
Targeting Metalloenzymes: The “Achilles’ Heel” of Viruses and Parasites





Abstract

1. Introduction
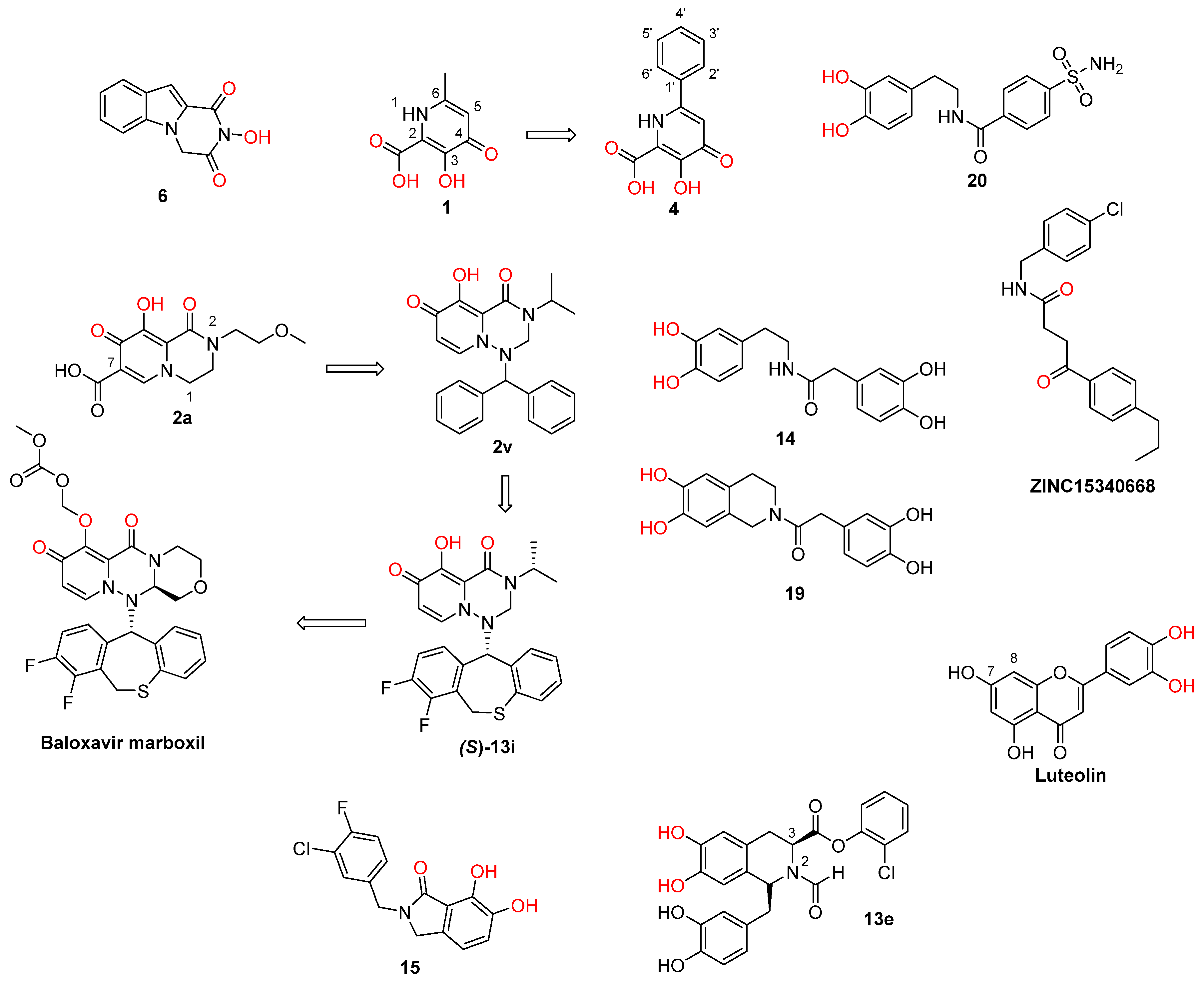
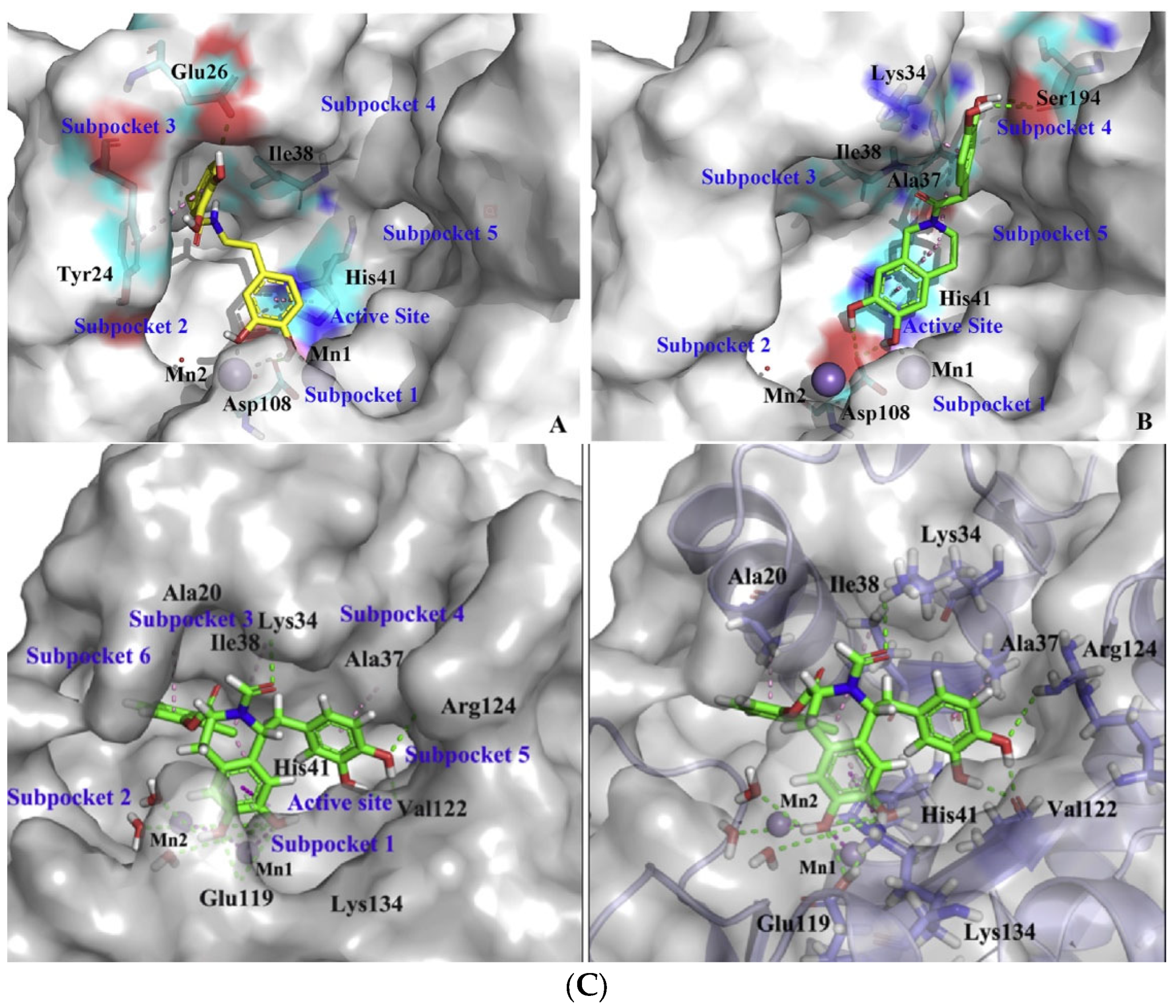
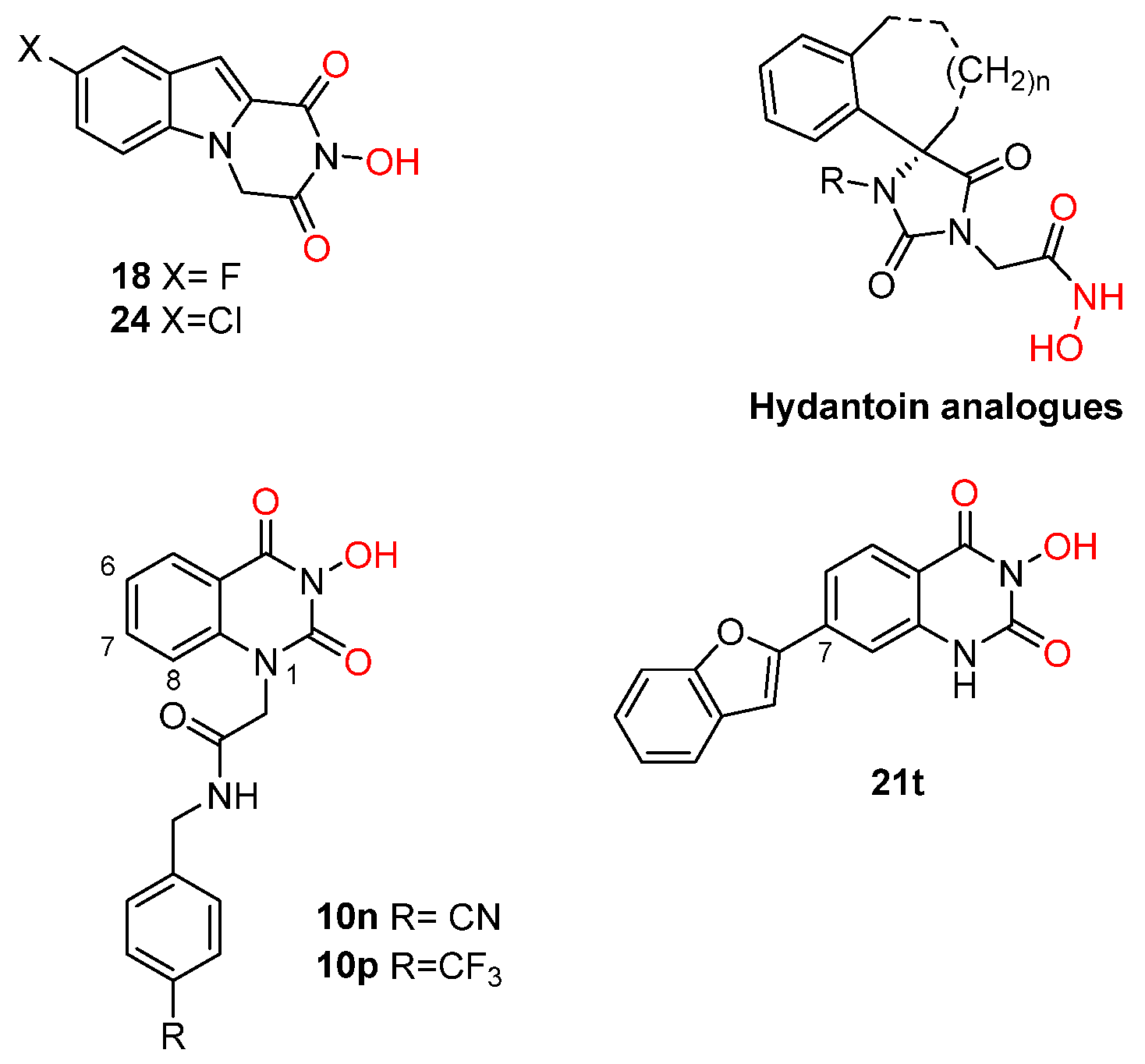
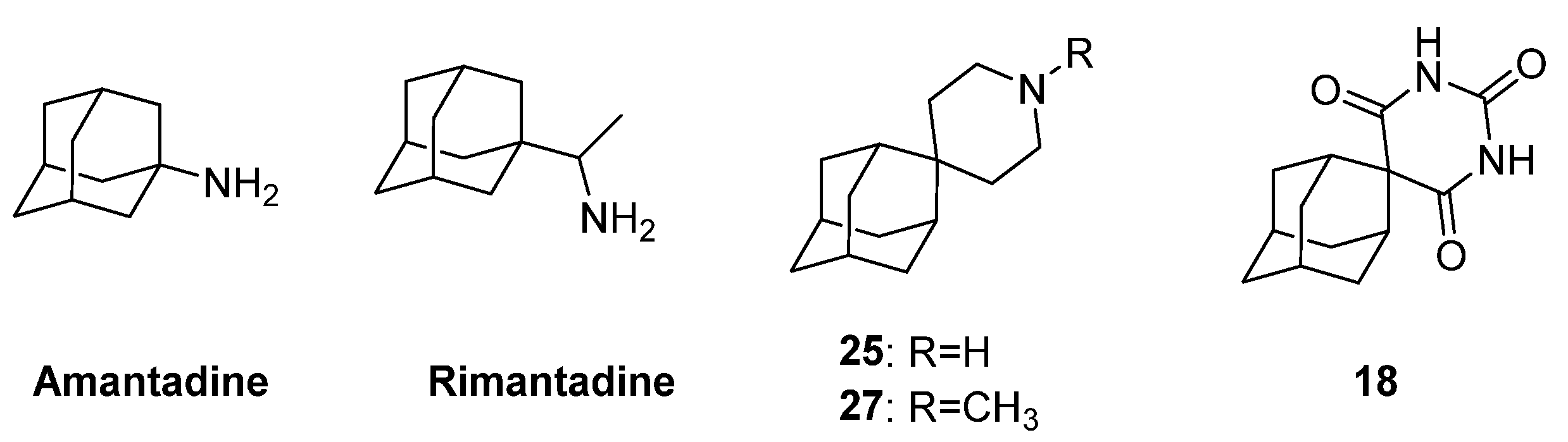
2. Influenza Virus RNA-Dependent RNA Polymerase PA N-Terminal Endonuclease Domain
3. Hepatitis B Virus—HBV Polymerase (Ribonuclease H)
4. Hepatitis C Virus Nonstructural Protein 5B (NS5B)
5. Trypanosoma brucei 6-Oxopurine Phosphoribosyltransferase (PRT)
6. Trypanosoma cruzi Carbonic Anhydrase (CA)
7. Human Immunodeficiency Virus (HIV) Integrase and Ribonuclease
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, A.Y.; Adamek, R.N.; Dick, B.L.; Credille, C.V.; Morrison, C.N.; Cohen, S.M. Targeting Metalloenzymes for Therapeutic Intervention. Chem. Rev. 2019, 119, 1323–1455. [Google Scholar] [CrossRef]
- Shi, W.; Chance, M.R. Metalloproteomics: Forward and Reverse Approaches in Metalloprotein Structural and Functional Characterization. Curr. Opin. Chem. Biol. 2011, 15, 144–148. [Google Scholar] [CrossRef]
- Putignano, V.; Rosato, A.; Banci, L.; Andreini, C. MetalPDB in 2018: A Database of Metal Sites in Biological Macromolecular Structures. Nucleic Acids Res. 2018, 46, D459–D464. [Google Scholar] [CrossRef]
- Day, J.A.; Cohen, S.M. Investigating the Selectivity of Metalloenzyme Inhibitors. J. Med. Chem. 2013, 56, 7997–8007. [Google Scholar] [CrossRef]
- Degtyarenko, K. Metalloproteins. In Encyclopedia of Genetics, Genomics, Proteomics and Bioinformatics; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2005; ISBN 978-0-470-01153-9. [Google Scholar]
- Maret, W. Metalloproteomics, Metalloproteomes, and the Annotation of Metalloproteins. Metallomics 2010, 2, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Rouffet, M.; Cohen, S.M. Emerging Trends in Metalloprotein Inhibition. Dalton Trans. 2011, 40, 3445–3454. [Google Scholar] [CrossRef]
- Javanian, M.; Barary, M.; Ghebrehewet, S.; Koppolu, V.; Vasigala, V.; Ebrahimpour, S. A Brief Review of Influenza Virus Infection. J. Med. Virol. 2021, 93, 4638–4646. [Google Scholar] [CrossRef] [PubMed]
- Clohisey, S.; Baillie, J.K. Host Susceptibility to Severe Influenza a Virus Infection. Crit. Care 2019, 23, 303. [Google Scholar] [CrossRef]
- Iuliano, A.D.; Roguski, K.M.; Chang, H.H.; Muscatello, D.J.; Palekar, R.; Tempia, S.; Cohen, C.; Gran, J.M.; Schanzer, D.; Cowling, B.J.; et al. Estimates of Global Seasonal Influenza-Associated Respiratory Mortality: A Modelling Study. Lancet 2018, 391, 1285–1300. [Google Scholar] [CrossRef]
- Troeger, C.E.; Blacker, B.F.; Khalil, I.A.; Zimsen, S.R.M.; Albertson, S.B.; Abate, D.; Abdela, J.; Adhikari, T.B.; Aghayan, S.A.; Agrawal, S.; et al. Mortality, Morbidity, and Hospitalisations Due to Influenza Lower Respiratory Tract Infections, 2017: An Analysis for the Global Burden of Disease Study 2017. Lancet Respir. Med. 2019, 7, 69–89. [Google Scholar] [CrossRef] [PubMed]
- Paget, J.; Spreeuwenberg, P.; Charu, V.; Taylor, R.J.; Iuliano, A.D.; Bresee, J.; Simonsen, L.; Viboud, C. Global Mortality Associated with Seasonal Influenza Epidemics: New Burden Estimates and Predictors from the GLaMOR Project. J. Glob. Health 2019, 9, 020421. [Google Scholar] [CrossRef]
- Lafond, K.E.; Porter, R.M.; Whaley, M.J.; Suizan, Z.; Ran, Z.; Aleem, M.A.; Thapa, B.; Sar, B.; Proschle, V.S.; Peng, Z.; et al. Global Burden of Influenza-Associated Lower Respiratory Tract Infections and Hospitalizations among Adults: A Systematic Review and Meta-Analysis. PLoS Med. 2021, 18, e1003550. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Webster, R.G.; Webby, R.J. Influenza Virus: Dealing with a Drifting and Shifting Pathogen. Viral Immunol. 2018, 31, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Paules, C.I.; Fauci, A.S. Influenza Vaccines: Good, but We Can Do Better. J. Infect. Dis. 2019, 219, S1–S4. [Google Scholar] [CrossRef] [PubMed]
- Tenforde, M.W.; Kondor, R.J.G.; Chung, J.R.; Zimmerman, R.K.; Nowalk, M.P.; Jackson, M.L.; Jackson, L.A.; Monto, A.S.; Martin, E.T.; Belongia, E.A.; et al. Effect of Antigenic Drift on Influenza Vaccine Effectiveness in the United States—2019–2020. Clin. Infect. Dis. 2021, 73, e4244–e4250. [Google Scholar] [CrossRef]
- Principi, N.; Camilloni, B.; Alunno, A.; Polinori, I.; Argentiero, A.; Esposito, S. Drugs for Influenza Treatment: Is There Significant News? Front. Med. 2019, 6, 109. [Google Scholar] [CrossRef]
- Gaitonde, D.Y. Influenza:Diagnosis and Treatment. Am. Fam. Physician 2019, 100, 8. [Google Scholar]
- O’Hanlon, R.; Shaw, M.L. Baloxavir Marboxil: The New Influenza Drug on the Market. Curr. Opin. Virol. 2019, 35, 14–18. [Google Scholar] [CrossRef]
- Jalily, P.H.; Duncan, M.C.; Fedida, D.; Wang, J.; Tietjen, I. Put a Cork in It: Plugging the M2 Viral Ion Channel to Sink Influenza. Antivir. Res. 2020, 178, 104780. [Google Scholar] [CrossRef]
- Walker, A.P.; Fodor, E. Interplay between Influenza Virus and the Host RNA Polymerase II Transcriptional Machinery. Trends Microbiol. 2019, 27, 398–407. [Google Scholar] [CrossRef]
- Sugiyama, K.; Obayashi, E.; Kawaguchi, A.; Suzuki, Y.; Tame, J.R.H.; Nagata, K.; Park, S.-Y. Structural Insight into the Essential PB1–PB2 Subunit Contact of the Influenza Virus RNA Polymerase. EMBO J. 2009, 28, 1803–1811. [Google Scholar] [CrossRef] [PubMed]
- Pflug, A.; Guilligay, D.; Reich, S.; Cusack, S. Structure of Influenza A Polymerase Bound to the Viral RNA Promoter. Nature 2014, 516, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Pflug, A.; Lukarska, M.; Resa-Infante, P.; Reich, S.; Cusack, S. Structural Insights into RNA Synthesis by the Influenza Virus Transcription-Replication Machine. Virus Res. 2017, 234, 103–117. [Google Scholar] [CrossRef]
- te Velthuis, A.J.W.; Fodor, E. Influenza Virus RNA Polymerase: Insights into the Mechanisms of Viral RNA Synthesis. Nat. Rev. Microbiol. 2016, 14, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Walker, A.P.; Carrique, L.; Keown, J.R.; Serna Martin, I.; Karia, D.; Sharps, J.; Hengrung, N.; Pardon, E.; Steyaert, J.; et al. Structures of Influenza A Virus RNA Polymerase Offer Insight into Viral Genome Replication. Nature 2019, 573, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lou, Z.; Bartlam, M.; Rao, Z. Structure-Function Studies of the Influenza Virus RNA Polymerase PA Subunit. Sci. China Ser. C 2009, 52, 450–458. [Google Scholar] [CrossRef]
- Nakazawa, M.; Kadowaki, S.; Watanabe, I.; Kadowaki, Y.; Takei, M.; Fukuda, H. PA Subunit of RNA Polymerase as a Promising Target for Anti-Influenza Virus Agents. Antivir. Res. 2008, 78, 194–201. [Google Scholar] [CrossRef]
- Jones, J.C.; Kumar, G.; Barman, S.; Najera, I.; White, S.W.; Webby, R.J.; Govorkova, E.A. Identification of the I38T PA Substitution as a Resistance Marker for Next-Generation Influenza Virus Endonuclease Inhibitors. mBio 2018, 9, e00430-18. [Google Scholar] [CrossRef]
- Zoidis, G.; Giannakopoulou, E.; Stevaert, A.; Frakolaki, E.; Myrianthopoulos, V.; Fytas, G.; Mavromara, P.; Mikros, E.; Bartenschlager, R.; Vassilaki, N.; et al. Novel Indole–Flutimide Heterocycles with Activity against Influenza PA Endonuclease and Hepatitis C Virus. Med. Chem. Commun. 2016, 7, 447–456. [Google Scholar] [CrossRef]
- Credille, C.V.; Dick, B.L.; Morrison, C.N.; Stokes, R.W.; Adamek, R.N.; Wu, N.C.; Wilson, I.A.; Cohen, S.M. Structure–Activity Relationships in Metal-Binding Pharmacophores for Influenza Endonuclease. J. Med. Chem. 2018, 61, 10206–10217. [Google Scholar] [CrossRef]
- Credille, C.V.; Morrison, C.N.; Stokes, R.W.; Dick, B.L.; Feng, Y.; Sun, J.; Chen, Y.; Cohen, S.M. SAR Exploration of Tight-Binding Inhibitors of Influenza Virus PA Endonuclease. J. Med. Chem. 2019, 62, 9438–9449. [Google Scholar] [CrossRef] [PubMed]
- Ferro, S.; Gitto, R.; Buemi, M.R.; Karamanou, S.; Stevaert, A.; Naesens, L.; De Luca, L. Identification of Influenza PA-Nter Endonuclease Inhibitors Using Pharmacophore- and Docking-Based Virtual Screening. Bioorg. Med. Chem. 2018, 26, 4544–4550. [Google Scholar] [CrossRef] [PubMed]
- Mohseni, S.S.; Nasri, F.; Davari, K.; Mirzaie, S.; Moradzadegan, A.; Abdi, F.; Farzaneh, F. Identification of Novel Inhibitor against Endonuclease Subunit of Influenza PH1N1 Polymerase: A Combined Molecular Docking, Molecular Dynamics, MMPBSA, QMMM and ADME Studies to Combat Influenza A Viruses. Comput. Biol. Chem. 2018, 77, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, M.; Akiyama, T.; Taoda, Y.; Takaya, K.; Takahashi-Kageyama, C.; Tomita, K.; Yasuo, K.; Hattori, K.; Shano, S.; Yoshida, R.; et al. Synthesis and SAR Study of Carbamoyl Pyridone Bicycle Derivatives as Potent Inhibitors of Influenza Cap-Dependent Endonuclease. J. Med. Chem. 2019, 62, 8101–8114. [Google Scholar] [CrossRef]
- Taoda, Y.; Miyagawa, M.; Akiyama, T.; Tomita, K.; Hasegawa, Y.; Yoshida, R.; Noshi, T.; Shishido, T.; Kawai, M. Dihydrodibenzothiepine: Promising Hydrophobic Pharmacophore in the Influenza Cap-Dependent Endonuclease Inhibitor. Bioorg. Med. Chem. Lett. 2020, 30, 127547. [Google Scholar] [CrossRef]
- Ivashchenko, A.A.; Mitkin, O.D.; Jones, J.C.; Nikitin, A.V.; Koryakova, A.G.; Karapetian, R.N.; Kravchenko, D.V.; Mochalov, S.V.; Ryakhovskiy, A.A.; Aladinskiy, V.; et al. Synthesis, Inhibitory Activity and Oral Dosing Formulation of AV5124, the Structural Analogue of Influenza Virus Endonuclease Inhibitor Baloxavir. J. Antimicrob. Chemother. 2021, 76, 1010–1018. [Google Scholar] [CrossRef]
- Liao, Y.; Ye, Y.; Li, S.; Zhuang, Y.; Chen, L.; Chen, J.; Cui, Z.; Huo, L.; Liu, S.; Song, G. Synthesis and SARs of Dopamine Derivatives as Potential Inhibitors of Influenza Virus PAN Endonuclease. Eur. J. Med. Chem. 2020, 189, 112048. [Google Scholar] [CrossRef]
- Zima, V.; Radilová, K.; Kožíšek, M.; Albiñana, C.B.; Karlukova, E.; Brynda, J.; Fanfrlík, J.; Flieger, M.; Hodek, J.; Weber, J.; et al. Unraveling the Anti-Influenza Effect of Flavonoids: Experimental Validation of Luteolin and Its Congeners as Potent Influenza Endonuclease Inhibitors. Eur. J. Med. Chem. 2020, 208, 112754. [Google Scholar] [CrossRef]
- Reiberger, R.; Radilová, K.; Kráľ, M.; Zima, V.; Majer, P.; Brynda, J.; Dračínský, M.; Konvalinka, J.; Kožíšek, M.; Machara, A. Synthesis and In Vitro Evaluation of C-7 and C-8 Luteolin Derivatives as Influenza Endonuclease Inhibitors. IJMS 2021, 22, 7735. [Google Scholar] [CrossRef]
- Rogolino, D.; Naesens, L.; Bartoli, J.; Carcelli, M.; De Luca, L.; Pelosi, G.; Stokes, R.W.; Van Berwaer, R.; Vittorio, S.; Stevaert, A.; et al. Exploration of the 2,3-Dihydroisoindole Pharmacophore for Inhibition of the Influenza Virus PA Endonuclease. Bioorg. Chem. 2021, 116, 105388. [Google Scholar] [CrossRef]
- Liu, Z.; Gu, S.; Zhu, X.; Liu, M.; Cao, Z.; Qiu, P.; Li, S.; Liu, S.; Song, G. Discovery and Optimization of New 6, 7-Dihydroxy-1, 2, 3, 4-Tetrahydroisoquinoline Derivatives as Potent Influenza Virus PAN Inhibitors. Eur. J. Med. Chem. 2022, 227, 113929. [Google Scholar] [CrossRef]
- Lin, C.-L.; Kao, J.-H. Natural History of Acute and Chronic Hepatitis B: The Role of HBV Genotypes and Mutants. Best Pract. Res. Clin. Gastroenterol. 2017, 31, 249–255. [Google Scholar] [CrossRef]
- Prifti, G.-M.; Moianos, D.; Giannakopoulou, E.; Pardali, V.; Tavis, J.; Zoidis, G. Recent Advances in Hepatitis B Treatment. Pharmaceuticals 2021, 14, 417. [Google Scholar] [CrossRef]
- Trépo, C.; Chan, H.L.Y.; Lok, A. Hepatitis B Virus Infection. Lancet 2014, 384, 2053–2063. [Google Scholar] [CrossRef]
- Yuen, M.-F.; Chen, D.-S.; Dusheiko, G.M.; Janssen, H.L.A.; Lau, D.T.Y.; Locarnini, S.A.; Peters, M.G.; Lai, C.-L. Hepatitis B Virus Infection. Nat. Rev. Dis. Primers 2018, 4, 18035. [Google Scholar] [CrossRef]
- Rehermann, B.; Nascimbeni, M. Immunology of Hepatitis B Virus and Hepatitis C Virus Infection. Nat. Rev. Immunol. 2005, 5, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.C.; Lomonosova, E.; Patel, J.A.; Li, Q.; Villa, J.A.; Gupta, A.K.; Morrison, L.A.; Bailly, F.; Cotelle, P.; Giannakopoulou, E.; et al. Inhibition of Hepatitis B Virus Replication by N -Hydroxyisoquinolinediones and Related Polyoxygenated Heterocycles. Antivir. Res. 2017, 143, 205–217. [Google Scholar] [CrossRef]
- Giannakopoulou, E.; Pardali, V.; Zoidis, G. Metal-Chelating Agents against Viruses and Parasites. Future Med. Chem. 2018, 10, 1283–1285. [Google Scholar] [CrossRef]
- Nowotny, M.; Yang, W. Stepwise Analyses of Metal Ions in RNase H Catalysis from Substrate Destabilization to Product Release. EMBO J. 2006, 25, 1924–1933. [Google Scholar] [CrossRef] [PubMed]
- Nowotny, M.; Gaidamakov, S.A.; Crouch, R.J.; Yang, W. Crystal Structures of RNase H Bound to an RNA/DNA Hybrid: Substrate Specificity and Metal-Dependent Catalysis. Cell 2005, 121, 1005–1016. [Google Scholar] [CrossRef] [PubMed]
- Hausen, P.; Stein, H. Ribonuclease H. An Enzyme Degrading the RNA Moiety of DNA-RNA Hybrids. Eur. J. Biochem. 1970, 14, 278–283. [Google Scholar] [CrossRef]
- Keller, W.; Crouch, R. Degradation of DNA RNA Hybrids by Ribonuclease H and DNA Polymerases of Cellular and Viral Origin. Proc. Natl. Acad. Sci. USA 1972, 69, 3360–3364. [Google Scholar] [CrossRef]
- Gerelsaikhan, T.; Tavis, J.E.; Bruss, V. Hepatitis B Virus Nucleocapsid Envelopment Does Not Occur without Genomic DNA Synthesis. J. Virol. 1996, 70, 4269–4274. [Google Scholar] [CrossRef]
- Tavis, J.E.; Zoidis, G.; Meyers, M.J.; Murelli, R.P. Chemical Approaches to Inhibiting the Hepatitis B Virus Ribonuclease H. ACS Infect. Dis. 2019, 5, 655–658. [Google Scholar] [CrossRef]
- Edwards, T.C.; Mani, N.; Dorsey, B.; Kakarla, R.; Rijnbrand, R.; Sofia, M.J.; Tavis, J.E. Inhibition of HBV Replication by N-Hydroxyisoquinolinedione and N-Hydroxypyridinedione Ribonuclease H Inhibitors. Antivir. Res. 2019, 164, 70–80. [Google Scholar] [CrossRef]
- Tramontano, E.; Corona, A.; Menéndez-Arias, L. Ribonuclease H, an Unexploited Target for Antiviral Intervention against HIV and Hepatitis B Virus. Antivir. Res. 2019, 171, 104613. [Google Scholar] [CrossRef]
- Hu, Y.; Cheng, X.; Cao, F.; Huang, A.; Tavis, J.E. β-Thujaplicinol Inhibits Hepatitis B Virus Replication by Blocking the Viral Ribonuclease H Activity. Antivir. Res. 2013, 99, 221–229. [Google Scholar] [CrossRef]
- Lomonosova, E.; Daw, J.; Garimallaprabhakaran, A.K.; Agyemang, N.B.; Ashani, Y.; Murelli, R.P.; Tavis, J.E. Efficacy and Cytotoxicity in Cell Culture of Novel α-Hydroxytropolone Inhibitors of Hepatitis B Virus Ribonuclease H. Antivir. Res. 2017, 144, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Lomonosova, E.; Cheng, X.; Moran, E.A.; Meyers, M.J.; Le Grice, S.F.J.; Thomas, C.J.; Jiang, J.; Meck, C.; Hirsch, D.R.; et al. Hydroxylated Tropolones Inhibit Hepatitis B Virus Replication by Blocking Viral Ribonuclease H Activity. Antimicrob. Agents Chemother. 2015, 59, 1070–1079. [Google Scholar] [CrossRef] [PubMed]
- Agyemang, N.B.; Kukla, C.R.; Edwards, T.C.; Li, Q.; Langen, M.K.; Schaal, A.; Franson, A.D.; Casals, A.G.; Donald, K.A.; Yu, A.J.; et al. Divergent Synthesis of a Thiolate-Based α-Hydroxytropolone Library with a Dynamic Bioactivity Profile. RSC Adv. 2019, 9, 34227–34234. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, A.J.; Abdelmessih, R.G.; Murelli, R.P. Amidation Strategy for Final-Step α-Hydroxytropolone Diversification. Tetrahedron Lett. 2018, 59, 3026–3028. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lomonosova, E.; Donlin, M.J.; Cao, F.; O’Dea, A.; Milleson, B.; Berkowitz, A.J.; Baucom, J.-C.; Stasiak, J.P.; Schiavone, D.V.; et al. Amide-Containing α-Hydroxytropolones as Inhibitors of Hepatitis B Virus Replication. Antivir. Res. 2020, 177, 104777. [Google Scholar] [CrossRef]
- Bak, E.; Miller, J.T.; Noronha, A.; Tavis, J.; Gallicchio, E.; Murelli, R.P.; Le Grice, S.F.J. 3,7-Dihydroxytropolones Inhibit Initiation of Hepatitis B Virus Minus-Strand DNA Synthesis. Molecules 2020, 25, 4434. [Google Scholar] [CrossRef] [PubMed]
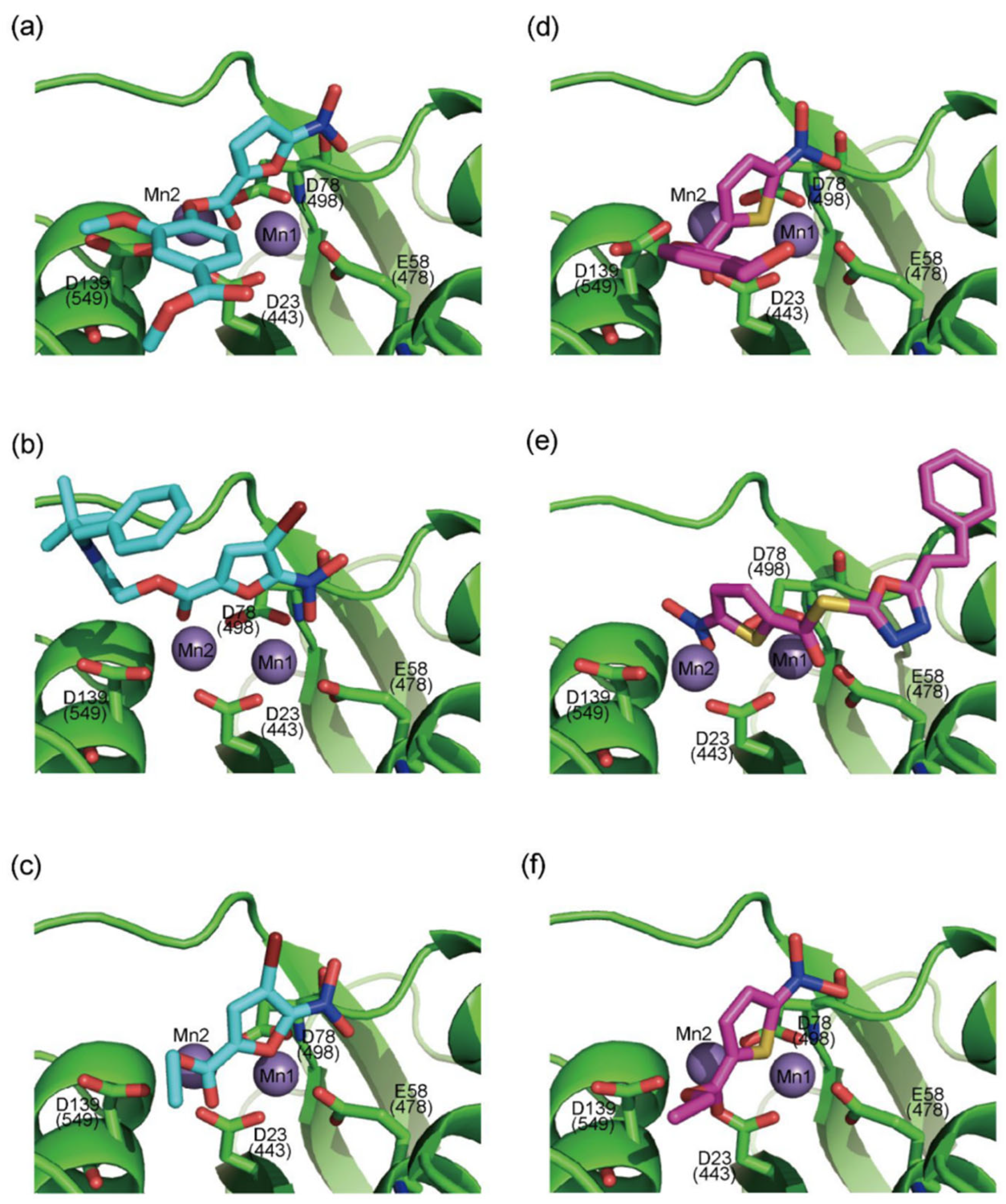
- Tavis, J.E.; Cheng, X.; Hu, Y.; Totten, M.; Cao, F.; Michailidis, E.; Aurora, R.; Meyers, M.J.; Jacobsen, E.J.; Parniak, M.A.; et al. The Hepatitis B Virus Ribonuclease H Is Sensitive to Inhibitors of the Human Immunodeficiency Virus Ribonuclease H and Integrase Enzymes. PLoS Pathog. 2013, 9, e1003125. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.W.; Lomonosova, E.; Moran, E.A.; Cheng, X.; Patel, K.B.; Bailly, F.; Cotelle, P.; Meyers, M.J.; Tavis, J.E. Hepatitis B Virus Replication Is Blocked by a 2-Hydroxyisoquinoline-1,3(2H,4H)-Dione (HID) Inhibitor of the Viral Ribonuclease H Activity. Antivir. Res. 2014, 108, 48–55. [Google Scholar] [CrossRef]
- Long, K.R.; Lomonosova, E.; Li, Q.; Ponzar, N.L.; Villa, J.A.; Touchette, E.; Rapp, S.; Liley, R.M.; Murelli, R.P.; Grigoryan, A.; et al. Efficacy of Hepatitis B Virus Ribonuclease H Inhibitors, a New Class of Replication Antagonists, in FRG Human Liver Chimeric Mice. Antivir. Res. 2018, 149, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Abuelizz, H.A.; Marzouk, M.; Bakheit, A.H.; Al-Salahi, R. Investigation of Some Benzoquinazoline and Quinazoline Derivatives as Novel Inhibitors of HCV-NS3/4A Protease: Biological, Molecular Docking and QSAR Studies. RSC Adv. 2020, 10, 35820–35830. [Google Scholar] [CrossRef]
- Ejeh, S.; Uzairu, A.; Shallangwa, G.A.; Abechi, S.E. Computational Insight to Design New Potential Hepatitis C Virus NS5B Polymerase Inhibitors with Drug-Likeness and Pharmacokinetic ADMET Parameters Predictions. Futur. J. Pharm. Sci. 2021, 7, 219. [Google Scholar] [CrossRef]
- Zając, M.; Muszalska, I.; Sobczak, A.; Dadej, A.; Tomczak, S.; Jelińska, A. Hepatitis C—New Drugs and Treatment Prospects. Eur. J. Med. Chem. 2019, 165, 225–249. [Google Scholar] [CrossRef]
- Hajarizadeh, B.; Grebely, J.; Dore, G.J. Epidemiology and Natural History of HCV Infection. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 553–562. [Google Scholar] [CrossRef]
- Li, W.; Si, H.; Li, Y.; Ge, C.; Song, F.; Ma, X.; Duan, Y.; Zhai, H. 3D-QSAR and Molecular Docking Studies on Designing Inhibitors of the Hepatitis C Virus NS5B Polymerase. J. Mol. Struct. 2016, 1117, 227–239. [Google Scholar] [CrossRef]
- World Health Organization. Accelerating Access to Hepatitis C Diagnostics and Treatment: Overcoming Barriers in Low-and Middle-Income Countries: Global Progress Report 2020; World Health Organization: Geneza, Switzerland, 2021.
- World Health Organization. Global Progress Report on HIV, Viral Hepatitis and Sexually Transmitted Infections, 2021: Accountability for the Global Health Sector Strategies 2016–2021: Actions for Impact; World Health Organization: Geneva, Switzerland, 2021; ISBN 9789240027077.
- World Health Organization. Progress Report on HIV, Viral Hepatitis and Sexually Transmitted Infections 2019: Accountability for the Global Health Sector Strategies, 2016–2021; World Health Organization: Geneva, Switzerland, 2019.
- Gutierrez, J.A.; Lawitz, E.J.; Poordad, F. Interferon-Free, Direct-Acting Antiviral Therapy for Chronic Hepatitis C. J. Viral. Hepat. 2015, 22, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Moradpour, D.; Penin, F.; Rice, C.M. Replication of Hepatitis C Virus. Nat. Rev. Microbiol. 2007, 5, 453–463. [Google Scholar] [CrossRef]
- Appleby, T.C.; Perry, J.K.; Murakami, E.; Barauskas, O.; Feng, J.; Cho, A.; Fox, D.; Wetmore, D.R.; McGrath, M.E.; Ray, A.S.; et al. Structural Basis for RNA Replication by the Hepatitis C Virus Polymerase. Science 2015, 347, 771–775. [Google Scholar] [CrossRef]
- Kao, C. An Update on Small Molecule Inhibitors of the HCV NS5B Polymerase: Effects on RNA Synthesis in Vitro and in Cultured Cells, and Potential Resistance in Viral Quasispecies. VAAT 2010, 73, 73–89. [Google Scholar] [CrossRef]
- Lesburg, C.A.; Cable, M.B.; Ferrari, E.; Hong, Z.; Mannarino, A.F.; Weber, P.C. Crystal Structure of the RNA-Dependent RNA Polymerase from Hepatitis C Virus Reveals a Fully Encircled Active Site. Nat. Struct. Mol. Biol. 1999, 6, 937–943. [Google Scholar] [CrossRef]
- Joyce, C.M.; Steitz, T.A. Polymerase Structures and Function: Variations on a Theme? J. Bacteriol. 1995, 177, 6321–6329. [Google Scholar] [CrossRef] [PubMed]
- Summa, V.; Petrocchi, A.; Pace, P.; Matassa, V.G.; De Francesco, R.; Altamura, S.; Tomei, L.; Koch, U.; Neuner, P. Discovery of α,γ-Diketo Acids as Potent Selective and Reversible Inhibitors of Hepatitis C Virus NS5b RNA-Dependent RNA Polymerase. J. Med. Chem. 2004, 47, 14–17. [Google Scholar] [CrossRef] [PubMed]
- Pace, P.; Nizi, E.; Pacini, B.; Pesci, S.; Matassa, V.; De Francesco, R.; Altamura, S.; Summa, V. The Monoethyl Ester of Meconic Acid Is an Active Site Inhibitor of HCV NS5B RNA-Dependent RNA Polymerase. Bioorg. Med. Chem. Lett. 2004, 14, 3257–3261. [Google Scholar] [CrossRef] [PubMed]
- Koch, U.; Attenni, B.; Malancona, S.; Colarusso, S.; Conte, I.; Di Filippo, M.; Harper, S.; Pacini, B.; Giomini, C.; Thomas, S.; et al. 2-(2-Thienyl)-5,6-Dihydroxy-4-Carboxypyrimidines as Inhibitors of the Hepatitis C Virus NS5B Polymerase: Discovery, SAR, Modeling, and Mutagenesis. J. Med. Chem. 2006, 49, 1693–1705. [Google Scholar] [CrossRef]
- Pacini, B.; Avolio, S.; Ercolani, C.; Koch, U.; Migliaccio, G.; Narjes, F.; Pacini, L.; Tomei, L.; Harper, S. 2-(3-Thienyl)-5,6-Dihydroxypyrimidine-4-Carboxylic Acids as Inhibitors of HCV NS5B RdRp. Bioorg. Med. Chem. Lett. 2009, 19, 6245–6249. [Google Scholar] [CrossRef]
- Chen, Y.-L.; Tang, J.; Kesler, M.J.; Sham, Y.Y.; Vince, R.; Geraghty, R.J.; Wang, Z. The Design, Synthesis and Biological Evaluations of C-6 or C-7 Substituted 2-Hydroxyisoquinoline-1,3-Diones as Inhibitors of Hepatitis C Virus. Bioorg. Med. Chem. 2012, 20, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Giannakopoulou, E.; Pardali, V.; Frakolaki, E.; Siozos, V.; Myrianthopoulos, V.; Mikros, E.; Taylor, M.C.; Kelly, J.M.; Vassilaki, N.; Zoidis, G. Scaffold Hybridization Strategy towards Potent Hydroxamate-Based Inhibitors of Flaviviridae Viruses and Trypanosoma Species. Med. Chem. Commun. 2019, 10, 991–1006. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Aimaiti, A.; Zhu, Z.; Zhou, L.; Ye, D. Discovery of Novel 3-Hydroxyquinazoline-2,4(1H,3H)-Dione Derivatives: A Series of Metal Ion Chelators with Potent Anti-HCV Activities. Int. J. Mol. Sci. 2022, 23, 5930. [Google Scholar] [CrossRef] [PubMed]
- Keating, J.; Yukich, J.O.; Sutherland, C.S.; Woods, G.; Tediosi, F. Human African Trypanosomiasis Prevention, Treatment and Control Costs: A Systematic Review. Acta Trop. 2015, 150, 4–13. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization; WHO Expert Committee on the Control and Surveillance of Human African Trypanosomiasis. Control and Surveillance of Human African Trypanosomiasis: Report of a WHO Expert Committee; WHO Technical Report Series 984; World Health Organization: Geneva, Switzerland, 2013; ISBN 9789240691728.
- Simarro, P.; Franco, J.; Diarra, A.; Jannin, J. Epidemiology of Human African Trypanosomiasis. CLEP 2014, 6, 257–275. [Google Scholar] [CrossRef]
- Munday, J.C.; Settimo, L.; de Koning, H.P. Transport Proteins Determine Drug Sensitivity and Resistance in a Protozoan Parasite, Trypanosoma Brucei. Front. Pharmacol. 2015, 6, 32. [Google Scholar] [CrossRef]
- Lüscher, A.; Lamprea-Burgunder, E.; Graf, F.E.; de Koning, H.P.; Mäser, P. Trypanosoma Brucei Adenine-Phosphoribosyltransferases Mediate Adenine Salvage and Aminopurinol Susceptibility but Not Adenine Toxicity. Int. J. Parasitol. Drugs Drug Resist. 2014, 4, 55–63. [Google Scholar] [CrossRef]
- Graf, F.E.; Baker, N.; Munday, J.C.; de Koning, H.P.; Horn, D.; Mäser, P. Chimerization at the AQP2–AQP3 Locus Is the Genetic Basis of Melarsoprol–Pentamidine Cross-Resistance in Clinical Trypanosoma Brucei Gambiense Isolates. Int. J. Parasitol. Drugs Drug Resist. 2015, 5, 65–68. [Google Scholar] [CrossRef]
- Mogk, S.; Meiwes, A.; Shtopel, S.; Schraermeyer, U.; Lazarus, M.; Kubata, B.; Wolburg, H.; Duszenko, M. Cyclical Appearance of African Trypanosomes in the Cerebrospinal Fluid: New Insights in How Trypanosomes Enter the CNS. PLoS ONE 2014, 9, e91372. [Google Scholar] [CrossRef]
- Doyle, M.A.; Gasser, R.B.; Woodcroft, B.J.; Hall, R.S.; Ralph, S.A. Drug Target Prediction and Prioritization: Using Orthology to Predict Essentiality in Parasite Genomes. BMC Genom. 2010, 11, 222. [Google Scholar] [CrossRef]
- Doleželová, E.; Terán, D.; Gahura, O.; Kotrbová, Z.; Procházková, M.; Keough, D.; Špaček, P.; Hocková, D.; Guddat, L.; Zíková, A. Evaluation of the Trypanosoma Brucei 6-Oxopurine Salvage Pathway as a Potential Target for Drug Discovery. PLoS Negl. Trop. Dis. 2018, 12, e0006301. [Google Scholar] [CrossRef] [PubMed]
- Terán, D.; Hocková, D.; Česnek, M.; Zíková, A.; Naesens, L.; Keough, D.T.; Guddat, L.W. Crystal Structures and Inhibition of Trypanosoma Brucei Hypoxanthine–Guanine Phosphoribosyltransferase. Sci. Rep. 2016, 6, 35894. [Google Scholar] [CrossRef]
- Terán, D.; Doleželová, E.; Keough, D.T.; Hocková, D.; Zíková, A.; Guddat, L.W. Crystal Structures of Trypanosoma Brucei Hypoxanthine—Guanine—Xanthine Phosphoribosyltransferase in Complex with IMP, GMP and XMP. FEBS J. 2019, 286, 4721–4736. [Google Scholar] [CrossRef]
- Kolocouris, N.; Zoidis, G.; Foscolos, G.B.; Fytas, G.; Prathalingham, S.R.; Kelly, J.M.; Naesens, L.; De Clercq, E. Design and Synthesis of Bioactive Adamantane Spiro Heterocycles. Bioorg. Med. Chem. Lett. 2007, 17, 4358–4362. [Google Scholar] [CrossRef]
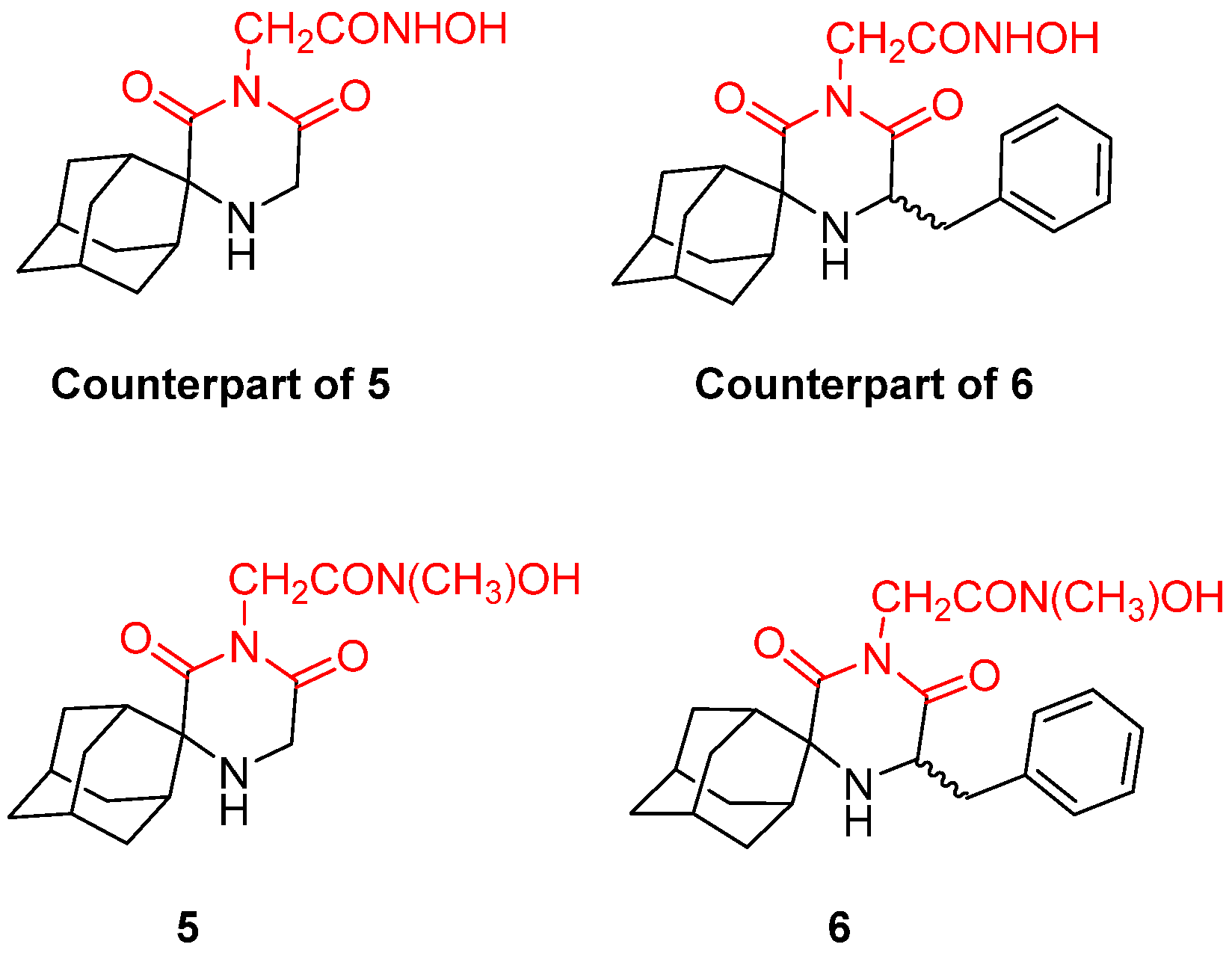
- Fytas, G.; Zoidis, G.; Taylor, M.C.; Kelly, J.M.; Tsatsaroni, A.; Tsotinis, A. Novel 2,6-Diketopiperazine-Derived Acetohydroxamic Acids as Promising Anti-Trypanosoma Brucei Agents. Future Med. Chem. 2019, 11, 1259–1266. [Google Scholar] [CrossRef]
- Zoidis, G.; Tsotinis, A.; Tsatsaroni, A.; Taylor, M.C.; Kelly, J.M.; Efstathiou, A.; Smirlis, D.; Fytas, G. Lipophilic Conformationally Constrained Spiro Carbocyclic 2,6-Diketopiperazine-1-Acetohydroxamic Acid Analogues as Trypanocidal and Leishmanicidal Agents: An Extended SAR Study. Chem. Biol. Drug Des. 2018, 91, 408–421. [Google Scholar] [CrossRef] [PubMed]
- Breidbach, T.; Scory, S.; Krauth-Siegel, R.L.; Steverding, D. Growth Inhibition of Bloodstream Forms of Trypanosoma Brucei by the Iron Chelator Deferoxamine. Int. J. Parasitol. 2002, 32, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.L.S.; Sodre, C.L.; Valle, R.S.; Silva, B.A.; Abi-chacra, E.A.; Silva, L.V.; Souza-Goncalves, A.L.; Sangenito, L.S.; Goncalves, D.S.; Souza, L.O.P.; et al. Antimicrobial Action of Chelating Agents: Repercussions on the Microorganism Development, Virulence and Pathogenesis. CMC 2012, 19, 2715–2737. [Google Scholar] [CrossRef]
- Rangel, M.; Moniz, T.; Silva, A.; Leite, A. Tuning the Anti(Myco)Bacterial Activity of 3-Hydroxy-4-Pyridinone Chelators through Fluorophores. Pharmaceuticals 2018, 11, 110. [Google Scholar] [CrossRef]
- Ellis, S.; Sexton, D.W.; Steverding, D. Trypanotoxic Activity of Thiosemicarbazone Iron Chelators. Exp. Parasitol. 2015, 150, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Aye, Y.; Long, M.J.C.; Stubbe, J. Mechanistic Studies of Semicarbazone Triapine Targeting Human Ribonucleotide Reductase in Vitro and in Mammalian Cells. J. Biol. Chem. 2012, 287, 35768–35778. [Google Scholar] [CrossRef] [PubMed]
- Control of Neglected Tropical Diseases. Available online: https://www.who.int/teams/control-of-neglected-tropical-diseases/overview (accessed on 3 March 2023).
- Zuma, A.A.; de Souza, W. Chagas Disease Chemotherapy: What Do We Know So Far? Curr. Pharm. Des. 2021, 27, 3963–3995. [Google Scholar] [CrossRef] [PubMed]
- Bonney, K.M. Chagas Disease in the 21st Century: A Public Health Success or an Emerging Threat? Parasite 2014, 21, 11. [Google Scholar] [CrossRef] [PubMed]
- Altcheh, J.; Moscatelli, G.; Moroni, S.; Garcia-Bournissen, F.; Freilij, H. Adverse Events after the Use of Benznidazole in Infants and Children with Chagas Disease. Pediatrics 2011, 127, e212–e218. [Google Scholar] [CrossRef]
- Lascano, F.; García Bournissen, F.; Altcheh, J. Review of Pharmacological Options for the Treatment of Chagas Disease. Br. J. Clin. Pharmacol. 2022, 88, 383–402. [Google Scholar] [CrossRef]
- Scarim, B.C.; Chin, M.C. Current Approaches to Drug Discovery for Chagas Disease: Methodological Advances. Comb. Chem. High Throughput Screen. 2019, 22, 509–520. [Google Scholar] [CrossRef]
- Villalta, F.; Rachakonda, G. Advances in Preclinical Approaches to Chagas Disease Drug Discovery. Expert Opin. Drug Discov. 2019, 14, 1161–1174. [Google Scholar] [CrossRef]
- Supuran, C.T. Inhibition of Carbonic Anhydrase from Trypanosoma Cruzi for the Management of Chagas Disease: An Underexplored Therapeutic Opportunity. Future Med. Chem. 2016, 8, 311–324. [Google Scholar] [CrossRef]
- Bonardi, A.; Parkkila, S.; Supuran, C.T. Inhibition Studies of the Protozoan α-Carbonic Anhydrase from Trypanosoma Cruzi with Phenols. J. Enzym. Inhib. Med. Chem. 2022, 37, 2417–2422. [Google Scholar] [CrossRef]
- Nocentini, A.; Cadoni, R.; Dumy, P.; Supuran, C.T.; Winum, J.-Y. Carbonic Anhydrases from Trypanosoma Cruzi and Leishmania Donovani Chagasi Are Inhibited by Benzoxaboroles. J. Enzym. Inhib. Med. Chem. 2018, 33, 286–289. [Google Scholar] [CrossRef]
- Güzel-Akdemir, Ö.; Akdemir, A.; Pan, P.; Vermelho, A.B.; Parkkila, S.; Scozzafava, A.; Capasso, C.; Supuran, C.T. A Class of Sulfonamides with Strong Inhibitory Action against the α-Carbonic Anhydrase from Trypanosoma Cruzi. J. Med. Chem. 2013, 56, 5773–5781. [Google Scholar] [CrossRef]
- Pan, P.; Vermelho, A.B.; Capaci Rodrigues, G.; Scozzafava, A.; Tolvanen, M.E.E.; Parkkila, S.; Capasso, C.; Supuran, C.T. Cloning, Characterization, and Sulfonamide and Thiol Inhibition Studies of an α-Carbonic Anhydrase from Trypanosoma Cruzi, the Causative Agent of Chagas Disease. J. Med. Chem. 2013, 56, 1761–1771. [Google Scholar] [CrossRef] [PubMed]
- Frost, S.C.; McKenna, R. (Eds.) Carbonic Anhydrase: Mechanism, Regulation, Links to Disease, and Industrial Applications; Subcellular Biochemistry; Springer: Dordrecht, The Netherlands, 2014; Volume 75, ISBN 978-94-007-7358-5. [Google Scholar]
- Supuran, C.T. Carbonic Anhydrases: Novel Therapeutic Applications for Inhibitors and Activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Alterio, V.; Di Fiore, A.; D’Ambrosio, K.; Supuran, C.T.; De Simone, G. Multiple Binding Modes of Inhibitors to Carbonic Anhydrases: How to Design Specific Drugs Targeting 15 Different Isoforms? Chem. Rev. 2012, 112, 4421–4468. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic Anhydrases: From Biomedical Applications of the Inhibitors and Activators to Biotechnological Use for CO2 Capture. J. Enzym. Inhib. Med. Chem. 2013, 28, 229–230. [Google Scholar] [CrossRef]
- Wang, Q.; Rosa, B.A.; Nare, B.; Powell, K.; Valente, S.; Rotili, D.; Mai, A.; Marshall, G.R.; Mitreva, M. Targeting Lysine Deacetylases (KDACs) in Parasites. PLoS Negl. Trop. Dis. 2015, 9, e0004026. [Google Scholar] [CrossRef]
- Gupta, S.P. (Ed.) Matrix Metalloproteinase Inhibitors: Specificity of Binding and Structure-Activity Relationships; Experientia Supplementum; Springer: Basel, Switzerland, 2012; Volume 103, ISBN 978-3-0348-0363-2. [Google Scholar]
- Rodrigues, G.C.; Feijó, D.F.; Bozza, M.T.; Pan, P.; Vullo, D.; Parkkila, S.; Supuran, C.T.; Capasso, C.; Aguiar, A.P.; Vermelho, A.B. Design, Synthesis, and Evaluation of Hydroxamic Acid Derivatives as Promising Agents for the Management of Chagas Disease. J. Med. Chem. 2014, 57, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Fytas, C.; Zoidis, G.; Tzoutzas, N.; Taylor, M.C.; Fytas, G.; Kelly, J.M. Novel Lipophilic Acetohydroxamic Acid Derivatives Based on Conformationally Constrained Spiro Carbocyclic 2,6-Diketopiperazine Scaffolds with Potent Trypanocidal Activity. J. Med. Chem. 2011, 54, 5250–5254. [Google Scholar] [CrossRef]
- Tsatsaroni, A.; Zoidis, G.; Zoumpoulakis, P.; Tsotinis, A.; Taylor, M.C.; Kelly, J.M.; Fytas, G. An E/Z Conformational Behaviour Study on the Trypanocidal Action of Lipophilic Spiro Carbocyclic 2,6-Diketopiperazine-1-Acetohydroxamic Acids. Tetrahedron Lett. 2013, 54, 3238–3240. [Google Scholar] [CrossRef]
- Alafeefy, A.M.; Ceruso, M.; Al-Jaber, N.A.; Parkkila, S.; Vermelho, A.B.; Supuran, C.T. A New Class of Quinazoline-Sulfonamides Acting as Efficient Inhibitors against the α-Carbonic Anhydrase from Trypanosoma Cruzi. J. Enzym. Inhib. Med. Chem. 2015, 30, 581–585. [Google Scholar] [CrossRef]
- Pan, P.; Vermelho, A.B.; Scozzafava, A.; Parkkila, S.; Capasso, C.; Supuran, C.T. Anion Inhibition Studies of the α-Carbonic Anhydrase from the Protozoan Pathogen Trypanosoma Cruzi, the Causative Agent of Chagas Disease. Bioorg. Med. Chem. 2013, 21, 4472–4476. [Google Scholar] [CrossRef]
- Debus, H. Ueber die Verbindungen der Sulfocarbaminsäure. Ann. Chem. Pharm. 1850, 73, 26–34. [Google Scholar] [CrossRef]
- Oliveira, J.W.D.F.; Rocha, H.A.O.; De Medeiros, W.M.T.Q.; Silva, M.S. Application of Dithiocarbamates as Potential New Antitrypanosomatids-Drugs: Approach Chemistry, Functional and Biological. Molecules 2019, 24, 2806. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, R.R.; Lane, J.E.; Carter, C.E.; Bogitsh, B.J.; Singh, P.K.; Zimmerman, L.J.; Molenda, J.J.; Jones, M.M. Chelating Agent Inhibition of Trypanosoma Cruzi Epimastigotes In Vitro. J. Inorg. Biochem. 1995, 60, 277–288. [Google Scholar] [CrossRef]
- Rogolino, D.; Carcelli, M.; Compari, C.; De Luca, L.; Ferro, S.; Fisicaro, E.; Rispoli, G.; Neamati, N.; Debyser, Z.; Christ, F.; et al. Diketoacid Chelating Ligands as Dual Inhibitors of HIV-1 Integration Process. Eur. J. Med. Chem. 2014, 78, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Carcelli, M.; Rogolino, D.; Gatti, A.; Pala, N.; Corona, A.; Caredda, A.; Tramontano, E.; Pannecouque, C.; Naesens, L.; Esposito, F. Chelation Motifs Affecting Metal-Dependent Viral Enzymes: N′-Acylhydrazone Ligands as Dual Target Inhibitors of HIV-1 Integrase and Reverse Transcriptase Ribonuclease H Domain. Front. Microbiol. 2017, 8, 440. [Google Scholar] [CrossRef] [PubMed]
- Nováková, L.; Pavlík, J.; Chrenková, L.; Martinec, O.; Červený, L. Current Antiviral Drugs and Their Analysis in Biological Materials—Part II: Antivirals against Hepatitis and HIV Viruses. J. Pharm. Biomed. Anal. 2018, 147, 378–399. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E.; Li, G. Approved Antiviral Drugs over the Past 50 Years. Clin. Microbiol. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef]
- Smerdon, S.J.; Jager, J.; Wang, J.; Kohlstaedt, L.A.; Chirino, A.J.; Friedman, J.M.; Rices, P.A.; Steitz, T.A. Structure of the Binding Site for Nonnucleoside Inhibitors of the Reverse Transcriptase of Human Immunodeficiency Virus Type 1. Proc. Nati. Acad. Sci. USA 1994, 91, 3911–3915. [Google Scholar] [CrossRef]
- Ding’, J.; Das’, K.; Zhang’, W.; Clark, A.; Lu, X.; Hsiou, Y.; Jacobo-Molinat, A.; Pauwels, R.; Moereels, H.; Koymans, L.; et al. Structure of HIV-1 Reverse Transcriptase in a Complex with the Non-Nucleoside Inhibitor Ot-APA R 95845 at 2.8 A Resolution. Structure 1995, 3, 365–379. [Google Scholar] [CrossRef]
- Wensing, A.M.J.; Van Maarseveen, N.M.; Nijhuis, M. Fifteen Years of HIV Protease Inhibitors: Raising the Barrier to Resistance. Antivir. Res. 2010, 85, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Matthews, T.; Salgo, M.; Greenberg, M.; Chung, J.; DeMasi, R.; Bolognesi, D. Enfuvirtide: The First Therapy to Inhibit the Entry of HIV-1 into Host CD4 Lymphocytes. Nat. Rev. Drug Discov. 2004, 3, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Dorr, P.; Westby, M.; Dobbs, S.; Griffin, P.; Irvine, B.; Macartney, M.; Mori, J.; Rickett, G.; Smith-Burchnell, C.; Napier, C.; et al. Maraviroc (UK-427,857), a Potent, Orally Bioavailable, and Selective Small-Molecule Inhibitor of Chemokine Receptor CCR5 with Broad-Spectrum Anti-Human Immunodeficiency Virus Type 1 Activity. Antimicrob. Agents Chemother. 2005, 49, 4721–4732. [Google Scholar] [CrossRef]
- Tramontano, E.; Esposito, F.; Badas, R.; Di Santo, R.; Costi, R.; La Colla, P. 6-[1-(4-Fluorophenyl)Methyl-1H-Pyrrol-2-Yl)]-2,4-Dioxo-5-Hexenoic Acid Ethyl Ester a Novel Diketo Acid Derivative Which Selectively Inhibits the HIV-1 Viral Replication in Cell Culture and the Ribonuclease H Activity in Vitro. Antivir. Res. 2005, 65, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Costi, R.; Métifiot, M.; Esposito, F.; Cuzzucoli Crucitti, G.; Pescatori, L.; Messore, A.; Scipione, L.; Tortorella, S.; Zinzula, L.; Novellino, E.; et al. 6-(1-Benzyl-1 H -Pyrrol-2-Yl)-2,4-Dioxo-5-Hexenoic Acids as Dual Inhibitors of Recombinant HIV-1 Integrase and Ribonuclease H, Synthesized by a Parallel Synthesis Approach. J. Med. Chem. 2013, 56, 8588–8598. [Google Scholar] [CrossRef]
- Costi, R.; Métifiot, M.; Chung, S.; Cuzzucoli Crucitti, G.; Maddali, K.; Pescatori, L.; Messore, A.; Madia, V.N.; Pupo, G.; Scipione, L.; et al. Basic Quinolinonyl Diketo Acid Derivatives as Inhibitors of HIV Integrase and Their Activity against RNase H Function of Reverse Transcriptase. J. Med. Chem. 2014, 57, 3223–3234. [Google Scholar] [CrossRef]
- Cuzzucoli Crucitti, G.; Métifiot, M.; Pescatori, L.; Messore, A.; Madia, V.N.; Pupo, G.; Saccoliti, F.; Scipione, L.; Tortorella, S.; Esposito, F.; et al. Structure–Activity Relationship of Pyrrolyl Diketo Acid Derivatives as Dual Inhibitors of HIV-1 Integrase and Reverse Transcriptase Ribonuclease H Domain. J. Med. Chem. 2015, 58, 1915–1928. [Google Scholar] [CrossRef]
- Budihas, S.R. Selective Inhibition of HIV-1 Reverse Transcriptase-Associated Ribonuclease H Activity by Hydroxylated Tropolones. Nucleic Acids Res. 2005, 33, 1249–1256. [Google Scholar] [CrossRef]
- Chung, S.; Himmel, D.M.; Jiang, J.-K.; Wojtak, K.; Bauman, J.D.; Rausch, J.W.; Wilson, J.A.; Beutler, J.A.; Thomas, C.J.; Arnold, E.; et al. Synthesis, Activity, and Structural Analysis of Novel α-Hydroxytropolone Inhibitors of Human Immunodeficiency Virus Reverse Transcriptase-Associated Ribonuclease H. J. Med. Chem. 2011, 54, 4462–4473. [Google Scholar] [CrossRef]
- Kirschberg, T.A.; Balakrishnan, M.; Squires, N.H.; Barnes, T.; Brendza, K.M.; Chen, X.; Eisenberg, E.J.; Jin, W.; Kutty, N.; Leavitt, S.; et al. RNase H Active Site Inhibitors of Human Immunodeficiency Virus Type 1 Reverse Transcriptase: Design, Biochemical Activity, and Structural Information. J. Med. Chem. 2009, 52, 5781–5784. [Google Scholar] [CrossRef]
- Summa, V.; Petrocchi, A.; Matassa, V.G.; Taliani, M.; Laufer, R.; De Francesco, R.; Altamura, S.; Pace, P. HCV NS5b RNA-Dependent RNA Polymerase Inhibitors: From α,γ-Diketoacids to 4,5-Dihydroxypyrimidine- or 3-Methyl-5- Hydroxypyrimidinonecarboxylic Acids. Design and Synthesis. J. Med. Chem. 2004, 47, 5336–5339. [Google Scholar] [CrossRef] [PubMed]
- Billamboz, M.; Bailly, F.; Lion, C.; Touati, N.; Vezin, H.; Calmels, C.; Andréola, M.-L.; Christ, F.; Debyser, Z.; Cotelle, P. Magnesium Chelating 2-Hydroxyisoquinoline-1,3(2 H,4 H)-Diones, as Inhibitors of HIV-1 Integrase and/or the HIV-1 Reverse Transcriptase Ribonuclease H Domain: Discovery of a Novel Selective Inhibitor of the Ribonuclease H Function. J. Med. Chem. 2011, 54, 1812–1824. [Google Scholar] [CrossRef] [PubMed]
- Vernekar, S.K.V.; Liu, Z.; Nagy, E.; Miller, L.; Kirby, K.A.; Wilson, D.J.; Kankanala, J.; Sarafianos, S.G.; Parniak, M.A.; Wang, Z. Design, Synthesis, Biochemical, and Antiviral Evaluations of C6 Benzyl and C6 Biarylmethyl Substituted 2-Hydroxylisoquinoline-1,3-Diones: Dual Inhibition against HIV Reverse Transcriptase-Associated RNase H and Polymerase with Antiviral Activities. J. Med. Chem. 2015, 58, 651–664. [Google Scholar] [CrossRef] [PubMed]
- Kirby, K.A.; Myshakina, N.A.; Christen, M.T.; Chen, Y.-L.; Schmidt, H.A.; Huber, A.D.; Xi, Z.; Kim, S.; Rao, R.K.; Kramer, S.T.; et al. A 2-Hydroxyisoquinoline-1,3-Dione Active-Site RNase H Inhibitor Binds in Multiple Modes to HIV-1 Reverse Transcriptase. Antimicrob. Agents Chemother. 2017, 61, e01351-17. [Google Scholar] [CrossRef]
- Kankanala, J.; Kirby, K.A.; Liu, F.; Miller, L.; Nagy, E.; Wilson, D.J.; Parniak, M.A.; Sarafianos, S.G.; Wang, Z. Design, Synthesis, and Biological Evaluations of Hydroxypyridonecarboxylic Acids as Inhibitors of HIV Reverse Transcriptase Associated RNase H. J. Med. Chem. 2016, 59, 5051–5062. [Google Scholar] [CrossRef]
- Tang, J.; Do, H.T.; Huber, A.D.; Casey, M.C.; Kirby, K.A.; Wilson, D.J.; Kankanala, J.; Parniak, M.A.; Sarafianos, S.G.; Wang, Z. Pharmacophore-Based Design of Novel 3-Hydroxypyrimidine-2,4-Dione Subtypes as Inhibitors of HIV Reverse Transcriptase-Associated RNase H: Tolerance of a Nonflexible Linker. Eur. J. Med. Chem. 2019, 166, 390–399. [Google Scholar] [CrossRef]
- Wang, L.; Tang, J.; Huber, A.D.; Casey, M.C.; Kirby, K.A.; Wilson, D.J.; Kankanala, J.; Parniak, M.A.; Sarafianos, S.G.; Wang, Z. 6-Biphenylmethyl-3-Hydroxypyrimidine-2,4-Diones Potently and Selectively Inhibited HIV Reverse Transcriptase-Associated RNase H. Eur. J. Med. Chem. 2018, 156, 680–691. [Google Scholar] [CrossRef]
- Fuji, H.; Urano, E.; Futahashi, Y.; Hamatake, M.; Tatsumi, J.; Hoshino, T.; Morikawa, Y.; Yamamoto, N.; Komano, J. Derivatives of 5-Nitro-Furan-2-Carboxylic Acid Carbamoylmethyl Ester Inhibit RNase H Activity Associated with HIV-1 Reverse Transcriptase. J. Med. Chem. 2009, 52, 1380–1387. [Google Scholar] [CrossRef]
- Yanagita, H.; Urano, E.; Matsumoto, K.; Ichikawa, R.; Takaesu, Y.; Ogata, M.; Murakami, T.; Wu, H.; Chiba, J.; Komano, J.; et al. Structural and Biochemical Study on the Inhibitory Activity of Derivatives of 5-Nitro-Furan-2-Carboxylic Acid for RNase H Function of HIV-1 Reverse Transcriptase. Bioorg. Med. Chem. 2011, 19, 816–825. [Google Scholar] [CrossRef]
- Yanagita, H.; Fudo, S.; Urano, E.; Ichikawa, R.; Ogata, M.; Yokota, M.; Murakami, T.; Wu, H.; Chiba, J.; Komano, J.; et al. Structural Modulation Study of Inhibitory Compounds for Ribonuclease H Activity of Human Immunodeficiency Virus Type 1 Reverse Transcriptase. Chem. Pharm. Bull. 2012, 60, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Komukai, Y.; Usami, K.; Guo, Y.; Qiao, X.; Nukaga, M.; Hoshino, T. Computational and Crystallographic Analysis of Binding Structures of Inhibitory Compounds for HIV-1 RNase H Activity. J. Chem. Inf. Model. 2022, 62, 6762–6774. [Google Scholar] [CrossRef] [PubMed]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
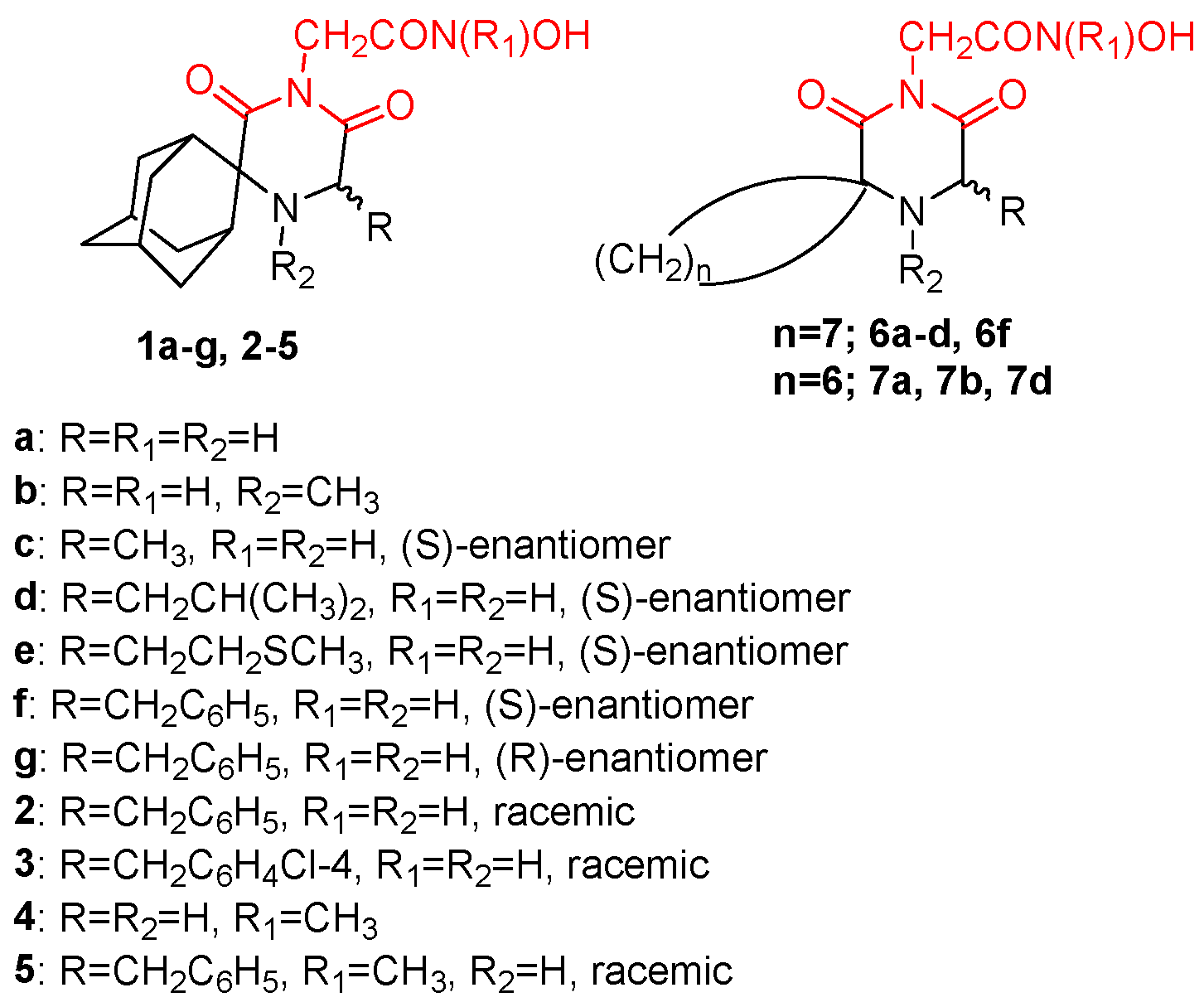{kind=link}
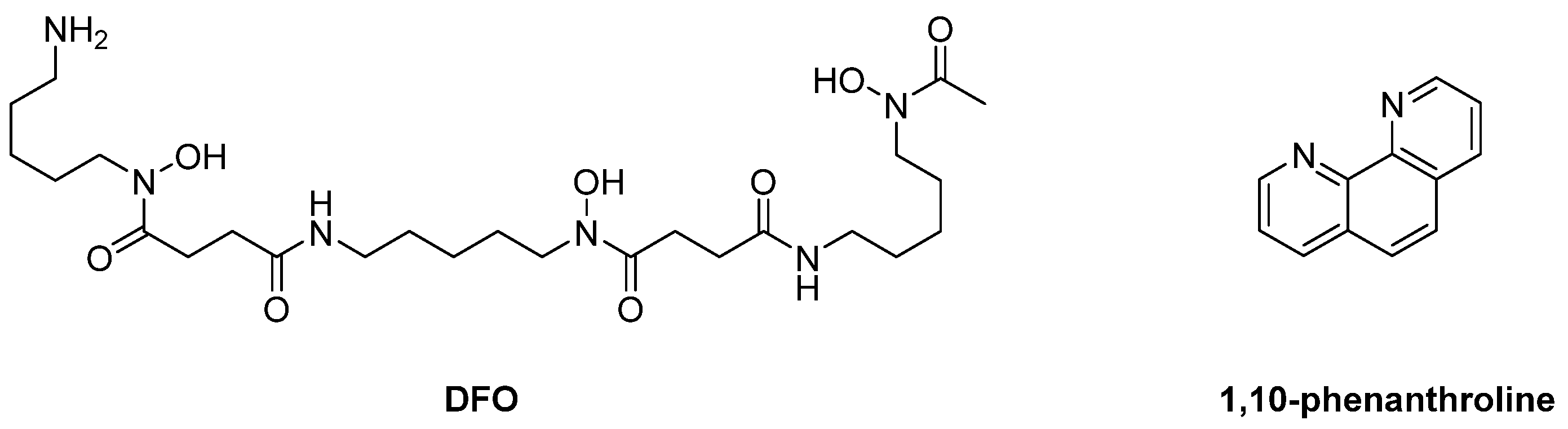{kind=link}
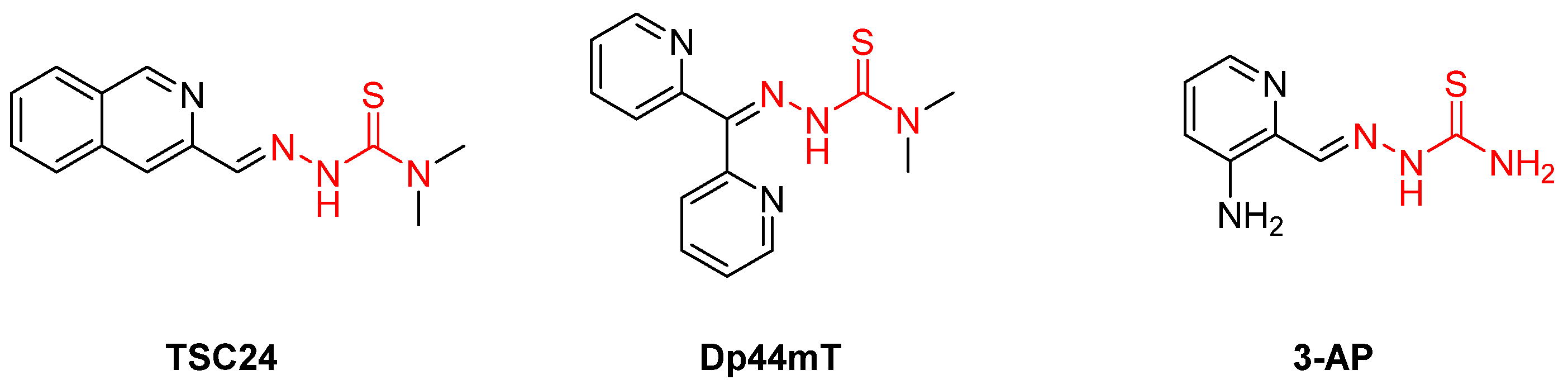{kind=link}
{kind=link}
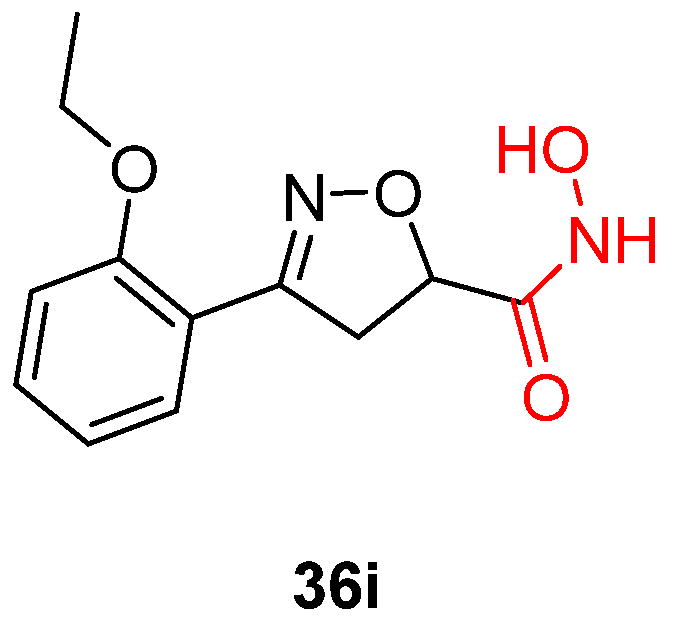{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| α-Hydroxytropolones | |||||
|---|---|---|---|---|---|
 α-hydroxytropolones |  β-thujaplicinol [60] EC50 = 1.0 μΜ CC50 = 25 μΜ TI = 25 |  [60] EC50 = 0.34 μΜ CC50 = 32 μΜ TI = 94 |  [59] EC50 = 0.5 μΜ CC50 = >77 μΜ TI = >154 | ||
| N-hydroxyimides | |||||
 [48] HID EC50 = 1.4 μΜ CC50 = 99 μΜ TI = 71 |  [55] HNO EC50 = 3.4 μΜ CC50 = 7.1 μΜ TI = 2 |  [55] N-hydroxypyrimidinedione EC50 = 5.5 μΜ CC50 = >100 μΜ TI = >18 | |||
 208 [48] HPD EC50 = 0.69 μΜ CC50 = 15 μΜ TI = 22 |  A23 [56] HPD EC50 = 0.11 μΜ CC50 = 33 μΜ TI = 300 |  A24 [56] HPD EC50 = 0.29 μΜ CC50 = 102 μΜ TI = 352 | |||
| Diketo Acids (DKAs) | |||
|---|---|---|---|
 |  |  | |
 |  |  | |
| α-hydroxytropolones | |||
 | |||
| Hydroxypyrimidine and hydroxypyridone carboxylic acids | |||
 |  |  | |
 | |||
| N-hydroxyimides | |||
 |  |  | |
 | |||
| N-hydroxypyrimidinediones (HPDs) | |||
 |  |  | |
| N-hydroxynaphthyridine (HNOs) | |||
 |  |  | |
| N-hydroxypyridopyrimidinones | N-hydroxypyridopyrazines | ||
 |  | ||
| N′-acylhydrazones | |||
 |  | ||
| 5-nitro-furan-2-carboxylic acid derivatives | |||
 |  | ||
 |  |  | |
 |  |  | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moianos, D.; Prifti, G.-M.; Makri, M.; Zoidis, G. Targeting Metalloenzymes: The “Achilles’ Heel” of Viruses and Parasites. Pharmaceuticals 2023, 16, 901. https://doi.org/10.3390/ph16060901
Moianos D, Prifti G-M, Makri M, Zoidis G. Targeting Metalloenzymes: The “Achilles’ Heel” of Viruses and Parasites. Pharmaceuticals. 2023; 16(6):901. https://doi.org/10.3390/ph16060901
Chicago/Turabian StyleMoianos, Dimitrios, Georgia-Myrto Prifti, Maria Makri, and Grigoris Zoidis. 2023. "Targeting Metalloenzymes: The “Achilles’ Heel” of Viruses and Parasites" Pharmaceuticals 16, no. 6: 901. https://doi.org/10.3390/ph16060901
APA StyleMoianos, D., Prifti, G.-M., Makri, M., & Zoidis, G. (2023). Targeting Metalloenzymes: The “Achilles’ Heel” of Viruses and Parasites. Pharmaceuticals, 16(6), 901. https://doi.org/10.3390/ph16060901