
Current Status and Challenges of Oncolytic Virotherapy for the Treatment of Glioblastoma


Abstract
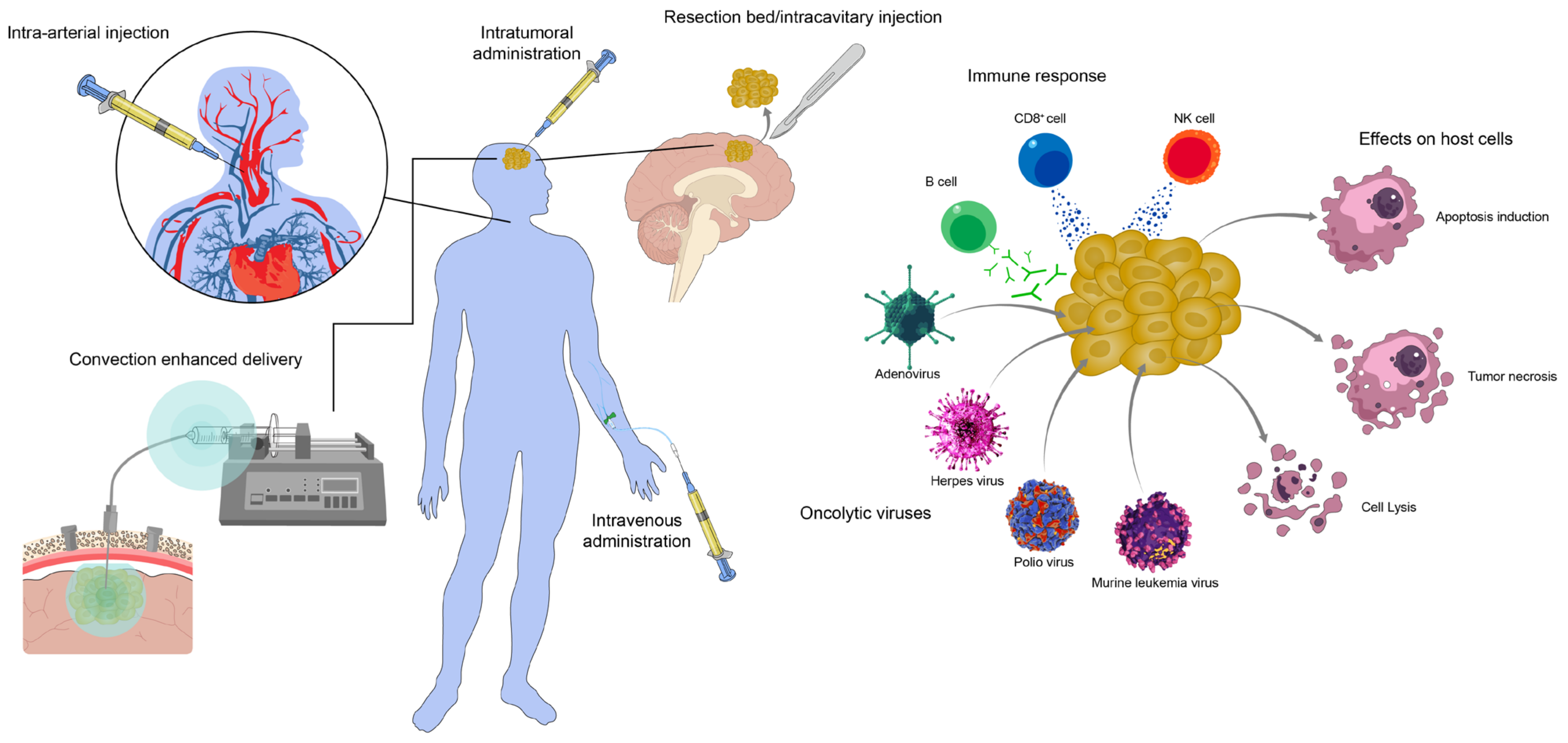
1. Introduction
2. Adenovirus-Based Therapies
2.1. Aglatimagene Besadenovec
2.2. Adenoviral RheoSwitch Therapeutic System Human Interleukin 12
2.3. Tasadenoturev
2.4. NSC-CRAd-S-pk7
3. Herpes Simplex Virus-Based Therapies
3.1. M032-HSV-1
3.2. HSV-1 Virus rQNestin34.5v.2
3.3. Teserpaturev
4. Polio Virus-Based Therapies
Recombinant Nonpathogenic Polio-Rhinovirus Chimera
5. Murine Leukemia Virus-Based Therapies
Vocimagene Amiretrorepvec
6. Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Price, M.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2015–2019. Neuro Oncol. 2022, 24 (Suppl. S5), v1–v95. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef]
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro Oncol. 2020, 22, 1073–1113. [Google Scholar] [CrossRef]
- Network, N.C.C. Central Nervous System Cancers (Version 1.2023). Available online: https://www.nccn.org/professionals/physician_gls/pdf/cns.pdf (accessed on 20 April 2023).
- Bagley, S.J.; Kothari, S.; Rahman, R.; Lee, E.Q.; Dunn, G.P.; Galanis, E.; Chang, S.M.; Nabors, L.B.; Ahluwalia, M.S.; Stupp, R.; et al. Glioblastoma Clinical Trials: Current Landscape and Opportunities for Improvement. Clin. Cancer Res. 2022, 28, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Sener, U.; Ruff, M.W.; Campian, J.L. Immunotherapy in Glioblastoma: Current Approaches and Future Perspectives. Int. J. Mol. Sci. 2022, 23, 7046. [Google Scholar] [CrossRef] [PubMed]
- Neth, B.J.; Webb, M.J.; Parney, I.F.; Sener, U.T. The Current Status, Challenges, and Future Potential of Therapeutic Vaccination in Glioblastoma. Pharmaceutics 2023, 15, 1134. [Google Scholar] [CrossRef]
- Martuza, R.L.; Malick, A.; Markert, J.M.; Ruffner, K.L.; Coen, D.M. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 1991, 252, 854–856. [Google Scholar] [CrossRef]
- Dock, G. The influence of complicating diseases upon leukaemia. Am. J. Med. Sci. 1904, 127, 563. [Google Scholar] [CrossRef]
- Larson, C.; Oronsky, B.; Scicinski, J.; Fanger, G.R.; Stirn, M.; Oronsky, A.; Reid, T.R. Going viral: A review of replication-selective oncolytic adenoviruses. Oncotarget 2015, 6, 19976–19989. [Google Scholar] [CrossRef]
- Gey, G. Tissue culture studies of the proliferative capacity of cervical carcinoma and normal epithelium. Cancer Res. 1952, 12, 264–265. [Google Scholar]
- Weller, T.H.; Robbins, F.C.; Enders, J.F. Cultivation of poliomyelitis virus in cultures of human foreskin and embryonic tissues. Proc. Soc. Exp. Biol. Med. 1949, 72, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Kelly, E.; Russell, S.J. History of oncolytic viruses: Genesis to genetic engineering. Mol. Ther. 2007, 15, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Santos Apolonio, J.; Lima de Souza Goncalves, V.; Cordeiro Santos, M.L.; Silva Luz, M.; Silva Souza, J.V.; Rocha Pinheiro, S.L.; de Souza, W.R.; Sande Loureiro, M.; de Melo, F.F. Oncolytic virus therapy in cancer: A current review. World J. Virol. 2021, 10, 229–255. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Xu, H.; Wang, J.; Wu, X.; Wen, W.; Liang, Y.; Wang, L.; Liu, F.; Du, X. Inhibition of breast cancer cells by targeting E2F-1 gene and expressing IL15 oncolytic adenovirus. Biosci. Rep. 2019, 39, BSR20190384. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, K.B.; Barra, N.G.; Davies, E.; Ashkar, A.A.; Lichty, B.D. Expressing human interleukin-15 from oncolytic vesicular stomatitis virus improves survival in a murine metastatic colon adenocarcinoma model through the enhancement of anti-tumor immunity. Cancer Gene Ther. 2012, 19, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.M.; Foreman, P.M.; Nabors, L.B.; Riley, K.O.; Gillespie, G.Y.; Markert, J.M. Design of a Phase I Clinical Trial to Evaluate M032, a Genetically Engineered HSV-1 Expressing IL-12, in Patients with Recurrent/Progressive Glioblastoma Multiforme, Anaplastic Astrocytoma, or Gliosarcoma. Hum. Gene Ther. Clin. Dev. 2016, 27, 69–78. [Google Scholar] [CrossRef]
- Wohlfahrt, M.E.; Beard, B.C.; Lieber, A.; Kiem, H.P. A capsid-modified, conditionally replicating oncolytic adenovirus vector expressing TRAIL Leads to enhanced cancer cell killing in human glioblastoma models. Cancer Res. 2007, 67, 8783–8790. [Google Scholar] [CrossRef]
- Barrett, J.A.; Cai, H.; Miao, J.; Khare, P.D.; Gonzalez, P.; Dalsing-Hernandez, J.; Sharma, G.; Chan, T.; Cooper, L.J.N.; Lebel, F. Regulated intratumoral expression of IL-12 using a RheoSwitch Therapeutic System((R)) (RTS((R))) gene switch as gene therapy for the treatment of glioma. Cancer Gene Ther. 2018, 25, 106–116. [Google Scholar] [CrossRef]
- Aguilar, L.K.; Guzik, B.W.; Aguilar-Cordova, E. Cytotoxic immunotherapy strategies for cancer: Mechanisms and clinical development. J. Cell. Biochem. 2011, 112, 1969–1977. [Google Scholar] [CrossRef]
- Fyfe, J.A.; Keller, P.M.; Furman, P.A.; Miller, R.L.; Elion, G.B. Thymidine kinase from herpes simplex virus phosphorylates the new antiviral compound, 9-(2-hydroxyethoxymethyl)guanine. J. Biol. Chem. 1978, 253, 8721–8727. [Google Scholar] [CrossRef] [PubMed]
- Eastham, J.A.; Chen, S.H.; Sehgal, I.; Yang, G.; Timme, T.L.; Hall, S.J.; Woo, S.L.; Thompson, T.C. Prostate cancer gene therapy: Herpes simplex virus thymidine kinase gene transduction followed by ganciclovir in mouse and human prostate cancer models. Hum. Gene Ther. 1996, 7, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Trask, T.W.; Trask, R.P.; Aguilar-Cordova, E.; Shine, H.D.; Wyde, P.R.; Goodman, J.C.; Hamilton, W.J.; Rojas-Martinez, A.; Chen, S.H.; Woo, S.L.; et al. Phase I study of adenoviral delivery of the HSV-tk gene and ganciclovir administration in patients with current malignant brain tumors. Mol. Ther. 2000, 1, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Sung, M.W.; Yeh, H.C.; Thung, S.N.; Schwartz, M.E.; Mandeli, J.P.; Chen, S.H.; Woo, S.L. Intratumoral adenovirus-mediated suicide gene transfer for hepatic metastases from colorectal adenocarcinoma: Results of a phase I clinical trial. Mol. Ther. 2001, 4, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Sterman, D.H.; Recio, A.; Vachani, A.; Sun, J.; Cheung, L.; DeLong, P.; Amin, K.M.; Litzky, L.A.; Wilson, J.M.; Kaiser, L.R.; et al. Long-term follow-up of patients with malignant pleural mesothelioma receiving high-dose adenovirus herpes simplex thymidine kinase/ganciclovir suicide gene therapy. Clin. Cancer Res. 2005, 11, 7444–7453. [Google Scholar] [CrossRef]
- Chevez-Barrios, P.; Chintagumpala, M.; Mieler, W.; Paysse, E.; Boniuk, M.; Kozinetz, C.; Hurwitz, M.Y.; Hurwitz, R.L. Response of retinoblastoma with vitreous tumor seeding to adenovirus-mediated delivery of thymidine kinase followed by ganciclovir. J. Clin. Oncol. 2005, 23, 7927–7935. [Google Scholar] [CrossRef]
- Hasenburg, A.; Tong, X.W.; Fischer, D.C.; Rojas-Martinez, A.; Nyberg-Hoffman, C.; Kaplan, A.L.; Kaufman, R.H.; Ramzy, I.; Aguilar-Cordova, E.; Kieback, D.G. Adenovirus-mediated thymidine kinase gene therapy in combination with topotecan for patients with recurrent ovarian cancer: 2.5-year follow-up. Gynecol. Oncol. 2001, 83, 549–554. [Google Scholar] [CrossRef]
- Bloomston, M.; Marsh, C.; Walker, J.; Coyle, W.; Marx, H.; Tahiri, S.; Cruz, C.M.; Aguilar, L.K.; Aguilar-Cordova, E.; Chung, V.M. Phase I trial of gene-mediated cytotoxic immunotherapy in combination with chemoradiation for locally advanced pancreatic cancer. J. Clin. Oncol. 2011, 29, 195. [Google Scholar] [CrossRef]
- Wheeler, L.A.; Manzanera, A.G.; Bell, S.D.; Cavaliere, R.; McGregor, J.M.; Grecula, J.C.; Newton, H.B.; Lo, S.S.; Badie, B.; Portnow, J.; et al. Phase II multicenter study of gene-mediated cytotoxic immunotherapy as adjuvant to surgical resection for newly diagnosed malignant glioma. Neuro Oncol. 2016, 18, 1137–1145. [Google Scholar] [CrossRef]
- Kieran, M.W.; Goumnerova, L.; Manley, P.; Chi, S.N.; Marcus, K.J.; Manzanera, A.G.; Polanco, M.L.S.; Guzik, B.W.; Aguilar-Cordova, E.; Diaz-Montero, C.M.; et al. Phase I study of gene-mediated cytotoxic immunotherapy with AdV-tk as adjuvant to surgery and radiation for pediatric malignant glioma and recurrent ependymoma. Neuro Oncol. 2019, 21, 537–546. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Aguilar, L.K.; Bell, S.D.; Kaur, B.; Hardcastle, J.; Cavaliere, R.; McGregor, J.; Lo, S.; Ray-Chaudhuri, A.; Chakravarti, A.; et al. Phase IB study of gene-mediated cytotoxic immunotherapy adjuvant to up-front surgery and intensive timing radiation for malignant glioma. J. Clin. Oncol. 2011, 29, 3611–3619. [Google Scholar] [CrossRef] [PubMed]
- Ji, N.; Weng, D.; Liu, C.; Gu, Z.; Chen, S.; Guo, Y.; Fan, Z.; Wang, X.; Chen, J.; Zhao, Y.; et al. Adenovirus-mediated delivery of herpes simplex virus thymidine kinase administration improves outcome of recurrent high-grade glioma. Oncotarget 2016, 7, 4369–4378. [Google Scholar] [CrossRef] [PubMed]
- Chiocca, E.A.; Yu, J.S.; Lukas, R.V.; Solomon, I.H.; Ligon, K.L.; Nakashima, H.; Triggs, D.A.; Reardon, D.A.; Wen, P.; Stopa, B.M.; et al. Regulatable interleukin-12 gene therapy in patients with recurrent high-grade glioma: Results of a phase 1 trial. Sci. Transl. Med. 2019, 11, eaaw5680. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Gelb, A.B.; Chen, C.C.; Rao, G.; Reardon, D.A.; Wen, P.Y.; Bi, W.L.; Peruzzi, P.; Amidei, C.; Triggs, D.; et al. Combined immunotherapy with controlled interleukin-12 gene therapy and immune checkpoint blockade in recurrent glioblastoma: An open-label, multi-institutional phase I trial. Neuro Oncol. 2022, 24, 951–963. [Google Scholar] [CrossRef] [PubMed]
- Lukas, R.V.; Kurz, S.C.; Yu, J.; Landolfi, J.C.; Rao, G.; Amidei, C.; Buck, J.Y.; Hadar, N.; Estupinan, T.; Miao, J.; et al. Survival of subjects with recurrent glioblastoma receiving intratumoral administration of controlled IL-12 with limited exposure to dexamethasone. J. Clin. Oncol. 2020, 38, 2564. [Google Scholar] [CrossRef]
- Lukas, R.; Oberheim-Bush, N.A.; Cavaliere, R.; Landolfi, J.; Yu, J.S.; Chen, C.; Cordova, C.; Amidei, C.; Buck, J.Y.; Hadar, N.; et al. Final results of controlled il-12 monotherapy and in combination with pd-1 inhibitor in adult subjects with recurrent glioblastoma. Neuro Oncol. 2021, 23, vi54. [Google Scholar] [CrossRef]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Yung, W.K.A.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D.; et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J. Clin. Oncol. 2018, 36, 1419–1427. [Google Scholar] [CrossRef]
- Lang, F.F.; Tran, N.D.; Puduvalli, V.K.; Elder, J.B.; Fink, K.L.; Conrad, C.A.; Yung, W.K.A.; Penas-Prado, M.; Gomez-Manzano, C.; Peterkin, J.; et al. Phase 1b open-label randomized study of the oncolytic adenovirus DNX-2401 administered with or without interferon gamma for recurrent glioblastoma. J. Clin. Oncol. 2017, 35, 2002. [Google Scholar] [CrossRef]
- Van Putten, E.H.P.; Kleijn, A.; van Beusechem, V.W.; Noske, D.; Lamers, C.H.J.; de Goede, A.L.; Idema, S.; Hoefnagel, D.; Kloezeman, J.J.; Fueyo, J.; et al. Convection Enhanced Delivery of the Oncolytic Adenovirus Delta24-RGD in Patients with Recurrent GBM: A Phase I Clinical Trial Including Correlative Studies. Clin. Cancer Res. 2022, 28, 1572–1585. [Google Scholar] [CrossRef]
- Fares, J.; Ahmed, A.U.; Ulasov, I.V.; Sonabend, A.M.; Miska, J.; Lee-Chang, C.; Balyasnikova, I.V.; Chandler, J.P.; Portnow, J.; Tate, M.C.; et al. Neural stem cell delivery of an oncolytic adenovirus in newly diagnosed malignant glioma: A first-in-human, phase 1, dose-escalation trial. Lancet Oncol. 2021, 22, 1103–1114. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Solomon, I.; Nakashima, H.; Lawler, S.E.; Triggs, D.; Zhang, A.; Grant, J.; Reardon, D.A.; Wen, P.Y.; Lee, E.Q.; et al. First-in-human CAN-3110 (ICP-34.5 expressing HSV-1 oncolytic virus) in patients with recurrent high-grade glioma. J. Clin. Oncol. 2021, 39, 2009. [Google Scholar] [CrossRef]
- Todo, T.; Ino, Y.; Ohtsu, H.; Shibahara, J.; Tanaka, M. A phase I/II study of triple-mutated oncolytic herpes virus G47∆ in patients with progressive glioblastoma. Nat. Commun. 2022, 13, 4119. [Google Scholar] [CrossRef] [PubMed]
- Todo, T.; Ito, H.; Ino, Y.; Ohtsu, H.; Ota, Y.; Shibahara, J.; Tanaka, M. Intratumoral oncolytic herpes virus G47∆ for residual or recurrent glioblastoma: A phase 2 trial. Nat. Med. 2022, 28, 1630–1639. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, A.; Gromeier, M.; Herndon, J.E., 2nd; Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; Friedman, H.S.; McSherry, F.; Muscat, A.M.; Nair, S.; et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N. Engl. J. Med. 2018, 379, 150–161. [Google Scholar] [CrossRef]
- Aghi, M.; Vogelbaum, M.A.; Kesari, S.; Chen, C.C.; Liau, L.M.; Piccioni, D.; Portnow, J.; Chang, S.; Robbins, J.M.; Boyce, T.; et al. Intratumoral Delivery of the Retroviral Replicating Vector (Rrv) Toca 511 in Subjects with Recurrent High Grade Glioma: Interim Report of Phase 1 Study (NCT 01156584). Neuro Oncol. 2014, 16, 8. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Landolfi, J.; Vogelbaum, M.A.; Ostertag, D.; Elder, J.B.; Bloomfield, S.; Carter, B.; Chen, C.C.; Kalkanis, S.N.; Kesari, S.; et al. Durable complete responses in some recurrent high-grade glioma patients treated with Toca 511 + Toca FC. Neuro Oncol. 2018, 20, 1383–1392. [Google Scholar] [CrossRef]
- Cloughesy, T.; Vogelbaum, M.A.; Ostertag, D.; Diego, O.; Jolly, D.J.; Ibanez, C.; Yagiz, K.; Robbins, J.; Chu, A.; Niethammer, A.; et al. Intravenous Toca 511 Delivery Leads to Viral DNA in Resected HGG. Neuro Oncol. 2015, 74, v74. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Petrecca, K.; Walbert, T.; Butowski, N.; Salacz, M.; Perry, J.; Damek, D.; Bota, D.; Bettegowda, C.; Zhu, J.J.; et al. Effect of Vocimagene Amiretrorepvec in Combination With Flucytosine vs Standard of Care on Survival Following Tumor Resection in Patients With Recurrent High-Grade Glioma: A Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1939–1946. [Google Scholar] [CrossRef]
- Lotze, M.T.; Matory, Y.L.; Ettinghausen, S.E.; Rayner, A.A.; Sharrow, S.O.; Seipp, C.A.; Custer, M.C.; Rosenberg, S.A. In vivo administration of purified human interleukin 2. II. Half life, immunologic effects, and expansion of peripheral lymphoid cells in vivo with recombinant IL 2. J. Immunol. 1985, 135, 2865–2875. [Google Scholar] [CrossRef]
- Vom Berg, J.; Vrohlings, M.; Haller, S.; Haimovici, A.; Kulig, P.; Sledzinska, A.; Weller, M.; Becher, B. Intratumoral IL-12 combined with CTLA-4 blockade elicits T cell-mediated glioma rejection. J. Exp. Med. 2013, 210, 2803–2811. [Google Scholar] [CrossRef]
- Leonard, J.P.; Sherman, M.L.; Fisher, G.L.; Buchanan, L.J.; Larsen, G.; Atkins, M.B.; Sosman, J.A.; Dutcher, J.P.; Vogelzang, N.J.; Ryan, J.L. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood 1997, 90, 2541–2548. [Google Scholar] [PubMed]
- Cai, H.; Sun, L.; Miao, J.; Krishman, S.; Lebel, F.; Barrett, J.A. Plasma Pharmacokinetics of Veledimex, a Small-Molecule Activator Ligand for a Proprietary Gene Therapy Promoter System, in Healthy Subjects. Clin. Pharmacol. Drug Dev. 2017, 6, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Dyson, N.; Guida, P.; McCall, C.; Harlow, E. Adenovirus E1A makes two distinct contacts with the retinoblastoma protein. J. Virol. 1992, 66, 4606–4611. [Google Scholar] [CrossRef] [PubMed]
- Flint, J.; Shenk, T. Viral transactivating proteins. Annu. Rev. Genet. 1997, 31, 177–212. [Google Scholar] [CrossRef] [PubMed]
- Tortosa, A.; Ino, Y.; Odell, N.; Swilley, S.; Sasaki, H.; Louis, D.N.; Henson, J.W. Molecular genetics of radiographically defined de novo glioblastoma multiforme. Neuropathol. Appl. Neurobiol. 2000, 26, 544–552. [Google Scholar] [CrossRef]
- Bello, L.; Francolini, M.; Marthyn, P.; Zhang, J.; Carroll, R.S.; Nikas, D.C.; Strasser, J.F.; Villani, R.; Cheresh, D.A.; Black, P.M. Alpha(v)beta3 and alpha(v)beta5 integrin expression in glioma periphery. Neurosurgery 2001, 49, 380–389, discussion 390. [Google Scholar] [CrossRef]
- Fueyo, J.; Alemany, R.; Gomez-Manzano, C.; Fuller, G.N.; Khan, A.; Conrad, C.A.; Liu, T.J.; Jiang, H.; Lemoine, M.G.; Suzuki, K.; et al. Preclinical characterization of the antiglioma activity of a tropism-enhanced adenovirus targeted to the retinoblastoma pathway. J. Natl. Cancer Inst. 2003, 95, 652–660. [Google Scholar] [CrossRef]
- Lal, S.; Lacroix, M.; Tofilon, P.; Fuller, G.N.; Sawaya, R.; Lang, F.F. An implantable guide-screw system for brain tumor studies in small animals. J. Neurosurg. 2000, 92, 326–333. [Google Scholar] [CrossRef]
- Gallego Perez-Larraya, J.; Garcia-Moure, M.; Labiano, S.; Patino-Garcia, A.; Dobbs, J.; Gonzalez-Huarriz, M.; Zalacain, M.; Marrodan, L.; Martinez-Velez, N.; Puigdelloses, M.; et al. Oncolytic DNX-2401 Virus for Pediatric Diffuse Intrinsic Pontine Glioma. N. Engl. J. Med. 2022, 386, 2471–2481. [Google Scholar] [CrossRef]
- Ulasov, I.V.; Zhu, Z.B.; Tyler, M.A.; Han, Y.; Rivera, A.A.; Khramtsov, A.; Curiel, D.T.; Lesniak, M.S. Survivin-driven and fiber-modified oncolytic adenovirus exhibits potent antitumor activity in established intracranial glioma. Hum. Gene Ther. 2007, 18, 589–602. [Google Scholar] [CrossRef]
- Asaoka, K.; Tada, M.; Sawamura, Y.; Ikeda, J.; Abe, H. Dependence of efficient adenoviral gene delivery in malignant glioma cells on the expression levels of the Coxsackievirus and adenovirus receptor. J. Neurosurg. 2000, 92, 1002–1008. [Google Scholar] [CrossRef] [PubMed]
- Van Houdt, W.J.; Haviv, Y.S.; Lu, B.; Wang, M.; Rivera, A.A.; Ulasov, I.V.; Lamfers, M.L.; Rein, D.; Lesniak, M.S.; Siegal, G.P.; et al. The human survivin promoter: A novel transcriptional targeting strategy for treatment of glioma. J. Neurosurg. 2006, 104, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Qiao, D.; Meyer, K.; Mundhenke, C.; Drew, S.A.; Friedl, A. Heparan sulfate proteoglycans as regulators of fibroblast growth factor-2 signaling in brain endothelial cells. Specific role for glypican-1 in glioma angiogenesis. J. Biol. Chem. 2003, 278, 16045–16053. [Google Scholar] [CrossRef] [PubMed]
- Staba, M.J.; Wickham, T.J.; Kovesdi, I.; Hallahan, D.E. Modifications of the fiber in adenovirus vectors increase tropism for malignant glioma models. Cancer Gene Ther. 2000, 7, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.U.; Thaci, B.; Tobias, A.L.; Auffinger, B.; Zhang, L.; Cheng, Y.; Kim, C.K.; Yunis, C.; Han, Y.; Alexiades, N.G.; et al. A preclinical evaluation of neural stem cell-based cell carrier for targeted antiglioma oncolytic virotherapy. J. Natl. Cancer Inst. 2013, 105, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.C.; Cassady, K.A.; Cody, J.J.; Parker, J.N.; Price, K.H.; Coleman, J.M.; Peggins, J.O.; Noker, P.E.; Powers, N.W.; Grimes, S.D.; et al. Evaluation of the safety and biodistribution of M032, an attenuated herpes simplex virus type 1 expressing hIL-12, after intracerebral administration to aotus nonhuman primates. Hum. Gene Ther. Clin. Dev. 2014, 25, 16–27. [Google Scholar] [CrossRef]
- Chou, J.; Kern, E.R.; Whitley, R.J.; Roizman, B. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science 1990, 250, 1262–1266. [Google Scholar] [CrossRef]
- Hellums, E.K.; Markert, J.M.; Parker, J.N.; He, B.; Perbal, B.; Roizman, B.; Whitley, R.J.; Langford, C.P.; Bharara, S.; Gillespie, G.Y. Increased efficacy of an interleukin-12-secreting herpes simplex virus in a syngeneic intracranial murine glioma model. Neuro Oncol. 2005, 7, 213–224. [Google Scholar] [CrossRef]
- Parker, J.N.; Gillespie, G.Y.; Love, C.E.; Randall, S.; Whitley, R.J.; Markert, J.M. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc. Natl. Acad. Sci. USA 2000, 97, 2208–2213. [Google Scholar] [CrossRef]
- Markert, J.M.; Liechty, P.G.; Wang, W.; Gaston, S.; Braz, E.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Lakeman, A.D.; Palmer, C.A.; et al. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Mol. Ther. 2009, 17, 199–207. [Google Scholar] [CrossRef]
- Markert, J.M.; Medlock, M.D.; Rabkin, S.D.; Gillespie, G.Y.; Todo, T.; Hunter, W.D.; Palmer, C.A.; Feigenbaum, F.; Tornatore, C.; Tufaro, F.; et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: Results of a phase I trial. Gene Ther. 2000, 7, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Markert, J.M.; Razdan, S.N.; Kuo, H.C.; Cantor, A.; Knoll, A.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Agee, B.S.; Coleman, J.M.; et al. A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol. Ther. 2014, 22, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Chiocca, E.A.; Nakashima, H.; Kasai, K.; Fernandez, S.A.; Oglesbee, M. Preclinical Toxicology of rQNestin34.5v.2: An Oncolytic Herpes Virus with Transcriptional Regulation of the ICP34.5 Neurovirulence Gene. Mol. Ther. Methods Clin. Dev. 2020, 17, 871–893. [Google Scholar] [CrossRef] [PubMed]
- Chung, R.Y.; Saeki, Y.; Chiocca, E.A. B-myb promoter retargeting of herpes simplex virus gamma34.5 gene-mediated virulence toward tumor and cycling cells. J. Virol. 1999, 73, 7556–7564. [Google Scholar] [CrossRef]
- Kambara, H.; Okano, H.; Chiocca, E.A.; Saeki, Y. An oncolytic HSV-1 mutant expressing ICP34.5 under control of a nestin promoter increases survival of animals even when symptomatic from a brain tumor. Cancer Res. 2005, 65, 2832–2839. [Google Scholar] [CrossRef]
- Todo, T.; Martuza, R.L.; Rabkin, S.D.; Johnson, P.A. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc. Natl. Acad. Sci. USA 2001, 98, 6396–6401. [Google Scholar] [CrossRef]
- Mineta, T.; Rabkin, S.D.; Yazaki, T.; Hunter, W.D.; Martuza, R.L. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat. Med. 1995, 1, 938–943. [Google Scholar] [CrossRef]
- Goldstein, D.J.; Weller, S.K. Herpes simplex virus type 1-induced ribonucleotide reductase activity is dispensable for virus growth and DNA synthesis: Isolation and characterization of an ICP6 lacZ insertion mutant. J. Virol. 1988, 62, 196–205. [Google Scholar] [CrossRef]
- Mavromara-Nazos, P.; Ackermann, M.; Roizman, B. Construction and properties of a viable herpes simplex virus 1 recombinant lacking coding sequences of the alpha 47 gene. J. Virol. 1986, 60, 807–812. [Google Scholar] [CrossRef]
- He, B.; Chou, J.; Brandimarti, R.; Mohr, I.; Gluzman, Y.; Roizman, B. Suppression of the phenotype of gamma(1)34.5- herpes simplex virus 1: Failure of activated RNA-dependent protein kinase to shut off protein synthesis is associated with a deletion in the domain of the alpha47 gene. J. Virol. 1997, 71, 6049–6054. [Google Scholar] [CrossRef]
- Fukuhara, H.; Martuza, R.L.; Rabkin, S.D.; Ito, Y.; Todo, T. Oncolytic herpes simplex virus vector g47delta in combination with androgen ablation for the treatment of human prostate adenocarcinoma. Clin. Cancer Res. 2005, 11, 7886–7890. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Martuza, R.L.; Rabkin, S.D. Intracarotid delivery of oncolytic HSV vector G47Delta to metastatic breast cancer in the brain. Gene Ther. 2005, 12, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, K.; Iwai, M.; Yajima, S.; Tanaka, M.; Yanagihara, K.; Seto, Y.; Todo, T. Efficacy of a Third-Generation Oncolytic Herpes Virus G47Delta in Advanced Stage Models of Human Gastric Cancer. Mol. Ther. Oncolytics 2020, 17, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Uchihashi, T.; Nakahara, H.; Fukuhara, H.; Iwai, M.; Ito, H.; Sugauchi, A.; Tanaka, M.; Kogo, M.; Todo, T. Oncolytic herpes virus G47Delta injected into tongue cancer swiftly traffics in lymphatics and suppresses metastasis. Mol. Ther. Oncolytics 2021, 22, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Yajima, S.; Sugawara, K.; Iwai, M.; Tanaka, M.; Seto, Y.; Todo, T. Efficacy and safety of a third-generation oncolytic herpes virus G47Delta in models of human esophageal carcinoma. Mol. Ther. Oncolytics 2021, 23, 402–411. [Google Scholar] [CrossRef]
- Gromeier, M.; Alexander, L.; Wimmer, E. Internal ribosomal entry site substitution eliminates neurovirulence in intergeneric poliovirus recombinants. Proc. Natl. Acad. Sci. USA 1996, 93, 2370–2375. [Google Scholar] [CrossRef]
- Gromeier, M.; Lachmann, S.; Rosenfeld, M.R.; Gutin, P.H.; Wimmer, E. Intergeneric poliovirus recombinants for the treatment of malignant glioma. Proc. Natl. Acad. Sci. USA 2000, 97, 6803–6808. [Google Scholar] [CrossRef]
- Chandramohan, V.; Bryant, J.D.; Piao, H.; Keir, S.T.; Lipp, E.S.; Lefaivre, M.; Perkinson, K.; Bigner, D.D.; Gromeier, M.; McLendon, R.E. Validation of an Immunohistochemistry Assay for Detection of CD155, the Poliovirus Receptor, in Malignant Gliomas. Arch. Pathol. Lab. Med. 2017, 141, 1697–1704. [Google Scholar] [CrossRef]
- Brown, M.C.; Holl, E.K.; Boczkowski, D.; Dobrikova, E.; Mosaheb, M.; Chandramohan, V.; Bigner, D.D.; Gromeier, M.; Nair, S.K. Cancer immunotherapy with recombinant poliovirus induces IFN-dominant activation of dendritic cells and tumor antigen-specific CTLs. Sci. Transl. Med. 2017, 9, eaan4220. [Google Scholar] [CrossRef]
- Ostertag, D.; Amundson, K.K.; Lopez Espinoza, F.; Martin, B.; Buckley, T.; Galvao da Silva, A.P.; Lin, A.H.; Valenta, D.T.; Perez, O.D.; Ibanez, C.E.; et al. Brain tumor eradication and prolonged survival from intratumoral conversion of 5-fluorocytosine to 5-fluorouracil using a nonlytic retroviral replicating vector. Neuro Oncol. 2012, 14, 145–159. [Google Scholar] [CrossRef]
- Dalba, C.; Klatzmann, D.; Logg, C.R.; Kasahara, N. Beyond oncolytic virotherapy: Replication-competent retrovirus vectors for selective and stable transduction of tumors. Curr. Gene Ther. 2005, 5, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Mullen, J.T.; Chandrasekhar, S.; Pawlik, T.M.; Yoon, S.S.; Tanabe, K.K. Multimodality therapy with a replication-conditional herpes simplex virus 1 mutant that expresses yeast cytosine deaminase for intratumoral conversion of 5-fluorocytosine to 5-fluorouracil. Cancer Res. 2001, 61, 5447–5452. [Google Scholar] [PubMed]
- Collins, S.A.; Shah, A.H.; Ostertag, D.; Kasahara, N.; Jolly, D.J. Clinical development of retroviral replicating vector Toca 511 for gene therapy of cancer. Expert. Opin. Biol. Ther. 2021, 21, 1199–1214. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, R.S.; Aghi, M.K.; Vogelbaum, M.A.; Bruce, J.N. Convection-enhanced drug delivery for glioblastoma: A review. J. Neurooncol. 2021, 151, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Friedmann-Morvinski, D. Glioblastoma heterogeneity and cancer cell plasticity. Crit. Rev. Oncog. 2014, 19, 327–336. [Google Scholar] [CrossRef]
- Becker, A.P.; Sells, B.E.; Haque, S.J.; Chakravarti, A. Tumor Heterogeneity in Glioblastomas: From Light Microscopy to Molecular Pathology. Cancers 2021, 13, 761. [Google Scholar] [CrossRef]
- Zhang, P.; Xia, Q.; Liu, L.; Li, S.; Dong, L. Current Opinion on Molecular Characterization for GBM Classification in Guiding Clinical Diagnosis, Prognosis, and Therapy. Front. Mol. Biosci. 2020, 7, 562798. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Behnan, J.; Finocchiaro, G.; Hanna, G. The landscape of the mesenchymal signature in brain tumours. Brain 2019, 142, 847–866. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, F. Advances and potential pitfalls of oncolytic viruses expressing immunomodulatory transgene therapy for malignant gliomas. Cell. Death Dis. 2020, 11, 485. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yao, Y.; Wang, H.; Guo, Y.; Zhang, H.; Chen, L. Effects of salidroside on glioma formation and growth inhibition together with improvement of tumor microenvironment. Chin. J. Cancer Res. 2013, 25, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.; Kumari Singh, D.; Panda, S.; Shiras, A. Extracellular Vesicles As Modulators of Tumor Microenvironment and Disease Progression in Glioma. Front. Oncol. 2017, 7, 144. [Google Scholar] [CrossRef]
- Moncayo, G.; Grzmil, M.; Smirnova, T.; Zmarz, P.; Huber, R.M.; Hynx, D.; Kohler, H.; Wang, Y.; Hotz, H.R.; Hynes, N.E.; et al. SYK inhibition blocks proliferation and migration of glioma cells and modifies the tumor microenvironment. Neuro Oncol. 2018, 20, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Nollen, E.A.A.; Rohrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov. 2019, 18, 379–401. [Google Scholar] [CrossRef] [PubMed]
- Woroniecka, K.; Chongsathidkiet, P.; Rhodin, K.; Kemeny, H.; Dechant, C.; Farber, S.H.; Elsamadicy, A.A.; Cui, X.; Koyama, S.; Jackson, C.; et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin. Cancer Res. 2018, 24, 4175–4186. [Google Scholar] [CrossRef] [PubMed]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Campian, J.L.; Ghosh, S.; Kapoor, V.; Yan, R.; Thotala, S.; Jash, A.; Hu, T.; Mahadevan, A.; Rifai, K.; Page, L.; et al. Long-Acting Recombinant Human Interleukin-7, NT-I7, Increases Cytotoxic CD8 T Cells and Enhances Survival in Mouse Glioma Models. Clin. Cancer Res. 2022, 28, 1229–1239. [Google Scholar] [CrossRef]
- Wojton, J.; Kaur, B. Impact of tumor microenvironment on oncolytic viral therapy. Cytokine Growth Factor Rev. 2010, 21, 127–134. [Google Scholar] [CrossRef]
- Berkey, S.E.; Thorne, S.H.; Bartlett, D.L. Oncolytic Virotherapy and the Tumor Microenvironment. Adv. Exp. Med. Biol. 2017, 1036, 157–172. [Google Scholar] [CrossRef]
- Achard, C.; Surendran, A.; Wedge, M.E.; Ungerechts, G.; Bell, J.; Ilkow, C.S. Lighting a Fire in the Tumor Microenvironment Using Oncolytic Immunotherapy. EBioMedicine 2018, 31, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Marelli, G.; Howells, A.; Lemoine, N.R.; Wang, Y. Oncolytic Viral Therapy and the Immune System: A Double-Edged Sword Against Cancer. Front. Immunol. 2018, 9, 866. [Google Scholar] [CrossRef] [PubMed]
- Lemos de Matos, A.; Franco, L.S.; McFadden, G. Oncolytic Viruses and the Immune System: The Dynamic Duo. Mol. Ther. Methods Clin. Dev. 2020, 17, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Prestwich, R.J.; Errington, F.; Diaz, R.M.; Pandha, H.S.; Harrington, K.J.; Melcher, A.A.; Vile, R.G. The case of oncolytic viruses versus the immune system: Waiting on the judgment of Solomon. Hum. Gene Ther. 2009, 20, 1119–1132. [Google Scholar] [CrossRef]
- Hwang, J.K.; Hong, J.; Yun, C.O. Oncolytic Viruses and Immune Checkpoint Inhibitors: Preclinical Developments to Clinical Trials. Int. J. Mol. Sci. 2020, 21, 8627. [Google Scholar] [CrossRef]
- Evgin, L.; Kottke, T.; Tonne, J.; Thompson, J.; Huff, A.L.; van Vloten, J.; Moore, M.; Michael, J.; Driscoll, C.; Pulido, J.; et al. Oncolytic virus-mediated expansion of dual-specific CAR T cells improves efficacy against solid tumors in mice. Sci. Transl. Med. 2022, 14, eabn2231. [Google Scholar] [CrossRef]
- Evgin, L.; Vile, R.G. Parking CAR T Cells in Tumours: Oncolytic Viruses as Valets or Vandals? Cancers 2021, 13, 1106. [Google Scholar] [CrossRef]


{kind=link}
| Virus | Clinical Trial Number | Phase | Dates | Status | Patient Population | Delivery | Dosing | Median OS (Months) | Range OS (Months) | Adverse Events | Citation |
|---|---|---|---|---|---|---|---|---|---|---|---|
| AdV-tk + GCV | N/A | I | - | Completed | Recurrent malignant glioma (9 GBM, 1 gliosarcoma, 3 AA) | Single IT inj | 2 × 109, 2 × 1010, 2 × 1011, 2 × 1012 vp | 16.8 * | 10.1–47.6 * | Seizure, hemiparesis, thrombocytopenia, hyponatremia, confusion, lethargy | Trask et al. (2000) [24] |
| AdV-tk + VCV | NCT00589875 | IIa | 10 January 2008–11 April 2017 | Completed | Malignant glioma (34 GBM, 2 AA/AO) | Resection Bed | 3 × 1011 vp | 16.7 *,§ | Not given | Fatigue, fever, headache, wound complication, seizure | Wheeler et al. (2016) [30] |
| AdV-tk + VCV | NCT00634231 | I | 12 March 2008–2 November 2021 | Completed | Pediatric malignant glioma (6 GBM, 1 AA, 1 recurrent ependymoma) | Resection Bed | 1 × 1011, 3 × 1011 vp | 15.9 * | 7.4–37.3 * | Fever, fatigue, nausea/vomiting, hyponatremia | Kieran et al. (2019) [31] |
| AdV-tk + VCV | NCT00751270 | Ib | 11 September 2008–4 March 2016 | Completed | Malignant glioma (10 GBM, 2 AA) | Resection Bed | 3 × 1010, 1 × 1011, 3 × 1011 vp | 10.9 * | 2.0–46.4 * | Fever, wound complication, nausea/vomiting, transaminitis, hyponatremia, confusion, headache | Chiocca et al. (2011) [32] |
| AdV-tk + GCV | NCT00870181 | II | 27 March 2009–25 June 2013 | Completed | Recurrent high-grade glioma (14 recurrent GBM, 8 AA/AO) | Intra-arterial cerebral infusion | Not given | 10.4 * | 2.1–54.9 * | Nausea/vomiting, vasospasm, transaminitis | Ji et al. (2016) [33] |
| AdV-tk + VCV | NCT03576612 | I | 3 July 2018–present | Active, not recruiting | Malignant glioma (36 patients allotted to study) | Resection Bed | Not given | - | - | - | - |
| Ad-RTS-hIL-12 + VDX | NCT02026271 | I | 1 January 2014–22 September 2021 | Completed | Recurrent/progressive GBM or grade III malignant glioma (28 GBM, 3 astrocytoma) | Resection bed inj | 2 × 1011 vp; 10, 20, 30, or 40 mg VDX | 12.7 (20 mg VDX arm) | Not given-30 | Lymphopenia, transaminitis, thrombocytopenia, hyponatremia, CRS, headache, confusion, aseptic meningitis | Chiocca et al. (2019) [34] |
| Ad-RTS-hIL-12 + VDX + Nivolumab | NCT03636477 | I | 17 August 2018–4 October 2021 | Completed | Recurrent or progressive GBM (21 patients enrolled) | Resection bed inj | 2 × 1011 vp; 10 or 20 mg VDX; 1 or 3 mg/kg nivolumab | 9.8 | 1–24 | Transaminitis, brain edema, cold type headache, lymphopenia, CRS | Chiocca et al. (2022) [35] |
| Ad-RTS-hIL-12 + VDX | NCT03679754 | I | 20 September 2018–22 September 2021 | Completed | Recurrent or progressive GBM (36 patients enrolled) | Resection bed inj | 2 × 1011 vp; 20 mg VDX | 16.2 (unifocal, ≤20 mg dex, n = 20) | Not published | Not published | (abstract) Lukas et al. (2020) [36] |
| Ad-RTS-hIL-12 + VDX + Cemiplimab-rwlc | NCT04006119 | II | 2 July 2019–11 November 2021 | Completed | Recurrent or progressive GBM (40 patients allotted to study) | Resection bed inj | 2 × 1011 vp; 20 mg VDX; 350 mg cemiplimab-rwlc | Not published | Not published | Not published | (abstract) Lukas et al. (2021) [37] |
| DNX-2401 | NCT00805376 | I | 9 December 2008–16 July 2018 | Completed | Recurrent malignant glioma (33 GBM, 2 AA, 2 gliosarcoma) | Single IT inj (Arm A), Single IT inj + resection bed inj (Arm B) | 1 × 107, 3 × 107, 1 × 108, 3 × 108, 1 × 109, 3 × 109, 1 × 1010, 3 × 1010 vp | 9.8 * | 2.3–57.9 * | Headache, speech disorder, hemiparesis, convulsion, muscular weakness, visual field defect | Lang et al. (2018) [38] |
| DNX-2401 | NCT01582516 | I/II | 20 April 2012–9 March 2015 | Completed | Recurrent GBM (20 patients allotted) | CED intra- and peritumorally | 1 × 107, 1 × 108, 1 × 109, 1 × 1010, 3 × 1010, 1 × 1011 vp | - | - | - | - |
| DNX-2401 + TMZ | NCT01956734 | I | 8 October 2013–24 October 2017 | Completed | First recurrent GBM (31 patients allotted) | IT or resection bed inj | 3 × 1010 vp; 150 mg/m2 TMZ | - | - | - | - |
| DNX-2401 + IFN-γ | NCT02197169 | Ib | 22 July 2014–16 July 2018 | Completed | Recurrent GBM or gliosarcoma (27 GB) | Single IT inj | 3 × 1010 vp; 50 mcg/m2 s.c. IFN-γ | Not published (OS 12 m of 33%, 18 m 22%) | Not published | Fatigue, headache, seizures | (abstract) Lang et al. (2017) [39] |
| DNX-2401 + Pembrolizumab | NCT02798406 | II | 14 June 2016–15 July 2021 | Completed | Recurrent GBM or gliosarcoma (49 patients allotted) | Single IT inj | 5 × 108, 5 × 109, 5 × 1010 vp; 200 mg pembrolizumab | - | - | - | - |
| DNX-2401 | - | I | - | Completed | Recurrent GBM (19 patients treated) | CED | 1 × 107, 1 × 108, 1 × 109, 1 × 1010, 3 × 1010 vp | 4.2 | 2.2–91.8 | Confusion, seizure, increased ICP, meningitis, hydrocephalus | van Putten et al. (2022) [40] |
| MSC loaded with DNX-2401 | NCT03896568 | I | 1 April 2019–present | Active, recruiting | Recurrent HGG (36 patients allotted) | Intra-arterial | Not given | - | - | - | - |
| NSC loaded with CRAd-Survivin-pk7 | NCT03072134 | I | 7 March 2017–20 January 2023 | Completed | Newly diagnosed malignant glioma (11 GBM, 1 AA) | Resection bed inj | 6.25 × 1010 vp in 5 × 107 NSCs, 1.25 × 1011 vp in 1 × 108 NSCs, 1.875 × 1011 in 1.5 × 108 NSCs | 18.4 | Not published | Meningitis, thromboembolic event, encephalopathy, cerebral edema, muscle weakness | Fares et al. (2021) [41] |
| NSC loaded with CRAd-Survivin-pk7 | NCT05139056 | I | 1 December 2021–present | Active, recruiting | Recurrent HGG (36 patients allotted) | Intracavitary post resection | Not given | - | - | - | - |
| M032-HSV-1 | NCT02062827 | I | 14 February 2014–present | Active, not recruiting | Recurrent or progressive GBM, AA, or gliosarcoma (24 patients allotted) | IT catheter infusion | 1 × 105, 1 × 106, 1 × 107, 1 × 108, 1 × 109 pfu | - | - | - | - |
| M032-HSV-1 + Pembrolizumab | NCT05084430 | I/II | 19 October 2021–present | Active, recruiting | Recurrent, progressive, or newly diagnosed GBM, AA, or gliosarcoma (28 patients allotted) | Not given | Not given; 200 mg pembrolizumab | - | - | - | - |
| rQNestin34.5v.2 (CAN-3110) | NCT03152318 | I | 15 May 2017–11 January 2023 | Active, recruiting | Recurrent malignant glioma (26 GBM, 1 AA, 3 AO) | Single IT inj | 1 × 106 at half-log increments up 1 × 1010 pfu | 13.25 | Not published | Not published | (abstract) Chiocca et al. (2021) [42] |
| G47∆ | UMIN000002661 | I/II | 23 October 2009–14 March 2019 | Completed | Recurrent GBM (13 patients) | IT inj, 2 doses | 3 × 108 or 1 × 109 pfu per dose | 30.5 (7.3 ǂ) | 3.2–143.9 ǂ | Headache, fever, vomiting, leukopenia, CN disorder, seizure | Todo et al. (2022) [43] |
| G47∆ | UMIN000015995 | II | 18 December 2014–26 June 2020 | Completed | Residual or recurrent GBM (19 patients) | IT inj up to 6 doses | 1 × 109 pfu per dose | 28.8 (20.2 ǂ) | 4.2–65.3 ǂ | Fever, vomiting, nausea, lymphocytopenia, leukopenia | Todo et al. (2022) [44] |
| PVSRIPO | NCT01491893 | I | 14 December 2011–28 September 2018 | Completed | Recurrent malignant glioma (61 GBM) | IT CED | 1 × 107, 5 × 107, 1 × 108, 3.3 × 108, 1 × 109, 3.3 × 109, 1 × 1010 TCID50 | 12.5 | 3.1–70.4 | Fatigue, gait disturbance, confusion, dysphagia, headache, paresthesia, pyramidal tract syndrome, seizure | Desjardins et al. (2018) [45] |
| PVSRIPO | NCT02986178 | II | 8 December 2016–present | Active, not recruiting | Recurrent malignant glioma (122 patients allotted) | IT CED | - | - | - | - | - |
| PVSRIPO | NCT03043391 | Ib | 6 February 2017–present | Active, not recruiting | Recurrent malignant glioma, pediatric (12 patients allotted) | IT CED | - | - | - | - | - |
| PVSRIPO + pembrolizumab | NCT04479241 | II | 21 July 2020–present | Active, not recruiting | Recurrent GBM (30 patients allotted) | IT CED | - | - | - | - | - |
| PVSRIPO | NCT04599647 | Expanded access | 23 October 2020–29 June 2022 | No longer available | N/A | IT CED | 5 × 107 TCID50 | - | - | - | - |
| Toca 511 + Toca FC | NCT01156584 | I | 5 July 2010–21 May 2018 | Completed | Recurrent HGG (36 patients at time of abstract, subtypes not published) | IT inj (24 patients) or CED (12 patients), Toca FC oral | 3.9 × 106 TU, half logs to a maximum of 1.5 × 109 TU; 120 mg/kg/day or 300 mg/kg/day Toca FC | Not published | Not published | - | (abstract) Aghi et al. (2014) [46] |
| Toca 511 + Toca FC | NCT01470794 | I | 11 November 2011–21 May 2018 | Completed | Recurrent HGG (46 GBM, 6 AA, 4 other) | Resection bed inj, Toca FC oral | Not given | 11.9 | Not published | Rash, mucositis, facial swelling, hemorrhagic enteritis, colitis, nausea, vomiting, diarrhea | Cloughesy et al. (2018) [47] |
| Toca 511 + Toca FC | NCT01985256 | I | 15 November 2013–21 May 2018 | Completed | Recurrent HGG (17 patients allotted) | IV + resection bed inj, Toca FC oral | 4.6 × 109 TU IV/day for 3 days, 9.5 × 109 TU IV/day for 5 days. (1.2 × 109 TU inj tumor bed); 220 mg/kg/day Toca FC | Not published | Not published | Not published | (abstract) Cloughesy et al. (2015) [48] |
| Toca 511 + Toca FC | NCT02414165 | II/III | 10 April 2015–7 February 2020 | Terminated | Recurrent GBM or AA (171 GBM, 30 AA) | Resection bed inj, Toca FC oral | 4 × 108 TU; Toca FC 220 mg/kg/d | 11.1 | Not published | Aphasia, hemiparesis, headache, seizure | Cloughesy et al. (2020) [49] |
| Toca 511 + Toca FC | NCT02598011 | Ib | 5 November 2015–30 March 2020 | Withdrawn | Recurrent HGG | Resection bed inj, Toca FC oral | - | - | - | - | - |
| Toca 511 + Toca FC | NCT04105374 | II/III | 26 September 2019–24 March 2020 | Withdrawn | Newly diagnosed GBM | Intracranial injection | - | - | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Webb, M.J.; Sener, U.; Vile, R.G. Current Status and Challenges of Oncolytic Virotherapy for the Treatment of Glioblastoma. Pharmaceuticals 2023, 16, 793. https://doi.org/10.3390/ph16060793
Webb MJ, Sener U, Vile RG. Current Status and Challenges of Oncolytic Virotherapy for the Treatment of Glioblastoma. Pharmaceuticals. 2023; 16(6):793. https://doi.org/10.3390/ph16060793
Chicago/Turabian StyleWebb, Mason J., Ugur Sener, and Richard G. Vile. 2023. "Current Status and Challenges of Oncolytic Virotherapy for the Treatment of Glioblastoma" Pharmaceuticals 16, no. 6: 793. https://doi.org/10.3390/ph16060793
APA StyleWebb, M. J., Sener, U., & Vile, R. G. (2023). Current Status and Challenges of Oncolytic Virotherapy for the Treatment of Glioblastoma. Pharmaceuticals, 16(6), 793. https://doi.org/10.3390/ph16060793