
Design, Synthesis, Biological Evaluation, and Molecular Docking Study of 4,6-Dimethyl-5-aryl/alkyl-2-[2-hydroxy-3-(4-substituted-1-piperazinyl)propyl]pyrrolo[3,4-c]pyrrole-1,3(2H,5H)-diones as Anti-Inflammatory Agents with Dual Inhibition of COX and LOX

 ,
,  ,
, 
Abstract
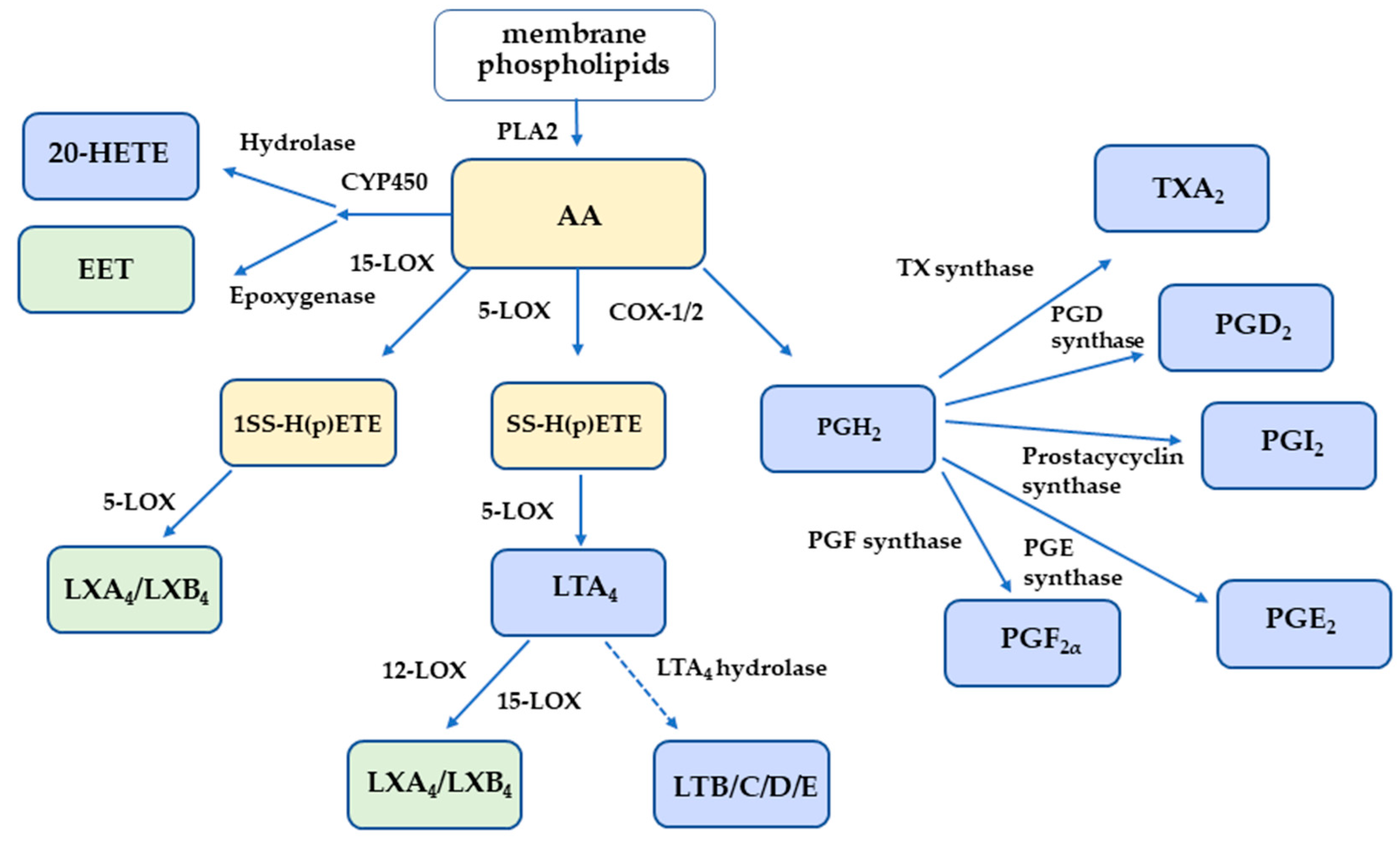
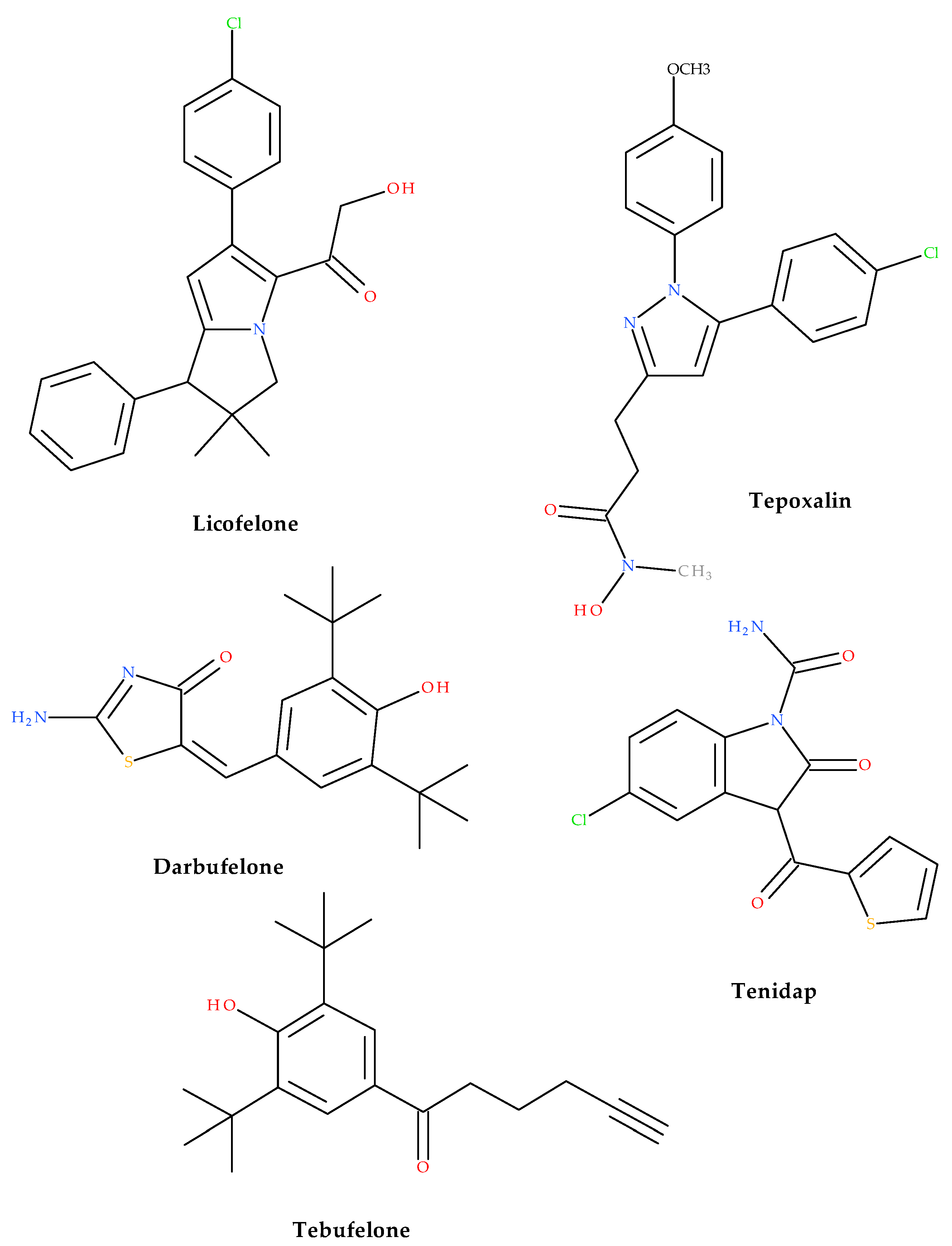
1. Introduction
2. Results and Discussion
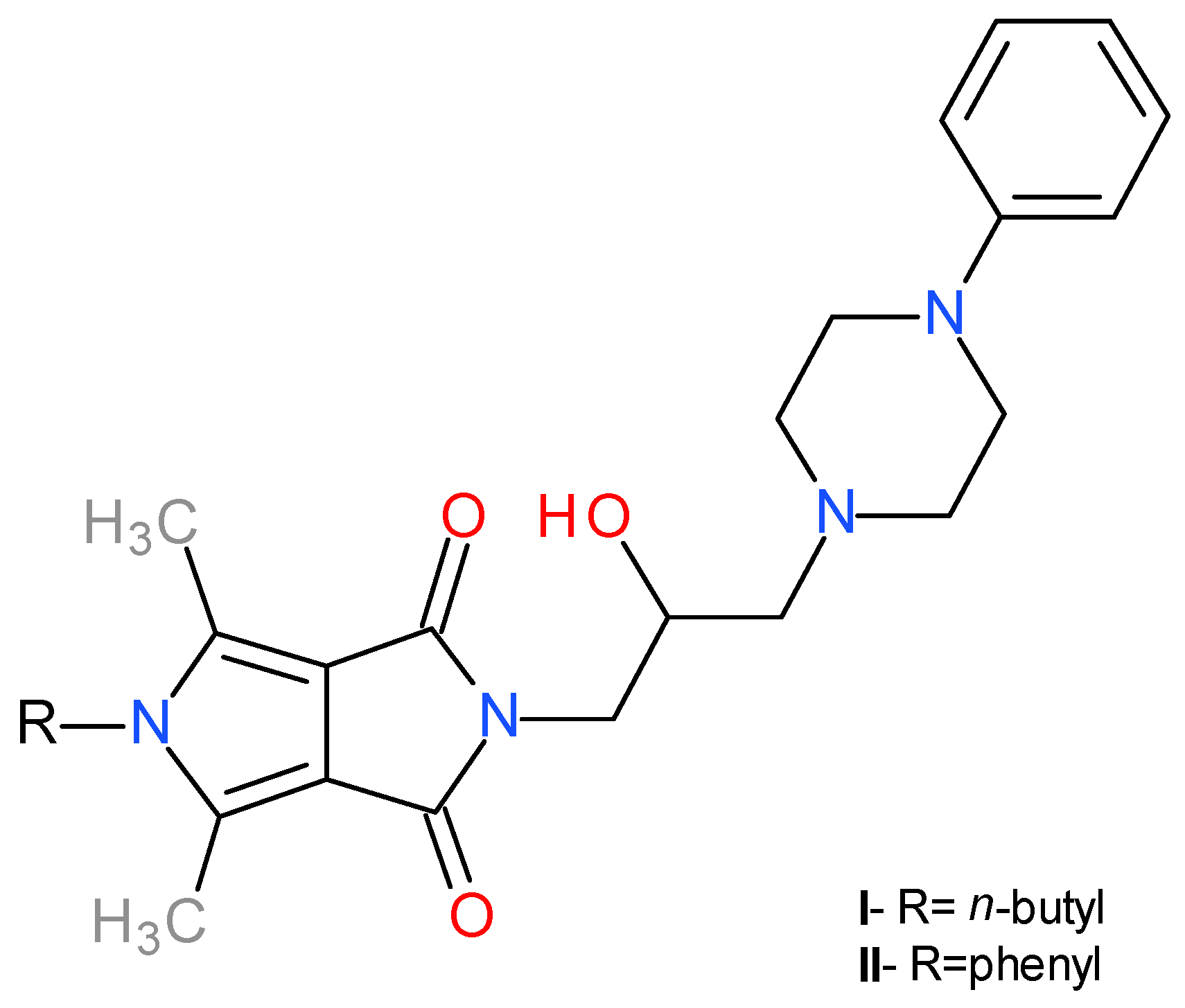
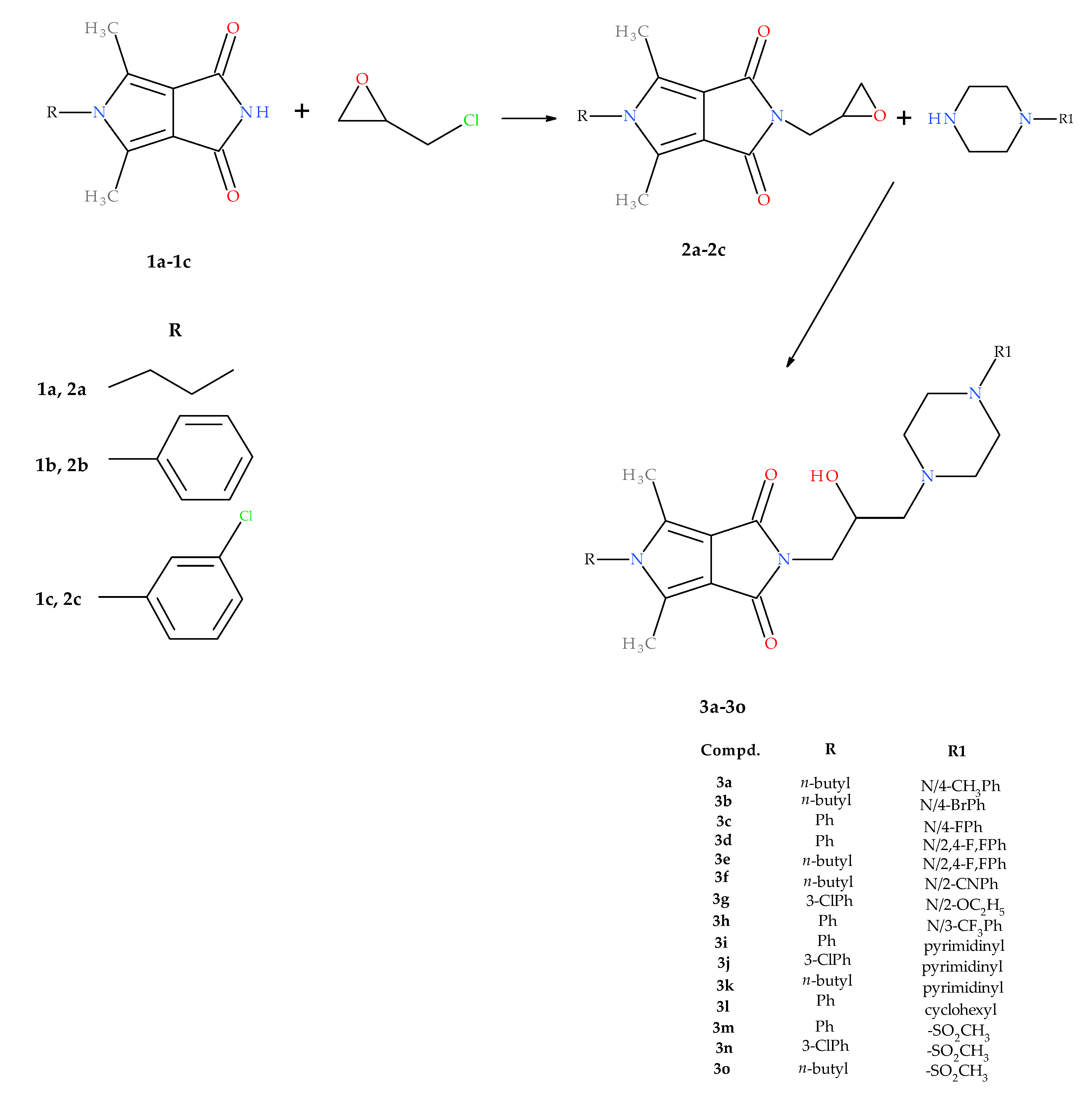
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. Cell Viability
2.2.2. COX-1 and COX-2 Inhibition Assay Results
2.2.3. LOX Inhibition Assay Results
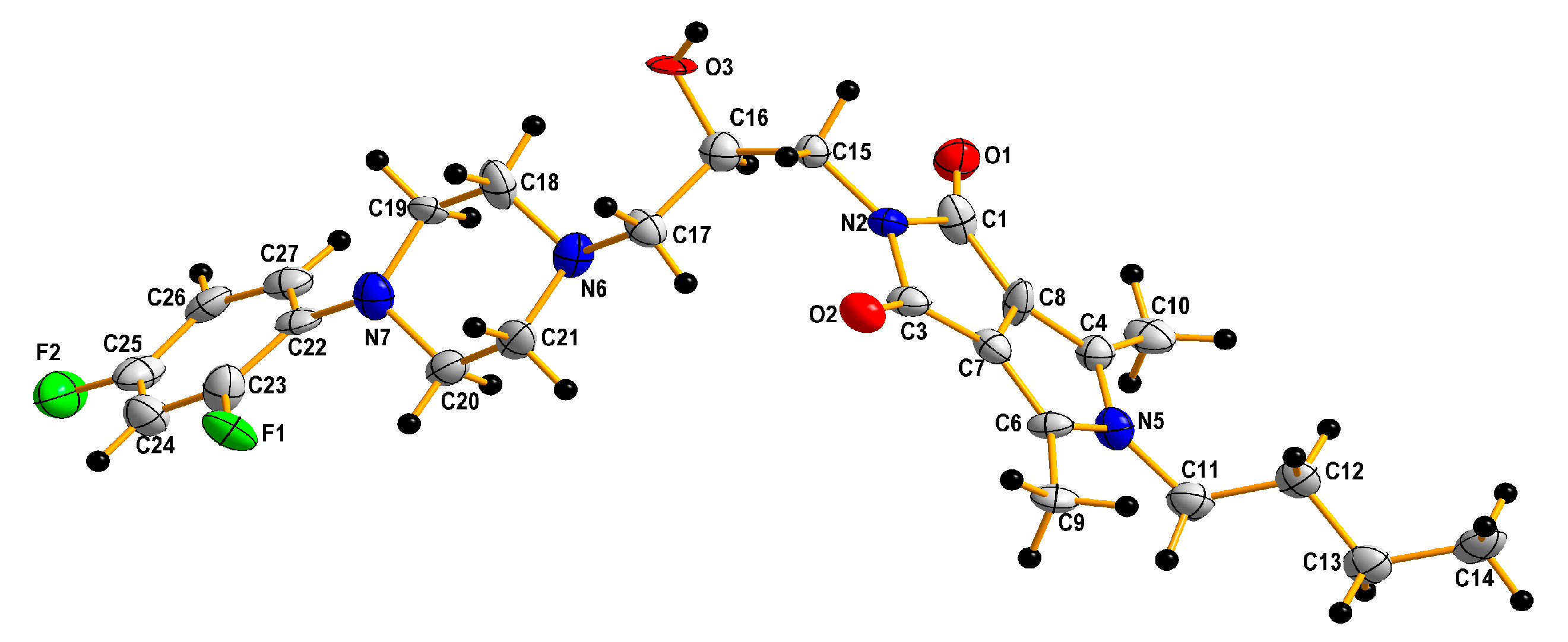
2.3. Computational Studies
2.3.1. The Analysis of Physicochemical and Pharmacokinetic Properties of Designed Compounds
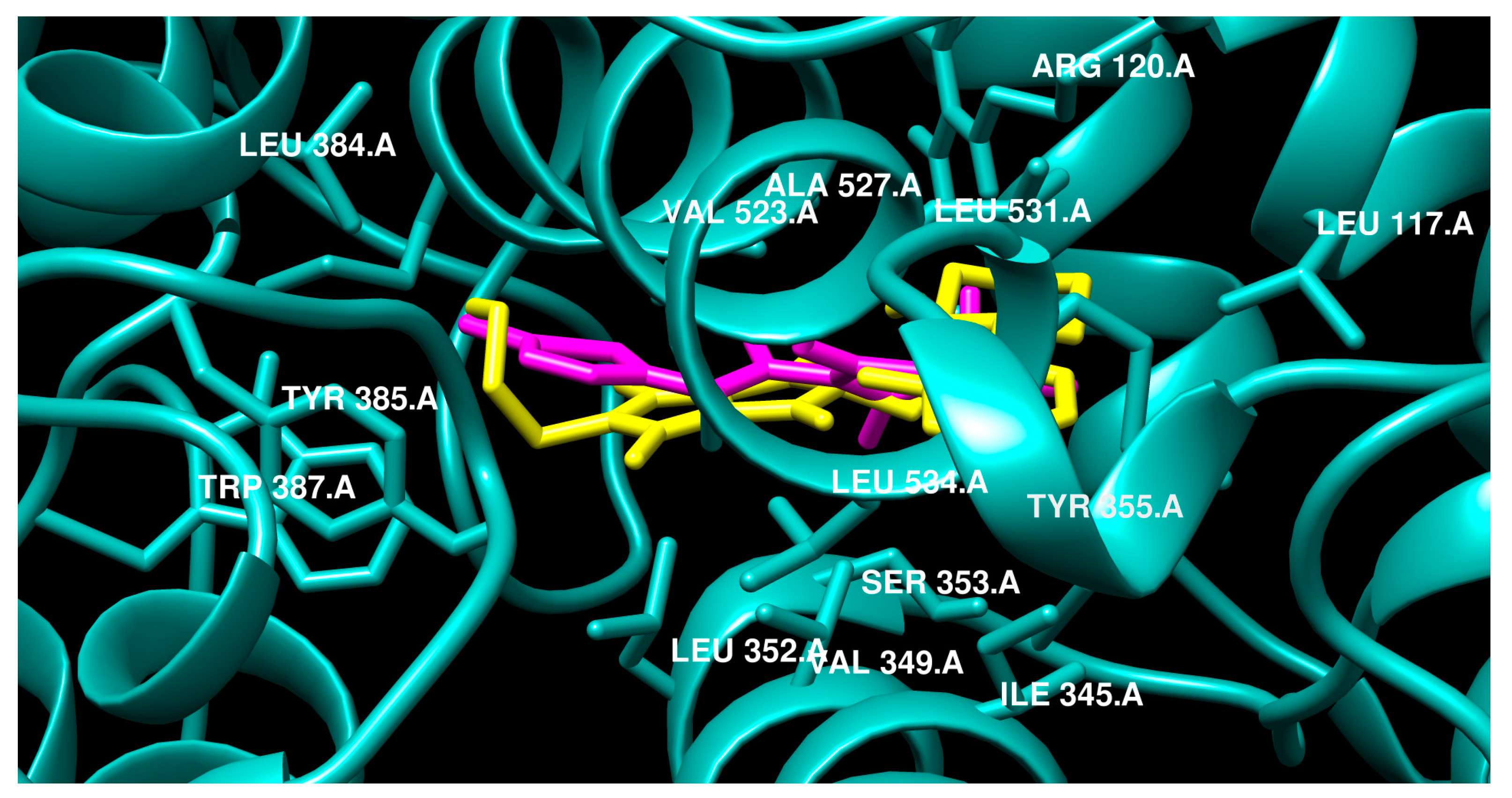
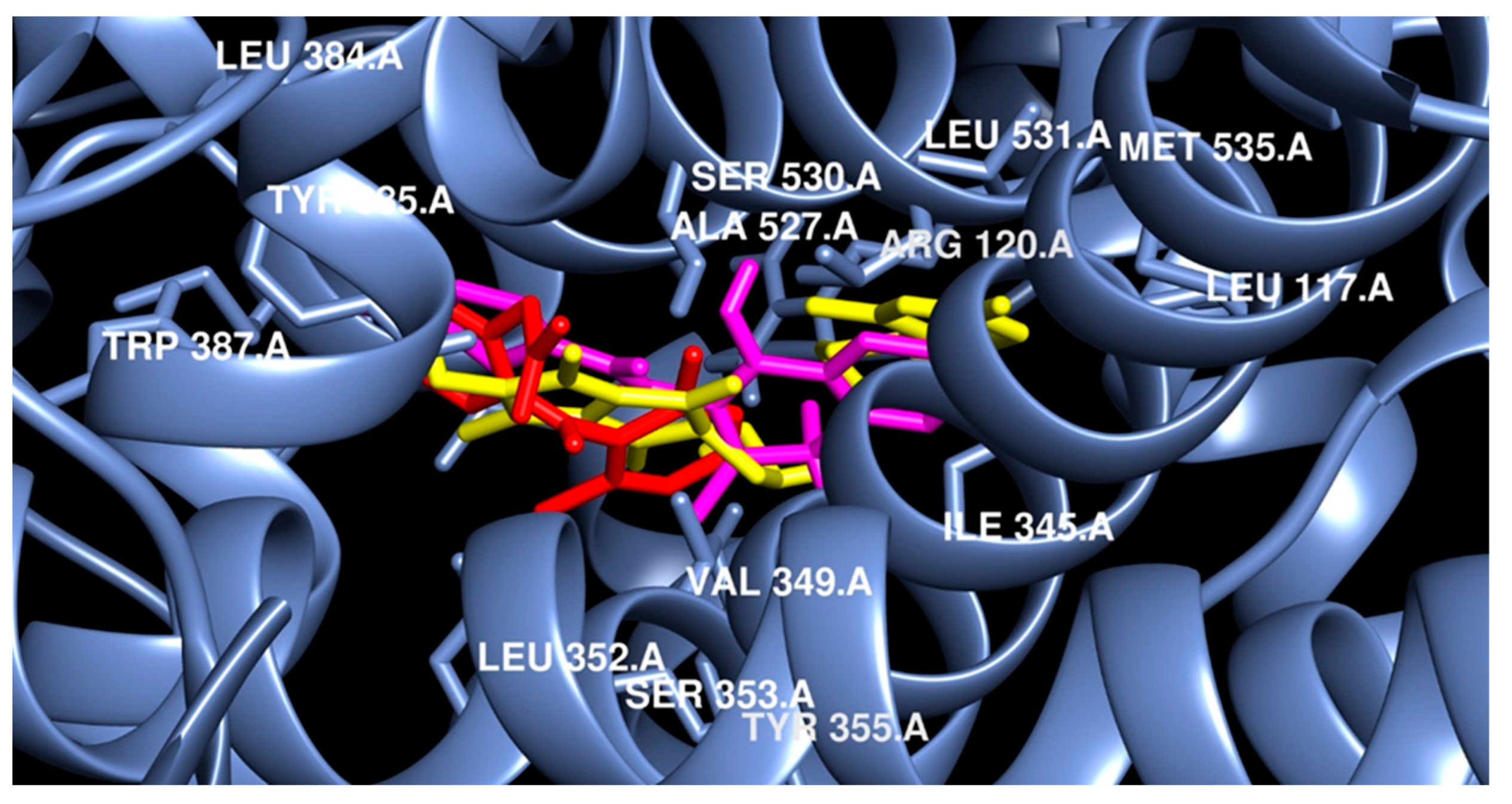
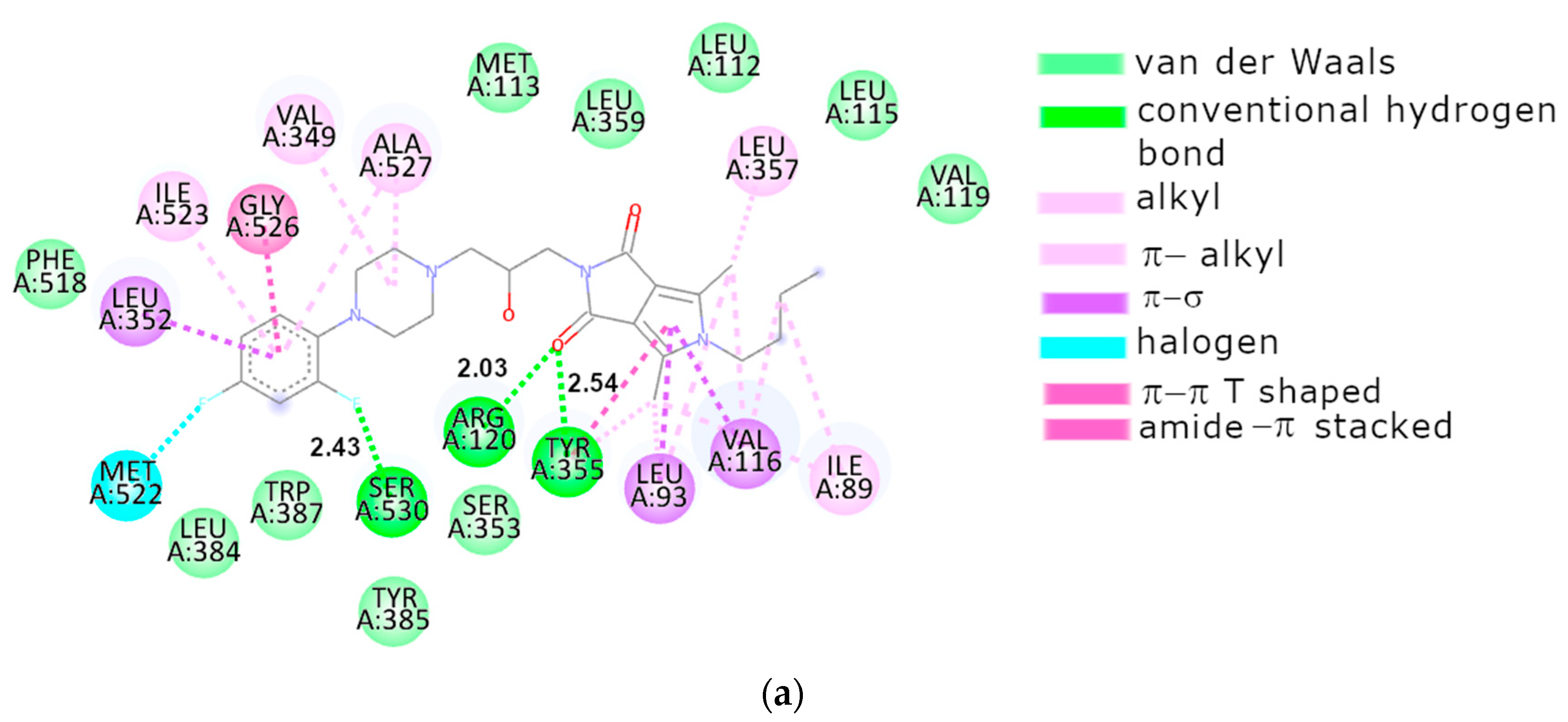
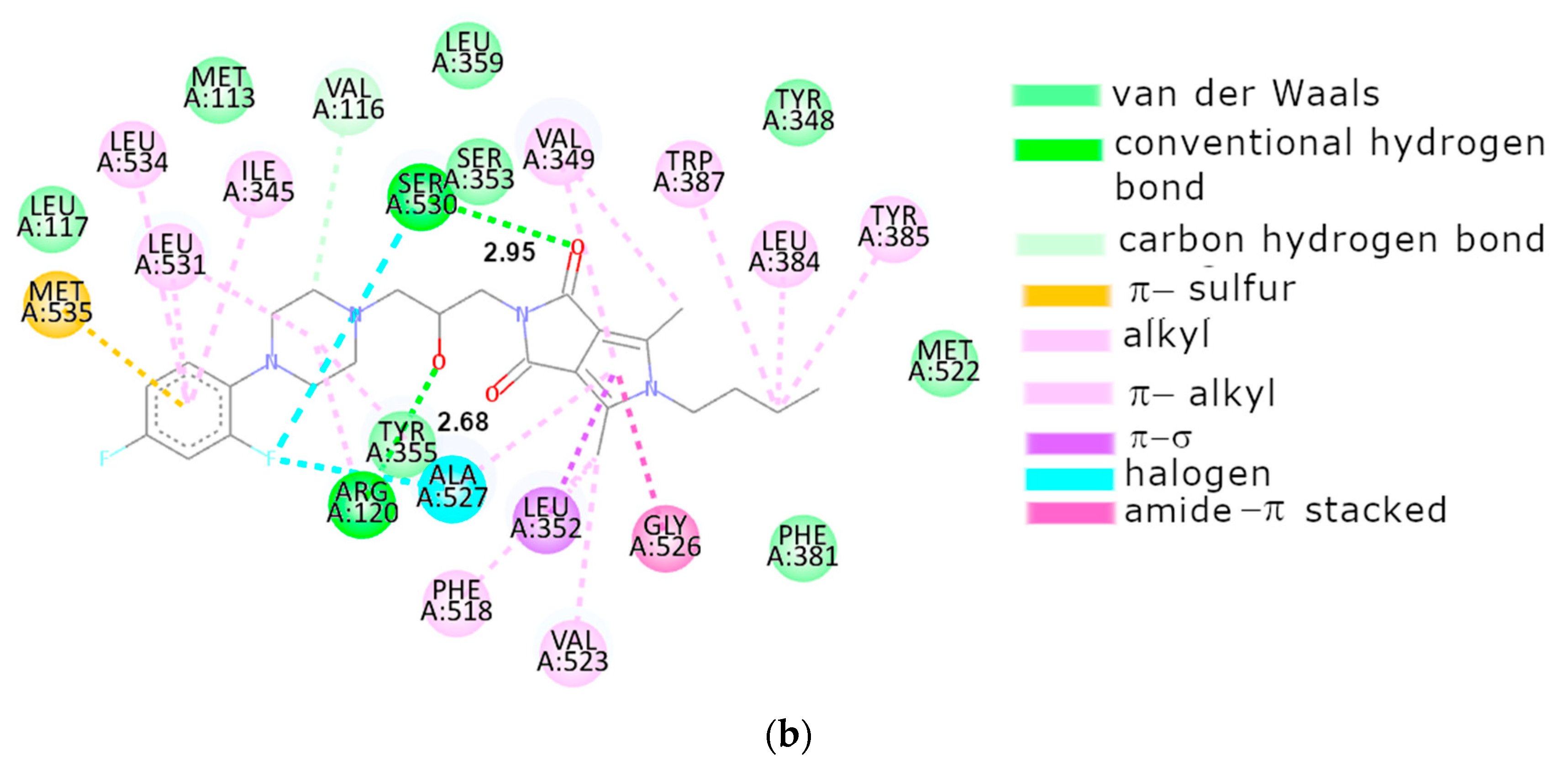
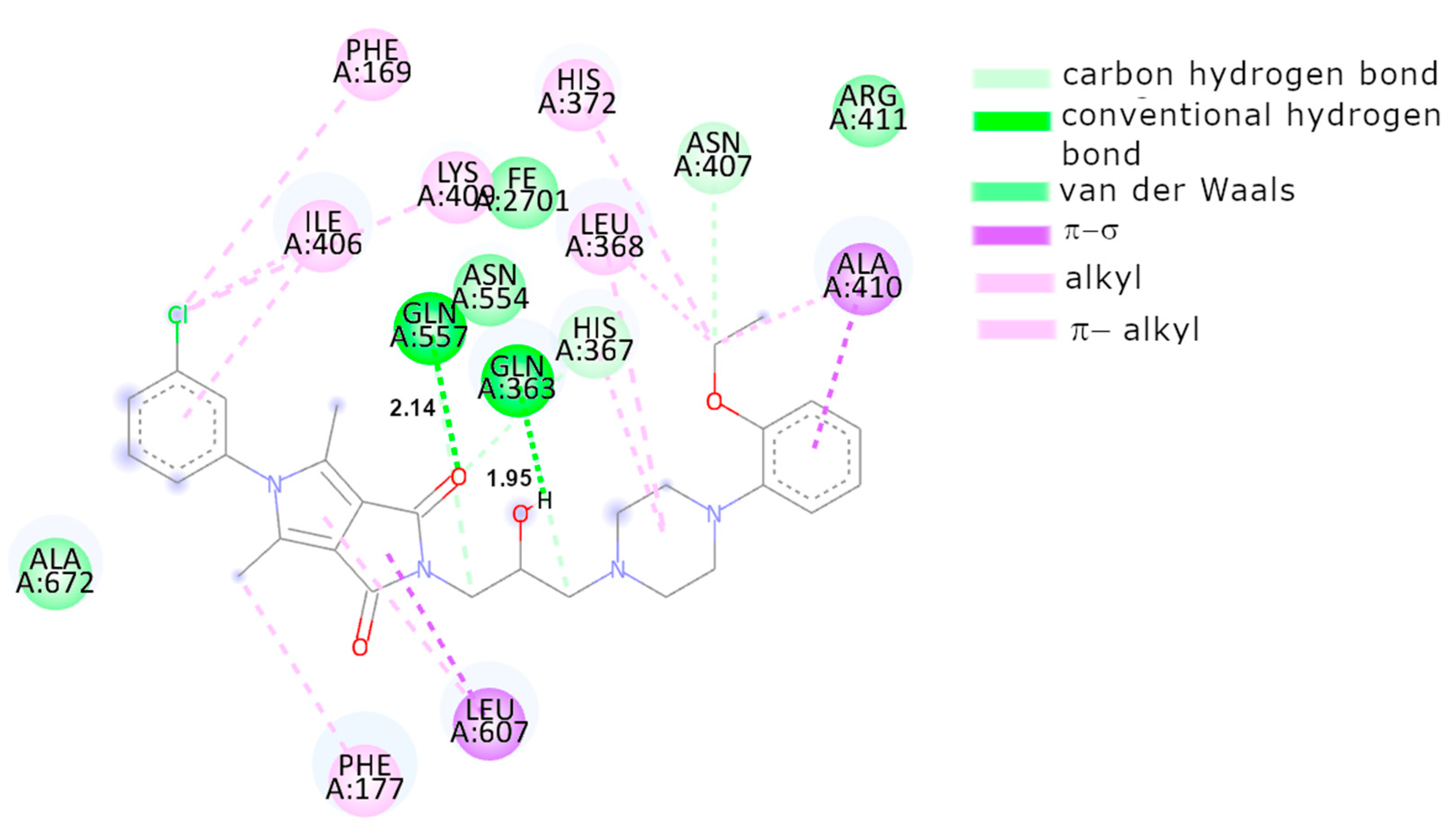
2.3.2. Molecular Docking
3. Discussion
4. Materials and Methods
4.1. Chemicals and Instruments
4.1.1. Synthesis of Compounds 2a–2c
- 2c: Yield 80%, m.p. 140–142 °C.
- 1H NMR (300 MHz, CDCl3) δ: 1.58 (s, 6H, 2,5xCH3), 2.69–2.71 (m, 1H, CH2), 2.78–2.80 (m, 1H, CH2), 3.11–3.22 (m, 1H, CH-oxiran), 3.63–3.70 (dd, 1H, CH2-oxiran), 3.87–3.93 (dd, 1H, CH2-oxiran), 7.13–7.15 (m, 1H, ArH), 7.26–7.28 (m, 1H, ArH), 7.52–7.54 (m, 2H, ArH)
- 13C NMR (75 MHz, CDCl3) δ: 165.64, 130.92, 129.99, 128.21, 126.21, 49.48, 46.20, 39.14, 11.86
- FT-IR (selected lines, γmax, cm−1): 1700 (C=O), 1760 (C=O)
- ESI-MS (m/z): calcd. for C26H37N4O3 [M+H]+: 453.5970; found: 453.2871
4.1.2. General Procedure for Preparation of 1-Substituted-2,5-dimethyl-N-[2-hydroxy-3-(4-substituted piperazine-1-yl)propyl]-3,4-pyrroledicarboximides 3a–3o
- 3a: from 2a and 1-(p-methylphenyl)piperazine. Yield 57%, m.p. 116–118 °C.
- 1H NMR (300 MHz, CDCl3) δ: 0.97 (t, 3H, CH3-butyl, J = 7.5 Hz), 1.35–1.42 (m, 2H, CH2-butyl), 1.60–1.65 (m, 2H, CH2-butyl), 2.26 (s, 3H, ArCH3), 2.39 (s, 6H, 4,6-CH3), 2.45 (d, 2H, CH2, Hγ-propyl, J = 6.6 Hz), 2.58–2.60 (m, 2H, CH2-piperazine), 2.75–2.76 (m, 2H, CH2-piperazine), 3.10–3.20 (m, 4H, N(CH2)2Ar), 3.64–3.67 (m, 2H, Npyrrole-CH2), 3.72–3.77 (m, 2H, CH2Hα-propyl), 398–4.02 (m, 1H, CH-Hβ-propyl), 6.81 (d, 2H ArH, J = 8.7 Hz), 7.04 (d, 2H, ArH, J = 8.4 Hz)
- 13C NMR (75 MHz, CDCl3) δ: 165.64, 149.16, 129.60, 129.20, 128.71, 116.37, 116.21, 65.84, 61.74, 53.32, 49.77, 43.83, 41.66, 32.46, 20.40, 20.02, 13.69, 11.37
- FT-IR (selected lines, γmax, cm−1): 1700 (C=O), 1760 (C=O), 3110 (OH)
- ESI-MS (m/z): calcd. for C26H37N4O3 [M+H]+: 453.5970; found: 453.2871
- 3b: from 2a and 1-(p-bromophenyl)piperazine. Yield 75%, m.p. 180–182 °C.
- 1H NMR (300 MHz, CDCl3) δ: 0.97 (t, 3H, CH3-butyl, J = 7.5 Hz), 1.34–1.42 (m, 2H CH2-butyl,), 1.57–1.64 (m, 2H, CH2-butyl,), 2.38 (s, 6H, 4,6-CH3), 2.45 (d, 2H, CH2 Hγ-propyl, J = 6.6 Hz), 2.55–2.59 (m, 2H, CH2-piperazine), 2.73–2.76 (m, 2H, CH2-piperazine), 3.12–3.15 (m, 4H, N(CH2)2Ar), 3.65–3.67 (m, 2H, Npyrrole-CH2), 3.72 (t, 2H, CH2 Hα-propyl J = 7.8 Hz), 3.98–4.02 (m, 1H, CH Hβ-propyl), 6.75 (d, 2H, ArH, J = 9 Hz), 7.30 (d, 2H, ArH, J = 9 Hz)
- 13C NMR (75 MHz, CDCl3) δ: 165.66, 150.26, 131.82, 128.76, 117.57, 116.19, 111.75, 66.06, 61.71, 53.13, 49.03, 43.83, 41.71, 32.46, 20.02, 13.68, 11.37
- FT-IR (selected lines, γmax, cm−1): 1690 (C=O), 1760 (C=O), 3110 (OH)
- ESI-MS (m/z): calcd. for C25H34BrN4O3 [M+H]+: 517.1814; found: 517.1789
- 3c: from 2b and 1-(p-fluorophenyl)piperazine. Yield 72%, m.p. 182–184 °C.
- 1H NMR (300 MHz, CDCl3) δ: 2.17 (s, 6H, 4,6–CH3), 2.49 (d, 2H, CH2 Hγ-propyl, J = 6.9 Hz), 2.59–2.62 (m, 2H, CH2-piperazine), 2.77–2.78 (m, 2H, CH2-piperazine), 3.09–3.10 (m, 4H, N(CH2)2Ar -), 3.70–3.73 (m, 2H, CH2 Hα-propyl), 4.03–4.07 (m, 1H, CH Hβ-propyl), 6.84–6.88 (m, 2H, ArH), 6.92–6.98 (m, 2H, ArH), 7.17–7.26 (m, 2H, ArH), 7.53–7.55 (m, 3H, ArH)
- 13C NMR (75 MHz, CDCl3) δ165.62, 158.74, 155.58, 147.94, 135.95, 130.06, 129.89, 127.83, 117.81, 117.71, 116.48, 115.62, 115.33, 65.88, 61.72, 53.32, 50.22, 41.82, 11.89
- FT-IR (selected lines, γmax, cm−1): 1680 (C=O), 1750 (C=O), 3110 (OH)
- ESI-MS (m/z): calcd. for C27H30FN4O3 [M+H]+: 477.5505; found: 477.2303
- 3d: from 2a and 2,4-difluorophenylpiperazine. Yield 69%, m.p. 143–145 °C.
- 1H NMR (300 MHz, CDCl3) δ: 2.20 (s, 6H, 4,6–CH3), 2.51–2.53 (m, 2H, CH2 Hγ-propyl), 2.62–2.67 (m, 2H, CH2-piperazine), 2.79–2.85 (m, 2H, CH2-piperazine), 3.00–3.15 (m, 4H, N(CH2)2Ar), 3.71–3.74 (m, 2H, CH2 Hα-propyl), 4.01–4.10 (m, 1H, CH Hβ-propyl), 6.78–6.95 (m, 3H, ArH), 7.20–7.28 (m, 2H, ArH), 7.54–7.57 (m, 3H, ArH)
- 13C NMR (75 MHz, CDCl3) δ: 165.62, 135.97, 130.07, 129.89, 129.56, 127.84, 119.52, 116.48, 110.52, 105.01, 104.68, 65.87, 61.72, 53.36, 50.99, 41.80, 11.89
- FT-IR (selected lines, γmax, cm−1): 1695 (C=O), 1750 (C=O), 3100 (OH)
- ESI-MS (m/z): calcd. for C27H29F2N4O3 [M+H]+: 495.5410; found: 495.2221
- 3e: from 2a and 2,4-difluorophenylpiperazine. Yield 76%, m.p. 139–141 °C.
- 1H NMR (300 MHz, CDCl3) δ: 0.97 (t, 3H, CH3-butyl, J = 7.2 Hz), 1.35–1.42 (m, 2H, CH2-butyl,), 1.57–1.65 (m, 2H, CH2-butyl), 2.39 (s, 6H, 4,6-CH3), 2.46 (d, 2H, CH2, Hγ-propyl, J = 6.3 Hz), 2.60–2.62 (m, 2H, CH2-piperazine), 2.75–2.77 (m, 2H, CH2-piperazine), 3.00–3.10 (m, 4H, N(CH2)2Ar), 3.65–3.67 (m, 2H, Npyrrole-CH2), 3.72–3.77 (m, 2H, CH2 Hα-propyl), 3.97–4.01 (m, 1H, CH Hβ-propyl), 6.75–6.84 (m, 2H ArH), 6.87–6.92 (m, 1H, ArH)
- 13C NMR (75 MHz, CDCl3) δ: 165.65, 136.64, 128.74, 119.50, 119.32, 116.20, 110.73, 110.45, 104.99, 104.66, 104.31, 65.96, 61.70, 53.35, 50.97, 43.83, 41.70, 32.47, 20.02, 13.68, 11.36
- FT-IR (selected lines, γmax, cm−1): 1700 (C=O), 1760 (C=O), 3140 (OH)
- ESI-MS (m/z): calcd. for C25H33F2N4O3 [M+H]+: 475.5513; found: 475.2527
- 3f: from 2a and 1-(o-cyjanophenyl)piperazine. Yield 79%, m.p. 154–156 °C.
- 1H NMR (300 MHz, CDCl3) δ: 0.98 (t, 3H, CH3-butyl, J = 7.5 Hz), 1.26–1.43 (m, 2H CH2-butyl,), 1.60–1.63 (m, 2H, CH2-butyl,), 2.39 (s, 6H, 4,6-CH3), 2.48 (d, 2H, CH2, Hγ-propyl, J = 6.6 Hz), 2.51–2.67 (m, 2H, CH2-piperazine), 2.80–2.82 (m, 2H, CH2-piperazine), 3.15–3.20 (m, 4H, N(CH2)2Ar), 3.66 (d, 2H, Npyrrole-CH2 J = 4.5 Hz), 3.75 (t, 2H, CH2 Hα-propyl, J = 7.8 Hz), 3.99–4.00 (m, 1H, CH-Hβ-propyl), 6.97–7.02 (m, 2H, ArH), 7.47 (t, 1H, ArH, J = 7.5 Hz), 7.53 (d, 1H, ArH, J = 7.8 Hz)
- 13C NMR (75 MHz, CDCl3) δ: 165.64, 155.64, 134.33, 133.74, 128.77, 121.73, 118.67, 118.39, 166.19, 106.05, 66.02, 61.66, 53.29, 51.59, 43.84, 41.65, 32.47, 20.02, 13.69, 11.37
- FT-IR (selected lines, γmax, cm−1): 1700 (C=O), 1760 (C=O), 3110 (OH)
- ESI-MS (m/z): calcd. for C26H34N5O3 [M+H]+: 464.5799; found: 464.2637
- 3g: from 2c and 1-(o-ethoxyphenyl)piperazine. Yield 65%, m.p. 129–131 °C.
- 1H NMR (300 MHz, CDCl3) δ: 1.42 (t, 3H, CH2CH3O), 2.19 (s, 6H, 4,6–CH3), 2.48–2.51 (m, 2H, CH2, Hγ-propyl), 2.60–2.65 (m, 2H, CH2-piperazine), 2.75–2.83 (m, 2H, CH2-piperazine), 3.01–3.15 (m, 4H, N(CH2)2Ar), 3.68–3.73 (m, 2H, CH2), 4.02–4.09 (m, 3H, CH+CH2 Hα-propyl+-Hβ-propyl), 6.83–6.85 (m, 1H, ArH), 6.90–6.93 (m, 3H, ArH), 7.11–7.13 (m, 1H, ArH), 7.20–7.24 (m, 1H, ArH), 7.49–7.52 (m, 2H, ArH)
- 13C NMR (75 MHz, CDCl3) δ: 165.37, 151.56, 141.32, 137.12, 135.60, 130.89, 129.94, 129.76, 128.22, 126.23, 122.73, 120.99, 118.15, 116.84, 112.53, 65.51, 63.56, 61.80, 53.54, 50.65, 41.80, 14.92, 11.87
- FT-IR (selected lines, γmax, cm−1): 1680 (C=O), 1750 (C=O), 3010 (OH)
- ESI-MS (m/z): calcd. for C29H34ClN4O4 [M+H]+: 537.2268; found: 537.2248
- 3h: from 2b and 1-(m-trifluoromethylphenyl)piperazine. Yield 77%, m.p. 135–138 °C.
- 1H NMR (300 MHz, CDCl3) δ: 2.17 (s, 6H, 4,6–CH3), 2.49 (d, 2H, CH2 Hγ-propyl, J = 6.6 Hz), 2.61–2.62 (m, 2H, CH2-piperazine), 2.70–2.79 (m, 2H, CH2-piperazine), 3.17–3.20 (m, 4H, N(CH2)2Ar), 3.70–3.73 (m, 2H, CH2 Hα-propyl), 4.00–4.10 (m, 1H, CH Hβ-propyl), 6.85–6.90 (m, 2H, ArH), 6.91–6.93 (m, 2H, ArH), 7.17–7.23 (m, 2H, ArH), 7.26–7.28 (m, 2H, ArH), 7.53–7.55 (m, 3H ArH)
- FT-IR (selected lines, γmax, cm−1): 1680 (C=O), 1750 (C=O), 3090 (OH)
- ESI-MS (m/z): calcd. for C28H30F3N4O3 [M+H]+: 527.5580; found: 527.8769
- 4,6-dimethyl-5-phenyl-2-[2-hydroxy-3-(4-pyrimidynylpiperazin-1-yl)propyl]pyrrolo[3,4-c]pyrrole-1,3(2H,5H)-dione (3i).
- 3i: from 2b and 1-pyrimidynylpiperazine. Yield 65%, m.p. 165–167 °C.
- 1H NMR (300 MHz, CDCl3) δ: 2.17 (s, 6H, 4,6–CH3), 2.48–2.50 (m, 4H, N(CH2)2Ar), 2.65–2.70 (m, 2H, CH2 Hα-propyl), 3.70–3.74 (m, 2H, CH2 Hβ-propyl), 3.80–3.90 (m, 2H, CH2), 4.00–4.09 (m, 1H, CH), 6.47–6.50 (m, 1H, ArH), 7.19–7.22 (m, 2H, ArH), 7.53–7.56 (m, 3H, ArH), 8.29 (d, 2H, ArH, J = 4.8 Hz)
- 13C NMR (75 MHz, CDCl3) δ: 165.61, 161.70, 157.69, 130.06, 129.89, 129.55, 127.84, 116.48, 109.88, 65.85, 61.92, 53.20, 43.79, 41.76, 11.88
- FT-IR (selected lines, γmax, cm−1): 1680 (C=O), 1750 (C=O), 3490 (OH)
- ESI-MS (m/z): calcd. for C25H29N6O3 [M+H]+: 461.5362; found: 461.2289
- 3j: from 2c and 1-pyrimidynylpiperazine. Yield 72%, m.p. 179–181 °C.
- 1H NMR (300 MHz, CDCl3) δ: 2.19 (s, 6H, 4,6–CH3), 2.47–2.52 (m, 4H, 2xCH2Hγpropyl+piperazine), 2.67–2.69 (m, 2H, CH2), 3.69–3.73 (m, 2H, Npyrrole-CH2), 3.80–3.81 (m, 4H, CH2 piperazine+CH2 Hα-propyl), 4.00–4.06 (m, 1H, CH Hβ-propyl), 6.48 (t, 2H, ArH, J = 4.8 Hz), 7.11–7.13 (m, 1H, ArH), 7.24–7.25 (m, 1H, ArH), 7.50–7.52 (m, 2H, ArH), 8.29 (d, 2H, ArH, J = 4.5 Hz)
- 13C NMR (75 MHz, CDCl3) δ: 165.38, 157.69, 137.09, 135.61, 130.90, 129.96, 129.84, 128.21, 126.21, 116.78, 109.91, 65.75, 61.90, 53.20, 43.77, 41.77, 11.86
- FT-IR (selected lines, γmax, cm−1): 1680 (C=O), 1750 (C=O), 3350 (OH)
- ESI-MS (m/z): calcd. for C25H28ClN6O3 [M+H]+: 495.9812; found: 495.1909
- 3k: from 2a and 1-pyrimidynylpiperazine. Yield 65%, m.p. 182–184 °C.
- 1H NMR (300 MHz, CDCl3) δ: 0.97 (t, 3H, CH3-butyl J = 7.2 Hz), 1.24–1.42 (m, 2H, CH2-butyl), 1.57–1.65 (m, 2H, CH2-butyl), 2.38 (s, 6H, 4,6-CH3), 2.44–2.49 (m, 4H, N(CH2)2Ar,), 2.64–2.68 (m, 2H, CH2-piperazine), 3.64–3.67 (m, 2H, CH2 Hα-propyl), 3.72–3.81 (m, 4H, CH2-piperazine + Npyrrole-CH2), 4.01–4.06 (m, 1H, CH Hβ-propyl), 6.47 (t, 1H, ArH, J = 4.8 Hz)), 7.11–7.13 (m, 1H, ArH), 7.50–7.52 (m, 2H, ArH), 7.24–7.26 (m, 1H, ArH), 8.28 (d, 2H, ArH, J = 7.8 Hz)
- 13C NMR (75 MHz, CDCl3) δ: 165.63, 161.67, 157.67, 128.73, 116.20, 109.86, 65.93, 61.91, 53.19, 43.83, 43.76, 41.67, 32.46, 20.01, 13.68, 11.36
- FT-IR (selected lines, γmax, cm−1): 1680 (C=O), 1750 (C=O), 3350 (OH)
- ESI-MS (m/z): calcd. for C23H33N6O3 [M+H]+: 441.5465; found: 441.2597
- 3l: from 2a and 1-cyclohexylpiperazine. Yield 70%, m.p. 111–113 °C.
- 1H NMR (300 MHz, CDCl3) δ: 1.19–1.22 (m, 5H, CH2), 1.35 (t, 2H, CH2, J = 6.9 Hz), 1.55–1.64 (m, 2H, CH2), 1.75–1.80 (m, 2H, CH2), 1.81–1.90 (m, 2H, CH2), 2.20 (s, 6H, 4,6–CH3), 2.38–2.43 (m, 4H, N(CH2)2Ar), 2.65–2.70 (m, 5H, CH2CH2CH), 3.64–3.69 (m, 2H, CH2 Hα-propyl), 3.90–4.05 (m, 1H, CH Hβ-propyl), 4.32–4.34 (m, 1H, CH), 7.16–7.22 (m, 2H, ArH), 7.49–7.55 (m, 3H, ArH)
- 13C NMR (75 MHz, CDCl3) δ: 165.56, 136.00, 129.96, 129.87, 129.65, 129.51, 127.85, 116.51, 66.60, 65.56, 63.51, 61.68, 53.76, 49.03, 41.77, 28.94, 26.28, 25.85, 13.62, 12.84, 11.88
- FT-IR (selected lines, γmax, cm−1): 1680 (C=O), 1760 (C=O), 3310 (OH)
- ESI-MS (m/z): calcd. for C27H37N4O3 [M+H]+: 465.6077 found: 465.2863
- 3m: from 2b and N-methylsulphonylpiperazine. Yield 72%, m.p. 136–138 °C.
- 1H NMR (300 MHz, CDCl3) δ: 2.15 (s, 6H, 4,6–CH3), 2.46 (d, 2H, CH2 Hγ-propyl, J = 6.6 Hz), 2.54–2.60 (m, 2H, CH2-piperazine), 2.68–2.71 (m, 2H, CH2-piperazine), 2.76 (s, 3H, CH3SO2), 3.21–3.24 (m, 4H, N(CH2)2Ar), 3.67 (d, 2H, CH2Hα-propyl J = 5.4 Hz), 3.97–4.00 (m, 1H, CH Hβ-propyl), 7.18–7.21 (m, 2H, ArH), 7.52–7.54 (m, 3H, ArH)
- 13C NMR (75 MHz, CDCl3) δ: 165.66, 135.85, 130.24, 129.92, 129.61, 127.79, 116.31, 65.55, 61.47, 52.68, 45.92, 41.87, 34.22, 11.89
- FT-IR (selected lines, γmax, cm−1): 1320 (SO2), 1680 (C=O), 1750 (C=O), 3155(OH)
- ESI-MS (m/z): calcd. for C22H29N4O5S [M+H]+: 461.5544; found: 461.1846
- 3n: from 2c and N-methylsulphonylpiperazine. Yield 79%, m.p. 119–122 °C.
- 1H NMR (300 MHz, CDCl3) δ: 2.18 (s, 6H, 4,6-CH3), 2.47 (d, 2H, CH2 Hγ-propyl, J = 6.6 Hz), 2.55–2.59 (m, 2H, CH2-piperazine), 2.68–2.73 (m, 2H, CH2-piperazine), 2.78 (s, 3H, CH3SO2), 3.24 (t, 4H, N(CH2)2Ar), 3.69 (d, 2H, CH2, J = 5.7 Hz), 3.75 (t, 2H, CH2 Hα-propyl, J = 7.8 Hz), 3.95–4.05 (m, 1H, CH Hβ-propyl), 7.11–7.13 (m, 1H, ArH), 7.23–7.24 (m, 1H, ArH), 7.50–7.53 (m, 2H, ArH)
- 13C NMR (75 MHz, CDCl3) δ: 165.48, 136.98, 135.68, 131.01, 130.08, 128.21, 126.18, 116.65, 66.51, 61.48, 52.68, 41.92, 34.26, 11.88
- FT-IR (selected lines, γmax, cm−1): 1320 (SO2), 1580 (C=O), 1690 (C=O),3350 (OH)
- ESI-MS (m/z): calcd. for C22H28ClN4O5S [M+H]+: 495.9995; found: 495.1466
- 3o: from 2a and N-methylsulphonylpiperazine. Yield 75%, m.p. 139–141 °C.
- 1H NMR (300 MHz, CDCl3) δ: 0.97 (t, 3H, CH3-butyl, J = 7.2 Hz), 1.37–1.42 (m, 2H, CH2-butyl), 1.60–1.65 (m, 2H, CH2-butyl), 2.38 (s, 6H, 4,6-CH3), 2.44 (d, 2H, CH2 Hγ-propyl, J = 6.6 Hz), 2.55–2.59 (m, 2H, CH2-piperazine), 2.66–2.70 (m, 2H, CH2-piperazine), 2.76 (s, 3H, CH3SO2), 3.22 (t, 4H, N(CH2)2Ar), 3.64 (d, 2H, Npyrrole-CH2 J = 5.1 Hz), 3.75 (t, 2H, CH2 Hα-propyl, J = 7.5 Hz), 3.90–4.00 (m, 1H, CH Hβ-propyl)
- 13C NMR (75 MHz, CDCl3) δ: 165.73, 128.97, 116.07, 66.68, 61.43, 52.67, 45.91, 43.87, 41.78, 34.19, 32.45, 20.01, 13.68, 11.37
- FT-IR (selected lines, γmax, cm−1): 1320 (SO2), 1690 (C=O), 1750 (C=O), 3380 (OH)
- ESI-MS (m/z): calcd. for C20H33N4O5S [M+H]+: 441.5681; found: 441.2178
4.2. Materials and Methods of Biological Evaluation
4.2.1. Cell Line and Culture Medium
4.2.2. Tested Compounds
4.2.3. SRB Assay
4.2.4. Cyclooxygenase Inhibition Assay
4.2.5. Lipoxygenase Inhibitor Screening Assay
4.3. Molecular Modeling (Methodology)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nathan, C. Points of Control in Inflammation. Nature 2002, 420, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Klil-Drori, A.J.; Ariel, A. 15-Lipoxygenases in cancer: A double-edged sword? Prostaglandins Other Lipid Mediat. 2013, 106, 16–22. [Google Scholar] [CrossRef]
- Willoughby, D.A.; Moore, A.R.; Colville-Nash, P.R. COX-1, COX-2, and COX-3 and the Future Treatment of Chronic Inflammatory Disease. Lancet 2000, 355, 646–648. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, M.; Bekeschus, S.; Weltmann, K.D.; von Woedtke, T.; Wende, K. Non-steroidal anti-inflammatory drugs: Recent advances in the use of synthetic COX-2 inhibitors. RSC Med. Chem. 2022, 13, 471–496. [Google Scholar] [CrossRef]
- Ruiz, J.G.; Lowenthal, D.T. NSAIDS and Nephrotoxicity in the Elderly. Geriatr. Nephrol. Urol. 1997, 7, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Kothayer, H.; Rezq, S.; Abdelkhalek, A.S.; Romero, D.G.; Elbaramawi, S.S. Triple targeting of mutant EGFRL858R/T790M, COX-2, and 15-LOX: Design and synthesis of novel quinazolinone tethered phenyl urea derivatives for anti-inflammatory and anticancer evaluation. J. Enzym. Inhib. Med. Chem. 2023, 38, 2199166. [Google Scholar] [CrossRef]
- Shang, J.L.; Cheng, Q.; Yang, W.F.; Zhang, M.; Cui, Y.; Wang, Y.F. Possible roles of COX-1 in learning and memory impairment induced by traumatic brain injury in mice. Braz. J. Med. Biol. Res. 2014, 47, 1050–1056. [Google Scholar] [CrossRef]
- Cowley, T.R.; Fahey, B.; O’Mara, S.M. COX-2, but not COX-1, activity is necessary for the induction of perforant path long-term potentiation and spatial learning in vivo. Eur. J. Neurosci. 2008, 27, 2999–3008. [Google Scholar] [CrossRef]
- Gupta, R.A.; Tejada, L.V.; Tong, B.J.; Das, S.K.; Morrow, J.D. Cyclooxygenase-1 is overexpressed and promotes angiogenic growth factor production in ovarian cancer. Cancer Res. 2003, 63, 906–911. [Google Scholar]
- Wallace, J.L. Distribution and Expression of Cyclooxygenase (COX) Isoenzymes, Their Physiological Roles, and the Categorization of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs). Am. J. Med. 1999, 107, 11–16. [Google Scholar] [CrossRef]
- Cannon, C.P.; Cannon, P.J. Physiology: COX-2 Inhibitors and Cardiovascular Risk. Science 2012, 336, 1386–1387. [Google Scholar] [CrossRef]
- Dogné, J.M.; Supuran, C.T.; Pratico, D. Adverse Cardiovascular Effects of the Coxibs. J. Med. Chem. 2005, 48, 2251–2257. [Google Scholar] [CrossRef]
- Mashima, R.; Okuyama, T. The Role of Lipoxygenases in Pathophysiology; New Insights and Future Perspectives. Redox Biol. 2015, 6, 297–310. [Google Scholar] [CrossRef]
- Singh, H.; Agrawal, D.K. Recent Advances in the Development of Active Hybrid Molecules in the Treatment of Cardiovascular Diseases. Bioorg. Med. Chem. 2022, 62, 116706. [Google Scholar] [CrossRef]
- Charlier, C.; Michaux, C. Dual Inhibition of Cyclooxygenase-2 (COX-2) and 5-Lipoxygenase (5-LOX) as a New Strategy to Provide Safer Non-Steroidal Anti-Inflammatory Drugs. Eur. J. Med. Chem. 2003, 38, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Orafaie, A.; Matin, M.M.; Sadeghian, H. The Importance of 15-Lipoxygenase Inhibitors in Cancer Treatment. Cancer Metastasis Rev. 2018, 37, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Aghasizadeh, M.; Moghaddam, T.; Bahrami, A.R.; Sadeghian, H.; Alavi, S.J.; Matin, M.M. 8-Geranyloxycarbostyril as a Potent 15-LOX-1 Inhibitor Showed Great Anti-Tumor Effects against Prostate Cancer. Life Sci. 2022, 293, 120272. [Google Scholar] [CrossRef] [PubMed]
- Wittwer, J.; Hersberger, M. The Two Faces of the 15-Lipoxygenase in Atherosclerosis. Prostaglandins Leukot. Essent. Fat. Acids 2007, 77, 67–77. [Google Scholar] [CrossRef]
- Kühn, H.; Römisch, I.; Belkner, J. The Role of Lipoxygenase-Isoforms in Atherogenesis. Mol. Nutr. Food Res. 2005, 49, 1014–1029. [Google Scholar] [CrossRef]
- AlFadly, E.D.; Elzahhar, P.A.; Tramarin, A.; Elkazaz, S.; Shaltout, H.; Abu-Serie, M.M.; Janockova, J.; Soukup, O.; Ghareeb, D.A.; El-Yazbi, A.F.; et al. Tackling Neuroinflammation and Cholinergic Deficit in Alzheimer’s Disease: Multi-Target Inhibitors of Cholinesterases, Cyclooxygenase-2 and 15-Lipoxygenase. Eur. J. Med. Chem. 2019, 167, 161–186. [Google Scholar] [CrossRef]
- Khoshneviszadeh, M.; Shahraki, O.; Khoshneviszadeh, M.; Foroumadi, A.; Firuzi, O.; Edraki, N.; Nadri, H.; Moradi, A.; Shafiee, A.; Miri, R. Structure-Based Design, Synthesis, Molecular Docking Study and Biological Evaluation of 1,2,4-Triazine Derivatives Acting as COX/15-LOX Inhibitors with Anti-Oxidant Activities. J. Enzym. Inhib. Med. Chem. 2016, 31, 1602–1611. [Google Scholar] [CrossRef]
- Boshra, A.N.; Abdu-Allah HH, M.; Mohammed, A.F.; Hayallah, A.M. Click Chemistry Synthesis, Biological Evaluation and Docking Study of Some Novel 2′-Hydroxychalcone-Triazole Hybrids as Potent Anti-Inflammatory Agents. Bioorg. Chem. 2020, 95, 103505. [Google Scholar] [CrossRef] [PubMed]
- Moussa, G.; Alaaeddine, R.; Alaeddine, L.M.; Nassra, R.; Belal AS, F.; Ismail, A.; El-Yazbi, A.F.; Abdel-Ghany, Y.S.; Hazzaa, A. Novel Click Modifiable Thioquinazolinones as Anti-Inflammatory Agents: Design, Synthesis, Biological Evaluation and Docking Study. Eur. J. Med. Chem. 2018, 144, 635–650. [Google Scholar] [CrossRef]
- Bashir, B.; Riaz, N.; Abida Ejaz, S.; Saleem, M.; Ashraf, M.; Iqbal, A.; Muzaffar, S.; Ejaz, S.; Aziz-ur-Rehman; Mohammad Kashif Mahmood, H.; et al. Assessing P-Tolyloxy-1,3,4-Oxadiazole Acetamides as Lipoxygenase Inhibitors Assisted by in Vitro and in Silico Studies. Bioorg. Chem. 2022, 129, 106144. [Google Scholar] [CrossRef] [PubMed]
- Ghanim, A.M.; Rezq, S.; Ibrahim, T.S.; Romero, D.G.; Kothayer, H. Novel 1,2,4-Triazine-Quinoline Hybrids: The Privileged Scaffolds as Potent Multi-Target Inhibitors of LPS-Induced Inflammatory Response via Dual COX-2 and 15-LOX Inhibition. Eur. J. Med. Chem. 2021, 219, 113457. [Google Scholar] [CrossRef]
- Omar, Y.M.; Abdu-Allah, H.H.M.; Abdel-Moty, S.G. Synthesis, Biological Evaluation and Docking Study of 1,3,4-Thiadiazole-Thiazolidinone Hybrids as Anti-Inflammatory Agents with Dual Inhibition of COX-2 and 15-LOX. Bioorg. Chem. 2018, 80, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Elzahhar, P.A.; Alaaeddine, R.; Ibrahim, T.M.; Nassra, R.; Ismail, A.; Chua, B.S.K.; Frkic, R.L.; Bruning, J.B.; Wallner, N.; Knape, T.; et al. Shooting Three Inflammatory Targets with a Single Bullet: Novel Multi-Targeting Anti-Inflammatory Glitazones. Eur. J. Med. Chem. 2019, 167, 562–582. [Google Scholar] [CrossRef]
- Elzahhar, P.A.; Abd El Wahab, S.M.; Elagawany, M.; Daabees, H.; Belal, A.S.F.; EL-Yazbi, A.F.; Eid, A.H.; Alaaeddine, R.; Hegazy, R.R.; Allam, R.M.; et al. Expanding the Anticancer Potential of 1,2,3-Triazoles via Simultaneously Targeting Cyclooxygenase-2, 15-Lipoxygenase and Tumor-Associated Carbonic Anhydrases. Eur. J. Med. Chem. 2020, 200, 112439. [Google Scholar] [CrossRef]
- Mphahlele, M.J.; Agbo, E.N.; Gildenhuys, S.; Setshedi, I.B. Exploring Biological Activity of 4-Oxo-4H-Furo [2,3-h]Chromene Derivatives as Potential Multi-Target-Directed Ligands Inhibiting Cholinesterases, β-Secretase, Cyclooxygenase-2, and Lipoxygenase-5/15. Biomolecules 2019, 9, 736. [Google Scholar] [CrossRef]
- Abdelrahman, M.H.; Youssif, B.G.M.; Abdelgawad, M.A.; Abdelazeem, A.H.; Ibrahim, H.M.; Moustafa, A.E.G.A.; Treamblu, L.; Bukhari, S.N.A. Synthesis, Biological Evaluation, Docking Study and Ulcerogenicity Profiling of Some Novel Quinoline-2-Carboxamides as Dual COXs/LOX Inhibitors Endowed with Anti-Inflammatory Activity. Eur. J. Med. Chem. 2017, 127, 972–985. [Google Scholar] [CrossRef]
- Jahng, Y.; Zhao, L.-X.; Moon, Y.-S.; Basnet, A.; Kim, E.; Wook Chang, H.; Kyung Ju, H.; Cheon Jeong, T.; Lee, E.-S. Simple Aromatic Compounds Containing Propenone Moiety Show Considerable Dual COX/5-LOX Inhibitory Activities. Bioorg. Med. Chem. Lett. 2004, 14, 2559–2562. [Google Scholar] [CrossRef]
- Moreau, A.; Chen, Q.H.; Praveen Rao, P.N.; Knaus, E.E. Design, Synthesis, and Biological Evaluation of (E)-3-(4-Methanesulfonylphenyl)-2-(Aryl)Acrylic Acids as Dual Inhibitors of Cyclooxygenases and Lipoxygenases. Bioorg. Med. Chem. 2006, 14, 7716–7727. [Google Scholar] [CrossRef]
- Abd El-Hameed, R.H.; Mahgoub, S.; El-Shanbaky, H.M.; Mohamed, M.S.; Ali, S.A. Utility of Novel 2-Furanones in Synthesis of Other Heterocyclic Compounds Having Anti-Inflammatory Activity with Dual COX2/LOX Inhibition. J. Enzym. Inhib. Med. Chem. 2021, 36, 977–986. [Google Scholar] [CrossRef]
- Sadiq, A.; Mahnashi, M.H.; Alyami, B.A.; Alqahtani, Y.S.; Alqarni, A.O.; Rashid, U. Tailoring the Substitution Pattern of Pyrrolidine-2,5-Dione for Discovery of New Structural Template for Dual COX/LOX Inhibition. Bioorg. Chem. 2021, 112, 104969. [Google Scholar] [CrossRef] [PubMed]
- Zerilli, T.; Ocheretyaner, E. Apremilast (Otezla): A New Oral Treatment for Adults with Psoriasis and Psoriatic Arthritis. Pharm. Ther. 2015, 40, 495–500. [Google Scholar]
- Anderson, K.C. Lenalidomide and Thalidomide: Mechanisms of Action—Similarities and Differences. In Seminars in Hematology; W.B. Saunders: Philadelphia, PA, USA, 2005; Volume 42, pp. S3–S8. [Google Scholar]
- Terpos, E.; Kanellias, N.; Christoulas, D.; Kastritis, E.; Dimopoulos, M.A. Pomalidomide: A Novel Drug to Treat Relapsed and Refractory Multiple Myeloma. OncoTargets Ther. 2013, 6, 531–538. [Google Scholar] [CrossRef]
- Knight, R. IMiDs: A Novel Class of Immunomodulators. Semin. Oncol. 2005, 32, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Amirshahrokhi, K. Thalidomide Reduces Glycerol-Induced Acute Kidney Injury by Inhibition of NF-ΚB, NLRP3 Inflammasome, COX-2 and Inflammatory Cytokines. Cytokine 2021, 144, 155574. [Google Scholar] [CrossRef] [PubMed]
- Fujita, J.; Mestre, J.R.; Zeldis, J.B.; Subbaramaiah, K.; Dannenberg, A.J. Thalidomide and Its Analogues Inhibit Lipopolysaccharide-Mediated Induction of Cyclooxygenase-2. Clin. Cancer Res. 2001, 7, 3349–3355. [Google Scholar]
- Jin, S.H.; Kim, T.I.; Yang, K.M.; Kim, W.H. Thalidomide Destabilizes Cyclooxygenase-2 MRNA by Inhibiting P38 Mitogen-Activated Protein Kinase and Cytoplasmic Shuttling of HuR. Eur. J. Pharmacol. 2007, 558, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Prado SR, T.; Cechinel-Filho, V.; Campos-Buzzi, F.; Corrêa, R.; Cadena SM, C.S.; De Oliveira MB, M. Biological Evaluation of Some Selected Cyclic Imides: Mitochondrial Effects and in Vitro Cytotoxicity. Z. Nat.-Sect. C J. Biosci. 2004, 59, 663–672. [Google Scholar]
- Sultana, K.; Khan, H.; Shahid, K. Synthesis, Characterization and In Vitro Antibacterial Evaluation of Sn, Sb, and Zn Coordination Complexes of 2-(2-Methoxyphenyl)-1H-Isoindole-1, 3(2h)-Dione. Int. J. Pharm. Sci. Rev. Res. 2014, 28, 1–5. [Google Scholar]
- Marulasiddaiah, R.; Kalkhambkar, R.G.; Kulkarni, M.V. Synthesis and Biological Evaluation of Cyclic Imides with Coumarins and Azacoumarins. Open J. Med. Chem. 2012, 2, 89–97. [Google Scholar] [CrossRef]
- Hargreaves, M.K.; Pritchard, J.G.; Dave, H.R. Cyclic Carboxylic Monoimides. Chem. Rev. 1970, 70, 439–469. [Google Scholar] [CrossRef]
- Malinka, W.; Sieklucka-Dziuba, M.; Rajtar, G.; Rejdak, R.; Rejdak, K.; Kleinrok, Z. Synthesis of Some N-Substituted 3,4-Pyrroledicarboximides as Potential CNS Depressive Agents. Pharmazie 2000, 55, 9–16. [Google Scholar]
- Śladowska, H.; Filipek, B.; Szkatuła, D.; Sabiniarz, A.; Kardasz, M.S.; Potoczek, J.; Sieklucka-Dziuba, M.; Rajtar, G.; Kleinrok, Z.S.; Lis, T. Investigations on the Synthesis and Pharmacological Properties of 4-Alkoxy-2-[2-Hydroxy-3-(4-Aryl-1-Piperazinyl)Propyl]-6-Methyl-1H-Pyrrolo[3,4-c]Pyridine-1,3(2H)-Diones. Farmaco 2002, 57, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Śladowska, H.; Sabiniarz, A.; Szkatuła, D.; Filipek, B.; Sapa, J. Synthesis and Properties of 4-Alkoxy-2-[2-Hydroxy-3-(4-o,m,p-Halogenoaryl-1-Piperazinyl)Propyl]-6-Methyl-1H-Pyrrolo-[3,4-c]Pyridine-1,3(2H)-Diones with Analgesic and Sedative Activities. Acta Pol. Pharm.-Drug Res. 2006, 63, 245–254. [Google Scholar]
- Dziubina, A.; Szkatuła, D.; Gdula-Argasińska, J.; Sapa, J. Synthesis and Antinociceptive Activity of Four 1 H-isoindolo-1,3(2 H)-diones. Arch. Pharm. 2022, 355, 2100423. [Google Scholar] [CrossRef]
- Sağlık, B.N.; Osmaniye, D.; Levent, S.; Çevik, U.A.; Çavuşoğlu, B.K.; Özkay, Y.; Kaplancıklı, Z.A. Design, synthesis and biological assessment of new selective COX-2 inhibitors including methyl sulfonyl moiety. Eur. J. Med. Chem. 2021, 209, 112918. [Google Scholar] [CrossRef]
- Malinka, W.; Bodalski, T. Synthesis of Some 1-Substituted-2,5-Dimethylpyrrole-3,4-Dicarboxyimides from Alpha,Beta-Diacetylsuccinate. Pol. J. Chem. 1994, 68, 297–307. [Google Scholar]
- Redzicka, A.; Szczukowski, Ł.; Kochel, A.; Wiatrak, B.; Gębczak, K.; Czyżnikowska, Ż. COX-1/COX-2 Inhibition Activities and Molecular Docking Study of Newly Designed and Synthesized Pyrrolo[3,4-c]Pyrrole Mannich Bases. Bioorg. Med. Chem. 2019, 27, 3918–3928. [Google Scholar] [CrossRef] [PubMed]
- Malinka, W.; Bodalski, T. Rearrangement of Some N-Substituted Pyrrolo(3,4-c)Pyrroles to Corresponding Pyrrolo(3,4-c)Pyridines. Pol. J. Chem. 1995, 69, 95–102. [Google Scholar] [CrossRef]
- CrysAlis PRO; Rigaku Oxford Diffraction Ltd.: Yarnton, UK, 2020.
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Pantaleão, S.Q.; Fernandes, P.O.; Gonçalves, J.E.; Maltarollo, V.G.; Honorio, K.M. Recent Advances in the Prediction of Pharmacokinetics Properties in Drug Design Studies: A Review. ChemMedChem 2022, 17, e202100542. [Google Scholar] [CrossRef]
- Dulsat, J.; López-Nieto, B.; Estrada-Tejedor, R.; Borrell, J.I. Evaluation of Free Online ADMET Tools for Academic or Small Biotech Environments. Molecules 2023, 28, 776. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef]
- SwissADME. Available online: http://www.swissadme.ch/ (accessed on 14 April 2023).
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- PubChem. Meloxicam. C14H13N3O4S2. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Meloxicam (accessed on 14 April 2023).
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Gautam, R.; Jachak, S.M.; Kumar, V.; Mohan, C.G. Synthesis, Biological Evaluation and Molecular Docking Studies of Stellatin Derivatives as Cyclooxygenase (COX-1, COX-2) Inhibitors and Anti-Inflammatory Agents. Bioorg. Med. Chem. Lett. 2011, 21, 1612–1616. [Google Scholar] [CrossRef]
- Kiefer, J.R.; Pawiitz, J.L.; Moreland, K.T.; Stegeman, R.A.; Hood, W.F.; Glerse, J.K.; Stevens, A.M.; Goodwin, D.C.; Rowlinson, S.W.; Marnett, L.J.; et al. Structural Insights into the Stereochemistry of the Cyclooxygenase Reaction. Nature 2000, 405, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Rowlinson, S.W.; Kiefer, J.R.; Prusakiewicz, J.J.; Pawlitz, J.L.; Kozak, K.R.; Kalgutkar, A.S.; Stallings, W.C.; Kurumbail, R.G.; Marnett, L.J. A Novel Mechanism of Cyclooxygenase-2 Inhibition Involving Interactions with Ser-530 and Tyr-385. J. Biol. Chem. 2003, 278, 45763–45769. [Google Scholar] [CrossRef] [PubMed]
- Meshram, D.; Bhardwaj, K.; Rathod, C.; Mahady, G.B.; Soni, K.K. The Role of Leukotrienes Inhibitors in the Management of Chronic Inflammatory Diseases. Recent Pat. Inflamm. Allergy Drug Discov. 2020, 14, 15–31. [Google Scholar] [CrossRef]
- Chen, F.; Ghosh, A.; Lin, J.; Zhang, C.; Pan, Y.; Thakur, A.; Singh, K.; Hong, H.; Tang, S. 5-lipoxygenase pathway and its downstream cysteinyl leukotrienes as potential therapeutic targets for Alzheimer’s disease. Brain Behav. Immun. 2020, 88, 844–855. [Google Scholar] [CrossRef]
- Citron, F.; Perelli, L.; Deem, A.K.; Genovese, G.; Viale, A. Leukotrienes, a potential target for Covid-19. Prostaglandins Leukot. Essent. Fat. Acids 2020, 161, 102174. [Google Scholar] [CrossRef] [PubMed]
- Bruno, F.; Spaziano, G.; Liparulo, A.; Roviezzo, F.; Nabavi, S.M.; Sureda, A.; Filosa, R.; D’Agostino, B. Recent advances in the search for novel 5-lipoxygenase inhibitors for the treatment of asthma. Eur. J. Med. Chem. 2018, 153, 65–72. [Google Scholar] [CrossRef]
- Garland, L.L.; Guillen-Rodriguez, J.; Hsu, C.-H.; Davis, L.E.; Szabo, E.; Husted, C.R.; Liu, H.; LeClerc, A.; Alekseyev, Y.O.; Liu, G.; et al. Clinical Study of Aspirin and Zileuton on Biomarkers of Tobacco-Related Carcinogenesis in Current Smokers. Cancers 2022, 14, 2893. [Google Scholar] [CrossRef]
- Lim, H.-J.; Park, J.; Um, J.-Y.; Lee, S.-S.; Kwak, H.-J. Zileuton, a 5-Lipoxygenase Inhibitor, Exerts Anti-Angiogenic Effect by Inducing Apoptosis of HUVEC via BK Channel Activation. Cells 2019, 8, 1182. [Google Scholar] [CrossRef]
- Boyce, J.A. The role of 15 lipoxygenase 1 in asthma comes into focus. J. Clin. Investig. 2022, 132, e155884. [Google Scholar] [CrossRef]
- Lees, P.; Toutain, P.-L.; Elliott, J.; Giraudel, J.M.; Pelligand, L.; King, J.N. Pharmacology, safety, efficacy and clinical uses of the COX-2 inhibitor robenacoxib. J. Vet. Pharmacol. Ther. 2022, 45, 325–351. [Google Scholar] [CrossRef] [PubMed]
- Vahedpour, T.; Kaur, J.; Hemmati, S.; Hamzeh-Mivehroud, M.; Alizadeh, A.A.; Wuest, F.; Dastmalchi, S. Synthesis and Biological Evaluation of 1,3,5-Trisubstituted 2-Pyrazolines as Novel Cyclooxygenase-2 Inhibitors with Antiproliferative Activity. Chem. Biodivers. 2021, 18, e2000832. [Google Scholar] [CrossRef] [PubMed]
- Ganduri, V.; Rajasekaran, K.; Duraiyarasan, S.; Adefuye, M.A.; Manjunatha, N. Colorectal Carcinoma, Cyclooxygenases, and COX Inhibitors. Cureus 2022, 14, e28579. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, K.R.A.; Abdelall, E.K.A.; Elshemy, H.A.H.; Philoppes, J.N.; Hassanein, E.H.M.; Kahk, N.M. Optimization of pyrazole-based compounds with 1,2,4-triazole-3-thiol moiety as selective COX-2 inhibitors cardioprotective drug candidates: Design, synthesis, cyclooxygenase inhibition, anti-inflammatory, ulcerogenicity, cardiovascular evaluation, and molecular modeling studies. Bioorg. Chem. 2021, 114, 105122. [Google Scholar] [PubMed]
- Moore, N. Coronary Risks Associated with Diclofenac and Other NSAIDs: An Update. Drug Saf. 2020, 43, 301–318. [Google Scholar] [CrossRef]
- Schmidt, M.; Sørensen, H.T.; Pedersen, L. Cardiovascular Risks of Diclofenac Versus Other Older COX-2 Inhibitors (Meloxicam and Etodolac) and Newer COX-2 Inhibitors (Celecoxib and Etoricoxib): A Series of Nationwide Emulated Trials. Drug Saf. 2022, 45, 983–994. [Google Scholar] [CrossRef]
- Gedawy, E.M.; Kassab, A.E.; El Kerdawy, A.M. Design, synthesis and biological evaluation of novel pyrazole sulfonamide derivatives as dual COX-2/5-LOX inhibitors. Eur. J. Med. Chem. 2020, 189, 112066. [Google Scholar] [CrossRef]
- Peregrym, K.; Szczukowski, Ł.; Wiatrak, B.; Potyrak, K.; Czyżnikowska, Ż.; Świątek, P. In Vitro and In Silico Evaluation of New 1,3,4-Oxadiazole Derivatives of Pyrrolo[3,4-d]Pyridazinone as Promising Cyclooxygenase Inhibitors. Int. J. Mol. Sci. 2021, 22, 9130. [Google Scholar] [CrossRef]
- Youssif, B.G.M.; Mohamed, M.F.A.; Al-Sanea, M.M.; Moustafa, A.H.; Abdelhamid, A.A.; Hesham Gomaa, A.M. Novel aryl carboximidamide and 3-aryl-1,2,4-oxadiazole analogues of naproxen as dual selective COX-2/15-LOX inhibitors: Design, synthesis and docking studies. Bioorg. Chem. 2019, 85, 577–584. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A. Gaussian 09 Citation; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Gilbert, N.C.; Rui, Z.; Neau, D.B.; Waight, M.T.; Bartlett, S.G.; Boeglin, W.E.; Brash, A.R.; Newcomer, M.E. Conversion of Human 5-lipoxygenase to a 15-lipoxygenase by a Point Mutation to Mimic Phosphorylation at Serine-663. FASEB J. 2012, 26, 3222–3229. [Google Scholar] [CrossRef] [PubMed]
- ázquez-Libera, J.L.; Durán-Verdugo, F.; Valdés-Jiménez, A.; Valdés-Jiménez, A.; Núñez-Vivanco, G.; Caballero, J. LigRMSD: A Web Server for Automatic Structure Matching and RMSD Calculations among Identical and Similar Compounds in Protein-Ligand Docking. Bioinformatics 2020, 36, 2912–2914. [Google Scholar] [CrossRef] [PubMed]
- Szczęśniak-Sięga, B.M.; Wiatrak, B.; Czyżnikowska, Ż.; Janczak, J.; Wiglusz, R.J.; Maniewska, J. Synthesis and Biological Evaluation as Well as in Silico Studies of Arylpiperazine-1,2-Benzothiazine Derivatives as Novel Anti-Inflammatory Agents. Bioorg. Chem. 2021, 106, 104476. [Google Scholar] [CrossRef]
- Chen, D.; Menche, G.; Power, T.D.; Sower, L.; Peterson, J.W.; Schein, C.H. Accounting for Ligand-Bound Metal Ions in Docking Small Molecules on Adenylyl Cyclase Toxins. Proteins Struct. Funct. Bioinform. 2007, 67, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- BIOVIA. Dassault Systèmes. Biovia Diccovery Studio Vizualizer, v21.1.0.20298; Dassault Systèmes: San Diego, CA, USA, 2020. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Empirical formula | C25 H32 F2 N4 O3 |
| Formula weight | 474.54 |
| Temperature | 100(2) K |
| Wavelength | 1.54184 Å |
| Crystal system | Monoclinic |
| Space group | P21/c |
| Unit cell dimensions | a = 17.842(4) Å |
| b = 16.1601(17) Å | |
| =99.92(3)° | |
| c = 8.237(2) Å | |
| Volume | 2339.5(8) Å3 |
| Z | 4 |
| Density (calculated) | 1.347 Mg/m3 |
| Absorption coefficient | 0.832 mm−1 |
| F(000) | 1008 |
| Crystal size | 0.08 × 0.05 × 0.05 mm3 |
| Theta range for data collection | 2.514 to 78.067°. |
| Index ranges | −22 ≤ h ≤ 22, −20 ≤ k ≤ 19, −8 ≤ l ≤ 10 |
| Reflections collected | 21868 |
| Independent reflections | 4834 [R(int) = 0.2042] |
| Completeness to theta = 67.684° | 99.9% |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 4834/0/310 |
| Goodness-of-fit on F2 | 1.096 |
| Final R indices [I > 2sigma(I)] | R1 = 0.2094, wR2 = 0.4749 |
| R indices (all data) | R1 = 0.2794, wR2 = 0.5065 |
| Extinction coefficient | n/a |
| Largest diff. peak and hole | 0.682 and −0.602 e.Å−3 |
| Compound | NHDF Cells | Cyclooxygenase Inhibition Assay | COX Selectivity Ratio | Lipoxygenase Inhibition Assay | |
|---|---|---|---|---|---|
| IC50 [µM] | IC50 [µM] | IC50(COX-2)/IC50(COX-1) | IC50 [µM] | ||
| COX-1 | COX-2 | LOX | |||
| 3a | 207.72 ± 0.11 | 82.05 ± 0.02 | 60.16 ± 0.08 | 0.73 | 13.94 ± 0.06 |
| 3b | 286.24 ± 0.13 | 79.10 ± 0.06 | 59.77 ± 0.05 | 0.76 | 13.82 ± 0.05 |
| 3c | 142.29 ± 0.09 | 78.61 ± 0.08 | 60.05 ± 0.03 | 0.76 | 12.72 ± 0.02 |
| 3d | 86.55 ± 0.05 | 82.23 ± 0.07 | 59.70 ± 0.03 | 0.73 | 14.32 ± 0.02 |
| 3e | 167.88 ± 0.08 | 88.71 ± 0.09 | 56.43 ± 0.03 | 0.64 | 13.02 ± 0.02 |
| 3f | 225.96 ± 0.09 | 89.00 ± 0.06 | 60.36 ± 0.06 | 0.68 | 13.15 ± 0.05 |
| 3g | 433.50 ± 0.14 | 86.47 ± 0.05 | 60.55 ± 0.09 | 0.70 | 12.80 ± 0.04 |
| 3h | 138.57 ± 0.11 | 79.01 ± 0.02 | 59.34 ± 0.04 | 0.75 | 13.70 ± 0.07 |
| 3i | 114.78 ± 0.08 | 82.17 ± 0.11 | 57.93 ± 0.02 | 0.71 | 13.98 ± 0.02 |
| 3j | 243.47 ± 0.05 | 76.74 ± 0.08 | 60.28 ± 0.02 | 0.79 | 14.25 ± 0.03 |
| 3k | 118.02 ± 0.07 | 83.60 ± 0.08 | 60.83 ± 0.01 | 0.73 | 14.32 ± 0.02 |
| 3l | 103.05 ± 0.06 | 75.50 ± 0.11 | 58.23 ± 0.10 | 0.77 | 14.00 ± 0.06 |
| 3m | 563.49 ± 0.16 | 76.59 ± 0.04 | 61.00 ± 0.04 | 0.80 | 13.85 ± 0.07 |
| 3n | 107.32 ± 0.10 | 84.93 ± 0.02 | 60.25 ± 0.07 | 0.71 | 13.44 ± 0.02 |
| 3o | 49.47 ± 0.05 | 69.56 ± 0.03 | 59.42 ± 0.06 | 0.85 | 13.75 ± 0.08 |
| Meloxicam | 174.23 ± (0.09) | 83.68 ± (0.03) | 57.14 ± (0.05) | 0.68 | - |
| Zileuton | - | - | - | 13.37 ± 0.03 | |
| Compd. | Lipinski Rules | Veber Rules | |||||
|---|---|---|---|---|---|---|---|
| MW ≤500 | LogP ≤5 | NHD a ≤5 | NHA b ≤10 | Violations of Rules | NBR c ≤10 | TPSA d ≤140 | |
| 3a | 452.59 | 4.41 | 1 | 4 | 0 | 8 | 70.71 |
| 3b | 517.46 | 4.40 | 1 | 4 | 1 | 8 | 70.71 |
| 3c | 476.54 | 3.98 | 1 | 5 | 0 | 6 | 70.71 |
| 3d | 494.53 | 3.98 | 1 | 6 | 0 | 6 | 70.71 |
| 3e | 474.54 | 4.41 | 1 | 6 | 0 | 8 | 70.71 |
| 3f | 518.01 | 4.17 | 0 | 5 | 1 | 6 | 94.50 |
| 3g | 537.05 | 4.68 | 1 | 5 | 1 | 8 | 79.94 |
| 3h | 526.55 | 4.34 | 1 | 7 | 1 | 7 | 70.71 |
| 3i | 460.53 | 3.67 | 1 | 6 | 0 | 6 | 96.49 |
| 3j | 494.97 | 3.44 | 1 | 6 | 0 | 6 | 96.49 |
| 3k | 440.54 | 3.60 | 1 | 6 | 0 | 8 | 96.49 |
| 3l | 444.61 | 4.43 | 1 | 5 | 0 | 8 | 70.71 |
| 3m | 460.55 | 2.93 | 1 | 7 | 0 | 6 | 113.23 |
| 3n | 511.03 | 3.95 | 1 | 7 | 1 | 6 | 102.23 |
| 3o | 440.56 | 3.25 | 1 | 7 | 0 | 8 | 113.23 |
| Parameter | Compound | |||||
|---|---|---|---|---|---|---|
| 3c | 3e | 3g | 3h | 3l | mxm | |
| HIA (Human Intestinal Absorption; 1—high probability of being HIA (HIA < 30%); 0—low probability of being HIA)) | 0.315 | 0.006 | 0.008 | 0.006 | 0.005 | 0.004 |
| Caco-2 Permeability; optimal higher than −5.15 log cm/s | −5.32 | −5.29 | −5.45 | −5.46 | −5.31 | −4.71 |
| MDCK Permeability of high passive permeability: >20 × 10−6 | 2.5 × 10−5 | 9 × 10−6 | 2.5 × 10−5 | 1.3 × 10−5 | 7.8 × 10−6 | 1.8 × 10−5 |
| Pgp-inhibitor | + | - | - | + | -- | - |
| Pgp-substrate | - | - | -- | -- | - | - |
| PPB (Plasma Protein Binding; Optimal <90%) | 84% | 81% | 96% | 92% | 82% | 99% |
| VD (Volume Distribution; 0.04–20 L/kg) | 1.3 | 1.9 | 1.0 | 2.4 | 0.9 | 0.4 |
| BBB Penetration (Blood-Brain Barrier Penetration; 1—high probability of penetration; 0—low probability of penetration) | 0.6 | 0.4 | 0.8 | 0.4 | 0.5 | 0.1 |
| Parameter | Compound | |||||
|---|---|---|---|---|---|---|
| 3c | 3e | 3g | 3h | 3l | mxm | |
| CYP1A2 inhibitor | No | No | No | No | No | No |
| CYP2C19 inhibitor | Yes | Yes | Yes | Yes | No | No |
| CYP2C9 inhibitor | Yes | Yes | Yes | Yes | No | Yes |
| CYP2D6 inhibitor | No | Yes | Yes | No | Yes | No |
| CYP3A4 inhibitor | Yes | Yes | Yes | Yes | No | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Redzicka, A.; Wiatrak, B.; Jęśkowiak-Kossakowska, I.; Kochel, A.; Płaczek, R.; Czyżnikowska, Ż. Design, Synthesis, Biological Evaluation, and Molecular Docking Study of 4,6-Dimethyl-5-aryl/alkyl-2-[2-hydroxy-3-(4-substituted-1-piperazinyl)propyl]pyrrolo[3,4-c]pyrrole-1,3(2H,5H)-diones as Anti-Inflammatory Agents with Dual Inhibition of COX and LOX. Pharmaceuticals 2023, 16, 804. https://doi.org/10.3390/ph16060804
Redzicka A, Wiatrak B, Jęśkowiak-Kossakowska I, Kochel A, Płaczek R, Czyżnikowska Ż. Design, Synthesis, Biological Evaluation, and Molecular Docking Study of 4,6-Dimethyl-5-aryl/alkyl-2-[2-hydroxy-3-(4-substituted-1-piperazinyl)propyl]pyrrolo[3,4-c]pyrrole-1,3(2H,5H)-diones as Anti-Inflammatory Agents with Dual Inhibition of COX and LOX. Pharmaceuticals. 2023; 16(6):804. https://doi.org/10.3390/ph16060804
Chicago/Turabian StyleRedzicka, Aleksandra, Benita Wiatrak, Izabela Jęśkowiak-Kossakowska, Andrzej Kochel, Remigiusz Płaczek, and Żaneta Czyżnikowska. 2023. "Design, Synthesis, Biological Evaluation, and Molecular Docking Study of 4,6-Dimethyl-5-aryl/alkyl-2-[2-hydroxy-3-(4-substituted-1-piperazinyl)propyl]pyrrolo[3,4-c]pyrrole-1,3(2H,5H)-diones as Anti-Inflammatory Agents with Dual Inhibition of COX and LOX" Pharmaceuticals 16, no. 6: 804. https://doi.org/10.3390/ph16060804
APA StyleRedzicka, A., Wiatrak, B., Jęśkowiak-Kossakowska, I., Kochel, A., Płaczek, R., & Czyżnikowska, Ż. (2023). Design, Synthesis, Biological Evaluation, and Molecular Docking Study of 4,6-Dimethyl-5-aryl/alkyl-2-[2-hydroxy-3-(4-substituted-1-piperazinyl)propyl]pyrrolo[3,4-c]pyrrole-1,3(2H,5H)-diones as Anti-Inflammatory Agents with Dual Inhibition of COX and LOX. Pharmaceuticals, 16(6), 804. https://doi.org/10.3390/ph16060804