Dang, L.H.; Nikawa, T.; Rokutan, K. Chronic inhibition of tumor cell-derived VEGF enhances the malignant phenotype of colorectal cancer cells. BMC Cancer 2013, 13, 229. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhou, L.; Reille-Seroussi, M.; Gagey-Eilstein, N.; Broussy, S.; Zhang, T.; Ji, L.; Vidal, M.; Liu, W.-Q. Identification of peptidic antagonists of vascular endothelial growth factor receptor 1 by scanning the binding epitopes of its ligands. J. Med. Chem. 2017, 60, 6598–6606. [Google Scholar] [CrossRef] [PubMed]
- Duhovny, D.; Nussinov, R.; Wolfson, H.J. Efficient unbound docking of rigid molecules. In International Workshop on Algorithms in Bioinformatics, Proceedings of the Second International Workshop, WABI 2002, Rome, Italy, 17–21 September 2002; Guigó, R., Gusfield, D., Eds.; Springer: Berlin/Heidelberg, Germany, 2002; pp. 185–200. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Van Zundert, G.; Rodrigues, J.; Trellet, M.; Schmitz, C.; Kastritis, P.; Karaca, E.; Melquiond, A.; van Dijk, M.; De Vries, S.; Bonvin, A. The HADDOCK2. 2 web server: User-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.; Boelens, R.; Bonvin, A.M. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Venetsanakos, E.; Mirza, A.; Fanton, C.; Romanov, S.R.; Tlsty, T.; McMahon, M. Induction of tubulogenesis in telomerase-immortalized human microvascular endothelial cells by glioblastoma cells. Exp. Cell Res. 2002, 273, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.; Eramian, D.; Shen, M.Y.; Pieper, U.; Sali, A. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinform. 2006, 15, 5.6.1–5.6.30. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Benkert, P.; Tosatto, S.C.; Schomburg, D. QMEAN: A comprehensive scoring function for model quality assessment. Proteins: Struct. Funct. Bioinform. 2008, 71, 261–277. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2021, 40, 843. [Google Scholar] [CrossRef]
- Tao, Y.; Rao, Z.-H.; Liu, S.-Q. Insight derived from molecular dynamics simulation into substrate-induced changes in protein motions of proteinase K. J. Biomol. Struct. Dyn. 2010, 28, 143–157. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PatchDock | |||||
|---|---|---|---|---|---|
| Complex | Score | Atomic Contact Energy (kcal·mol−1) | |||
| C-VGB3/VEGFR2 | 6572 | −209.19 | |||
| L-VGB3/VEGFR2 | 6280 | −40.61 | |||
| HADDOCK | |||||
| Complex | Total interaction (kcal·mol−1) | Van der Waals (kcal·mol−1) | Electrostatic (kcal·mol−1) | Desolvation (kcal·mol−1) | Buried surface area (°A2) |
| C-VGB3/VEGFR2 | −184 ± 26.9 | −40.5 ± 5.2 | −143.5 ± 21.7 | 0.9 ± 6.1 | 961.7 ± 33.0 |
| L-VGB3/VEGFR2 | −65.5 ± 15.8 | −32.3 ± 1.2 | −33.2 ± 14.6 | −9.4 ± 6.4 | 800.9 ± 67.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Namjoo, M.; Ghafouri, H.; Assareh, E.; Aref, A.R.; Mostafavi, E.; Hamrahi Mohsen, A.; Balalaie, S.; Broussy, S.; Asghari, S.M. A VEGFB-Based Peptidomimetic Inhibits VEGFR2-Mediated PI3K/Akt/mTOR and PLCγ/ERK Signaling and Elicits Apoptotic, Antiangiogenic, and Antitumor Activities. Pharmaceuticals 2023, 16, 906. https://doi.org/10.3390/ph16060906
Namjoo M, Ghafouri H, Assareh E, Aref AR, Mostafavi E, Hamrahi Mohsen A, Balalaie S, Broussy S, Asghari SM. A VEGFB-Based Peptidomimetic Inhibits VEGFR2-Mediated PI3K/Akt/mTOR and PLCγ/ERK Signaling and Elicits Apoptotic, Antiangiogenic, and Antitumor Activities. Pharmaceuticals. 2023; 16(6):906. https://doi.org/10.3390/ph16060906
Chicago/Turabian StyleNamjoo, Mohadeseh, Hossein Ghafouri, Elham Assareh, Amir Reza Aref, Ebrahim Mostafavi, Ali Hamrahi Mohsen, Saeed Balalaie, Sylvain Broussy, and S. Mohsen Asghari. 2023. "A VEGFB-Based Peptidomimetic Inhibits VEGFR2-Mediated PI3K/Akt/mTOR and PLCγ/ERK Signaling and Elicits Apoptotic, Antiangiogenic, and Antitumor Activities" Pharmaceuticals 16, no. 6: 906. https://doi.org/10.3390/ph16060906
APA StyleNamjoo, M., Ghafouri, H., Assareh, E., Aref, A. R., Mostafavi, E., Hamrahi Mohsen, A., Balalaie, S., Broussy, S., & Asghari, S. M. (2023). A VEGFB-Based Peptidomimetic Inhibits VEGFR2-Mediated PI3K/Akt/mTOR and PLCγ/ERK Signaling and Elicits Apoptotic, Antiangiogenic, and Antitumor Activities. Pharmaceuticals, 16(6), 906. https://doi.org/10.3390/ph16060906