

Design, Synthesis, and Evaluation of Niclosamide Analogs as Therapeutic Agents for Enzalutamide-Resistant Prostate Cancer

 ,
,
Abstract
1. Introduction
2. Results and Discussion
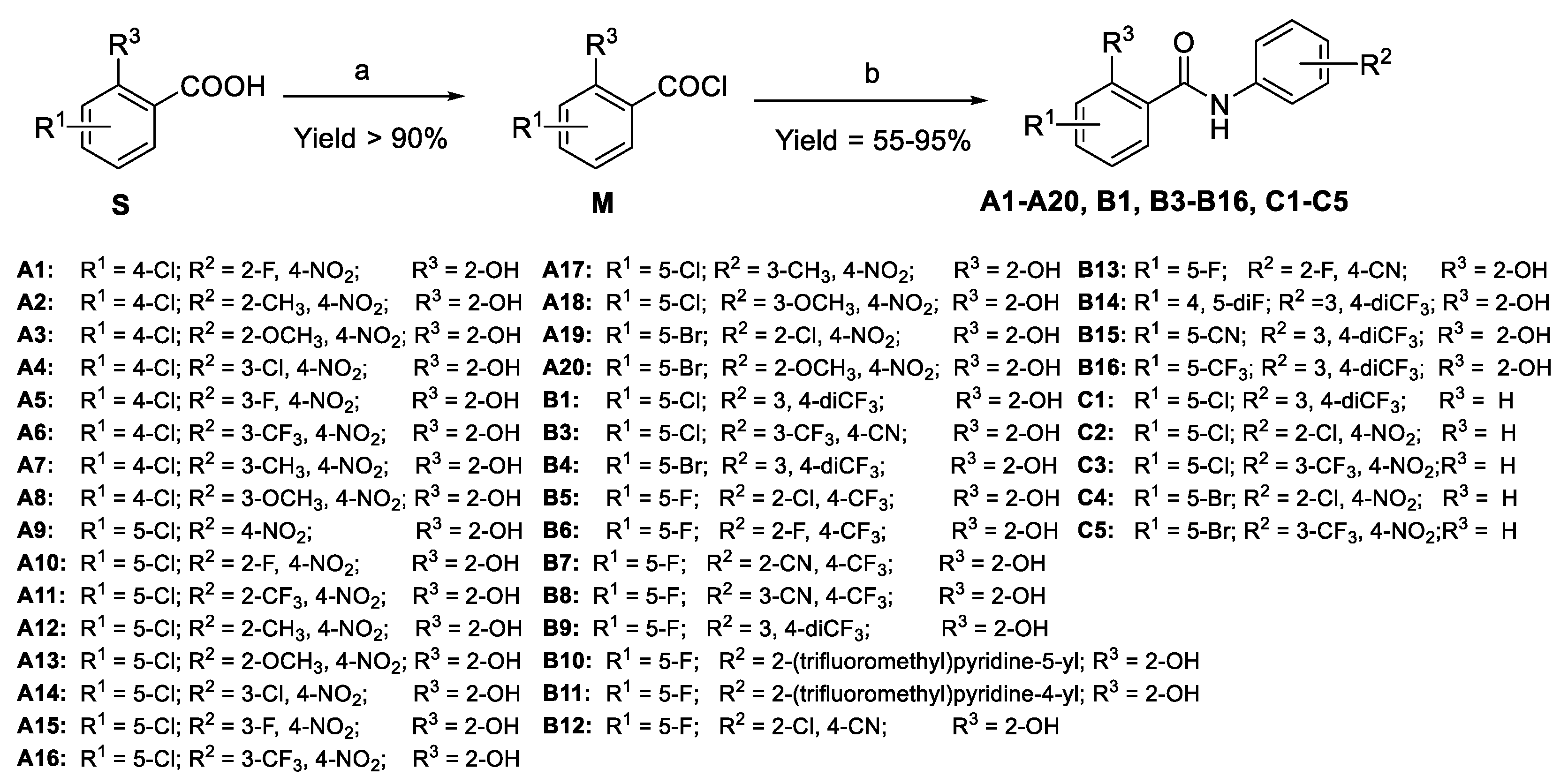
2.1. Chemistry
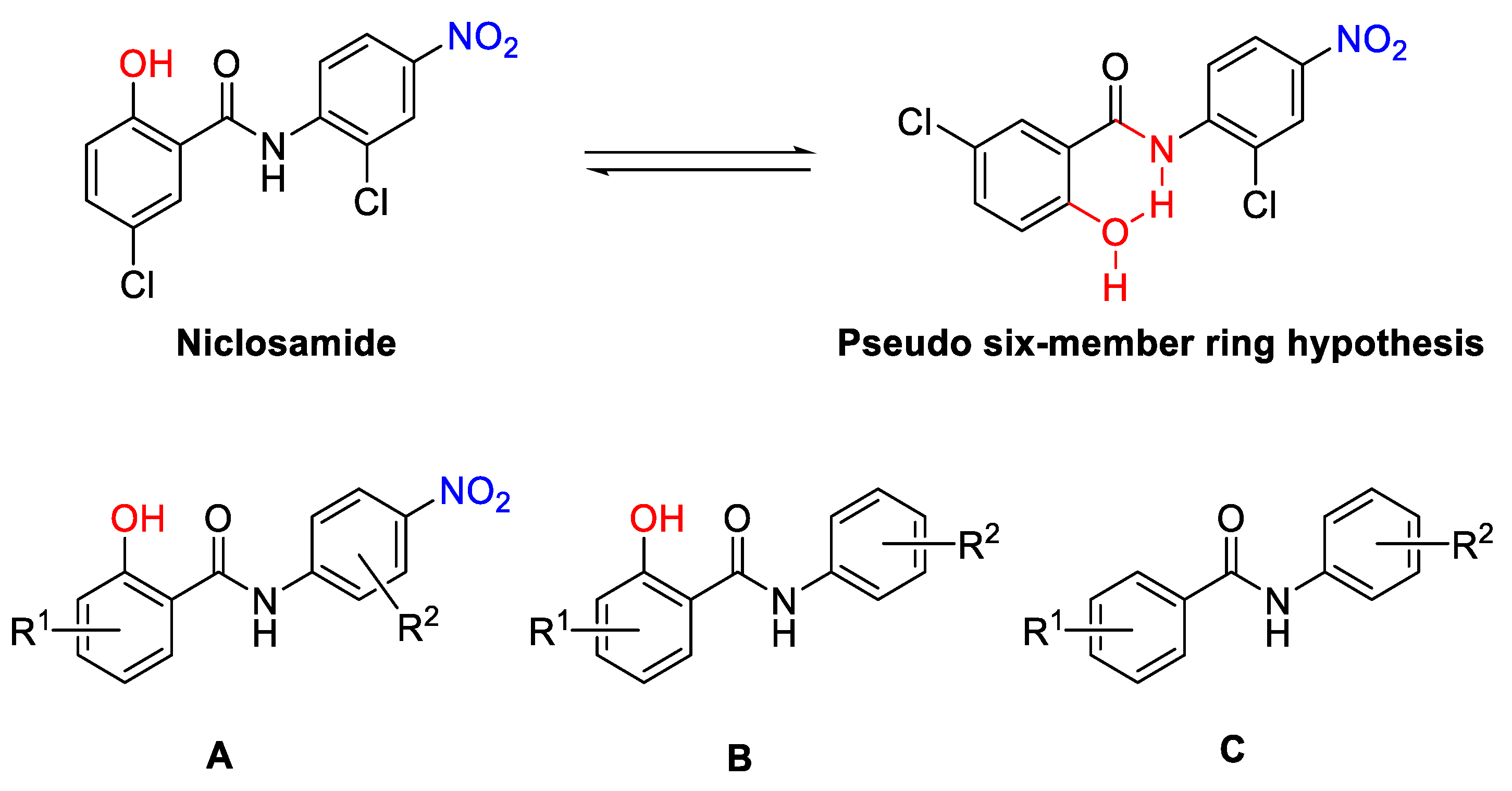
- Design
- Synthesis
2.2. Biological Screening
- Antiproliferative activities in enzalutamide resistant prostate cancer cells.
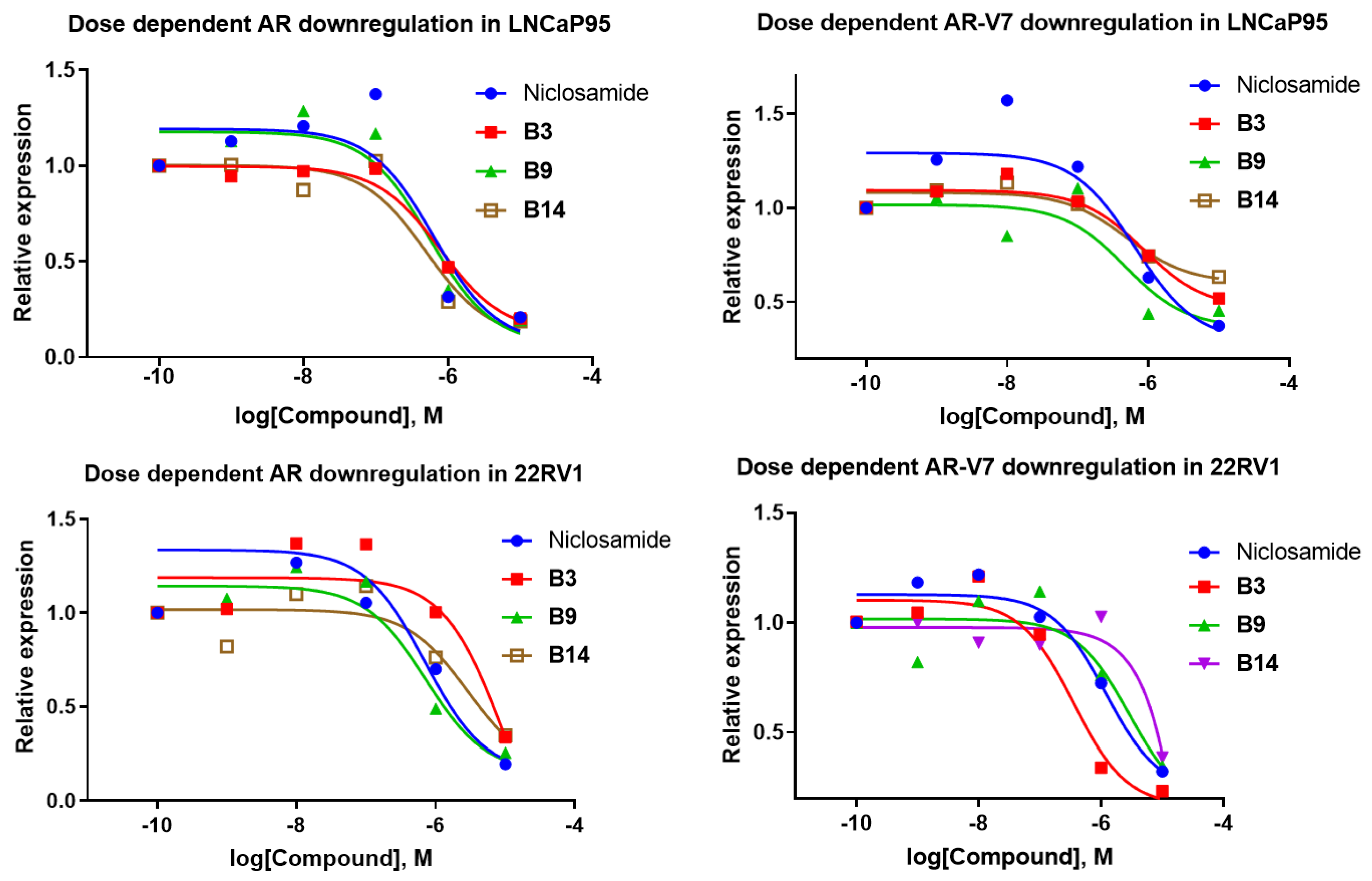
- Downregulation of AR-FL and AR-V7 in LNCaP95 and 22RV1 cells
2.3. Structure–Activity Relationship (SAR) Analysis
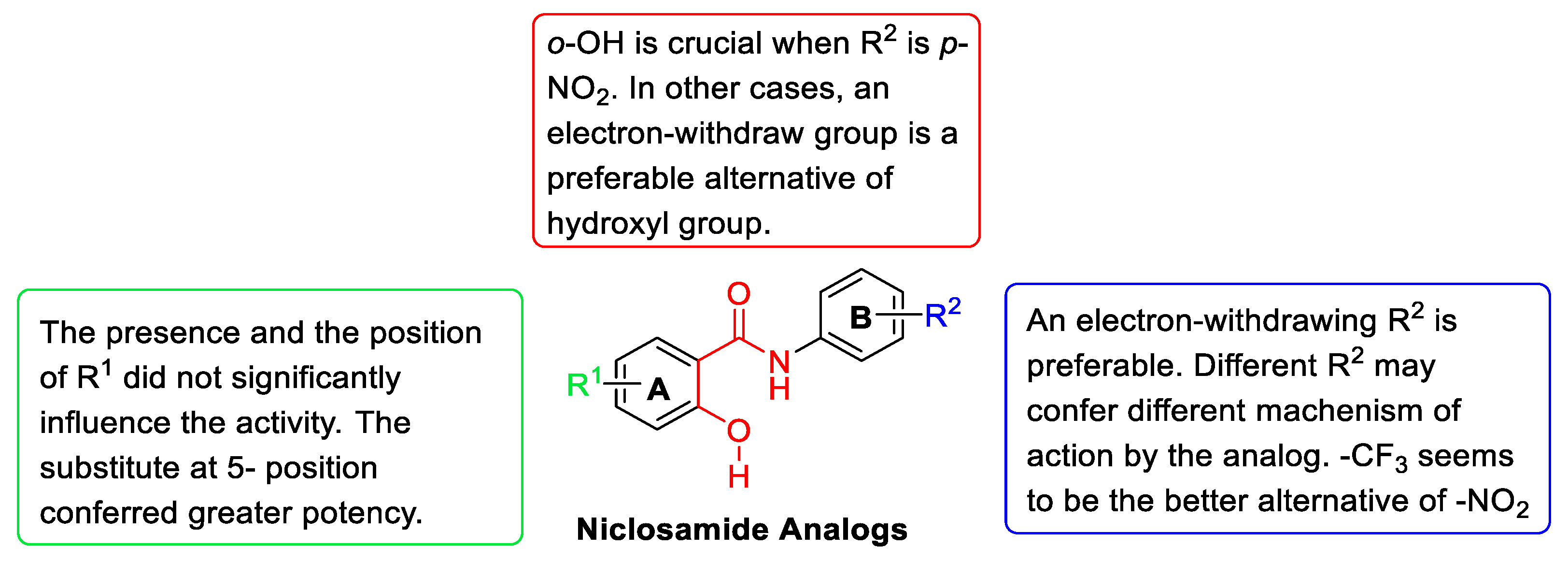
- The effect of substituents R1
- The effect of substituents R2
- The effect of o-OH on ring A
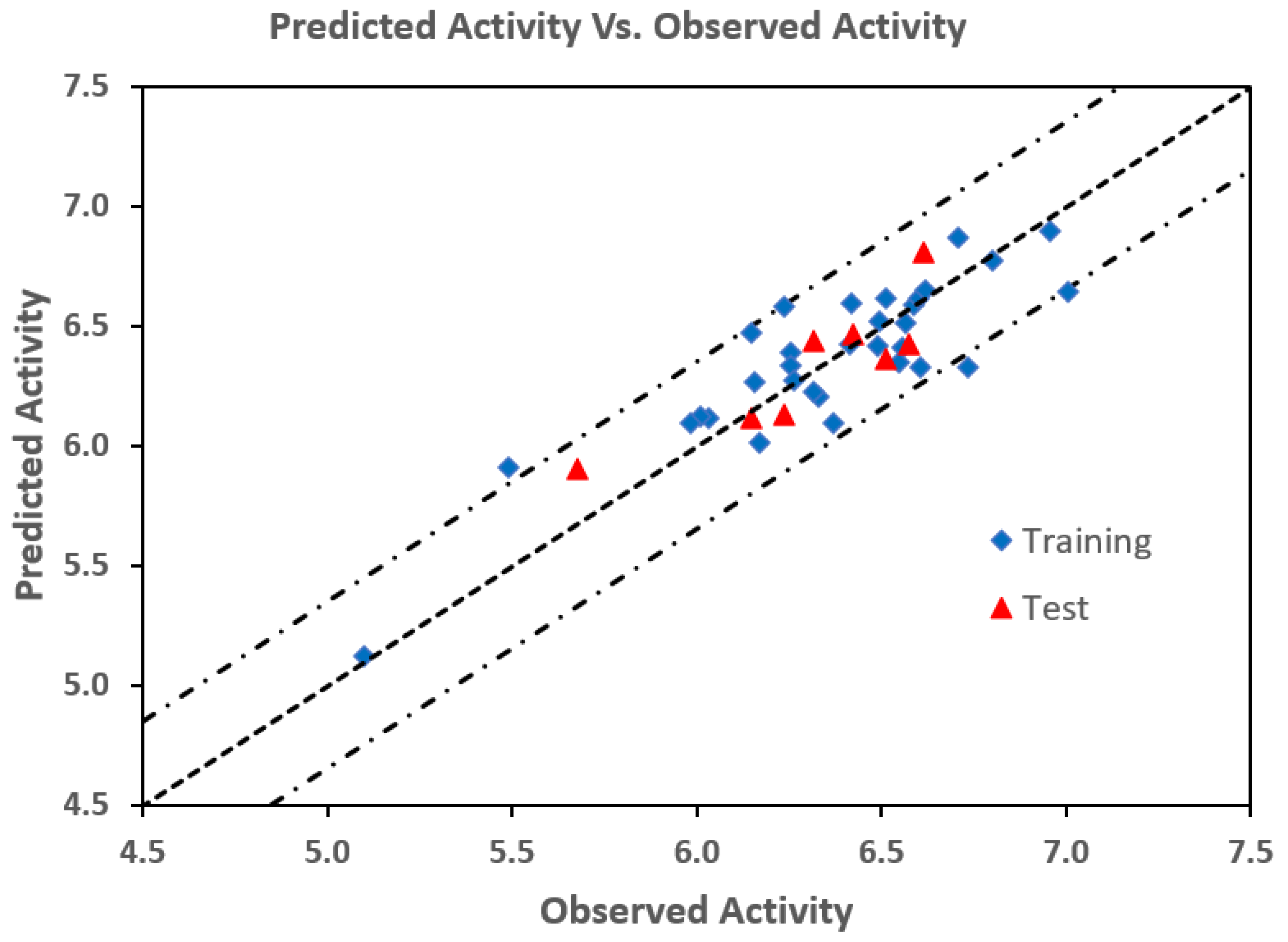
2.4. 3D-QSAR Analysis
3. Materials and Methods
3.1. General Information
3.2. General Procedures for the Preparation of A1–A20, B1–B16, and C1–C5
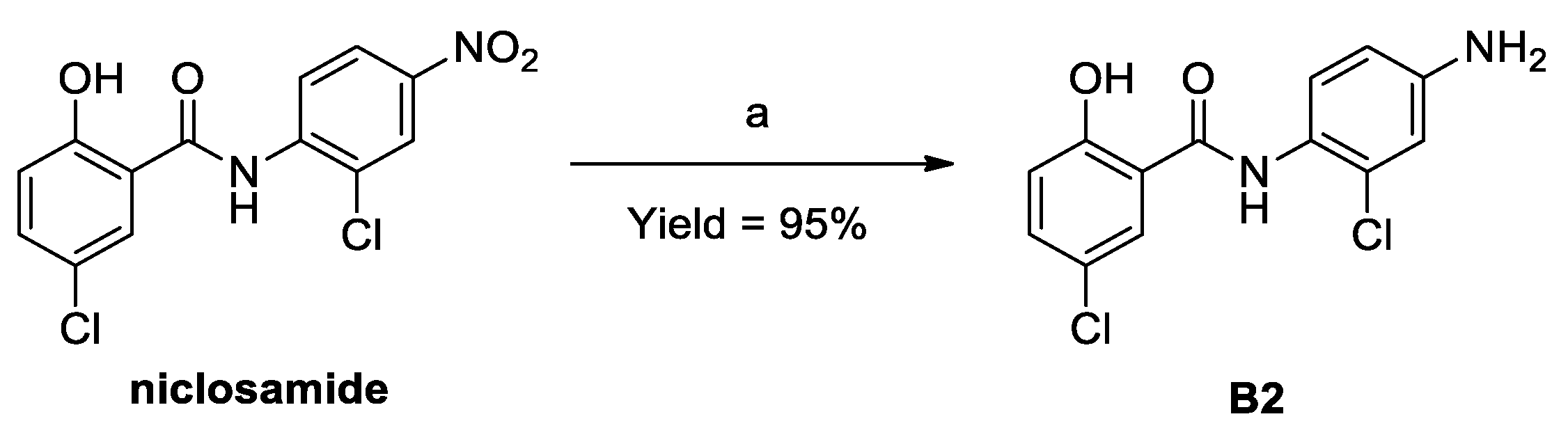
3.3. Procedures for the Preparation of B2
3.4. Procedures for the Preparation of C6–C8
3.5. Antiproliferative Assays
3.6. Western Blot Analysis
3.7. 3D-QSAR Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Attard, G.; Richards, J.; de Bono, J.S. New Strategies in Metastatic Prostate Cancer: Targeting the Androgen Receptor Signaling Pathway. Clin. Cancer Res. 2011, 17, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Carver, B.S. Strategies for targeting the androgen receptor axis in prostate cancer. Drug Discov. Today 2014, 19, 1493–1497. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Iczkowski, K.A.; Kilari, D.; See, W.; Nevalainen, M.T. Androgen receptor-dependent and -independent mechanisms driving prostate cancer progression: Opportunities for therapeutic targeting from multiple angles. Oncotarget 2017, 8, 3724–3745. [Google Scholar] [CrossRef]
- Lieberman, R. Androgen deprivation therapy for prostate cancer chemoprevention: Current status and future directions for agent development. Urology 2001, 58 (Suppl. S1), 83–90. [Google Scholar] [CrossRef]
- Dhawan, M.; Ryan, C.J. Utility of novel androgen receptor therapies in the real world: A nuanced approach. Urol. Oncol. Semin. Orig. Investig. 2016, 34, 340–347. [Google Scholar] [CrossRef]
- Crona, D.; Milowsky, M.; Whang, Y. Androgen receptor targeting drugs in castration-resistant prostate cancer and mechanisms of resistance. Clin. Pharmacol. Ther. 2015, 98, 582–589. [Google Scholar] [CrossRef]
- Modena, A.; Ciccarese, C.; Fantinel, E.; Bimbatti, D.; Tortora, G.; Massari, F. Metastatic castration-resistant prostate cancer: Targeting the mechanisms of resistance to abiraterone acetate and enzalutamide. Expert Rev. Anticancer. Ther. 2015, 15, 1037–1048. [Google Scholar] [CrossRef]
- Reichert, Z.R.; Hussain, M. Androgen Receptor and Beyond, Targeting Androgen Signaling in Castration-Resistant Prostate Cancer. Cancer J. 2016, 22, 326–329. [Google Scholar] [CrossRef]
- Graham, L.; Schweizer, M.T. Targeting persistent androgen receptor signaling in castration-resistant prostate cancer. Med. Oncol. 2016, 33, 44. [Google Scholar] [CrossRef]
- Imamura, Y.; Sadar, M.D. Androgen receptor targeted therapies in castration-resistant prostate cancer: Bench to clinic. Int. J. Urol. 2016, 23, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Mostaghel, E.A.; Marck, B.T.; Plymate, S.R.; Vessella, R.L.; Balk, S.; Matsumoto, A.M.; Nelson, P.S.; Montgomery, R.B. Resistance to CYP17A1 Inhibition with Abiraterone in Castration-Resistant Prostate Cancer: Induction of Steroidogenesis and Androgen Receptor Splice Variants. Clin. Cancer Res. 2011, 17, 5913–5925. [Google Scholar] [CrossRef]
- Galletti, G.; Leach, B.I.; Lam, L.; Tagawa, S.T. Mechanisms of resistance to systemic therapy in metastatic castration-resistant prostate cancer. Cancer Treat. Rev. 2017, 57, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Bubley, G.J.; Balk, S.P. Association Between Androgen Receptor Splice Variants and Prostate Cancer Resistance to Abiraterone and Enzalutamide. J. Clin. Oncol. 2017, 35, 2103–2105. [Google Scholar] [CrossRef]
- Li, Y.; Chan, S.C.; Brand, L.J.; Hwang, T.H.; Silverstein, K.A.; Dehm, S.M. Androgen Receptor Splice Variants Mediate Enzalutamide Resistance in Castration-Resistant Prostate Cancer Cell Lines. Cancer Res 2013, 73, 483–489. [Google Scholar] [CrossRef]
- Zhang, X.; Morrissey, C.; Sun, S.; Ketchandji, M.; Nelson, P.S.; True, L.D.; Vakar-Lopez, F.; Vessella, R.L.; Plymate, S.R. Androgen Receptor Variants Occur Frequently in Castration Resistant Prostate Cancer Metastases. PLoS ONE 2011, 6, e27970. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Sprenger, C.C.; Vessella, R.L.; Haugk, K.; Soriano, K.; Mostaghel, E.A.; Page, S.T.; Coleman, I.M.; Nguyen, H.M.; Sun, H.; et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J. Clin. Investig. 2010, 120, 2715–2730. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and Resistance to Enzalutamide and Abiraterone in Prostate Cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef]
- Pan, J.-X.; Ding, K.; Wang, C.-Y. Niclosamide, an old antihelminthic agent, demonstrates antitumor activity by blocking multiple signaling pathways of cancer stem cells. Chin. J. Cancer 2012, 31, 178–184. [Google Scholar] [CrossRef]
- Liu, C.; Lou, W.; Zhu, Y.; Nadiminty, N.; Schwartz, C.T.; Evans, C.P.; Gao, A.C. Niclosamide Inhibits Androgen Receptor Variants Expression and Overcomes Enzalutamide Resistance in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2014, 20, 3198–3210. [Google Scholar] [CrossRef]
- Liu, C.; Armstrong, C.; Zhu, Y.; Lou, W.; Gao, A.C. Niclosamide enhances abiraterone treatment via inhibition of androgen receptor variants in castration resistant prostate cancer. Oncotarget 2016, 7, 32210–32220. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, P.-K.; Roberts, M.J.; Arend, R.C.; Samant, R.S.; Buchsbaum, D.J. Multi-targeted therapy of cancer by niclosamide: A new application for an old drug. Cancer Lett. 2014, 349, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, J.K. The Merck Index, CD-ROM (Macintosh): An Encyclopedia of Chemicals, Drugs & Biologicals; Budavari, S., O’Neill, M., Smith, A., Heckelman, P., Kinneary, J., Eds.; Chapman & Hall: New York, NY, USA, 1997. [Google Scholar]
- Chang, Y.-W.; Yeh, T.-K.; Lin, K.-T.; Chen, W.-C.; Yao, H.-T.; Lan, S.-J.; Wu, Y.-S.; Hsieh, H.-P.; Chen, C.-M. Pharmacokinetics of anti-SARS-CoV agent niclosamide and its analogs in rats. J. Food Drug Anal. 2006, 14, 15. [Google Scholar] [CrossRef]
- Chen, H.; Yang, Z.; Ding, C.; Chu, L.; Zhang, Y.; Terry, K.; Liu, H.; Shen, Q.; Zhou, J. Discovery of O-Alkylamino-Tethered Niclosamide Derivatives as Potent and Orally Bioavailable Anticancer Agents. ACS Med. Chem. Lett. 2013, 4, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Acuña, U.M.; Fernandes, N.F.; Chettiar, S.; Li, P.-K.; De Blanco, E.C. Structure–Activity Relationship of Niclosamide Derivatives. Anticancer. Res. 2017, 37, 2839–2843. [Google Scholar] [CrossRef]
- Chen, W.; Mook, R.A., Jr.; Premont, R.T.; Wang, J. Niclosamide: Beyond an antihelminthic drug. Cell. Signal. 2018, 41, 89–96. [Google Scholar] [CrossRef]
- Mook, R.A.; Wang, J.; Ren, X.-R.; Chen, M.; Spasojevic, I.; Barak, L.S.; Lyerly, H.K.; Chen, W. Structure–activity studies of Wnt/β-catenin inhibition in the Niclosamide chemotype: Identification of derivatives with improved drug exposure. Bioorg. Med. Chem. 2015, 23, 5829–5838. [Google Scholar] [CrossRef]
- Li, S.-L.; Yan, J.; Zhang, Y.-Q.; Zhen, C.-L.; Liu, M.-Y.; Jin, J.; Gao, J.-L.; Xiao, X.-L.; Shen, X.; Tai, Y.; et al. Niclosamide ethanolamine inhibits artery constriction. Pharmacol. Res. 2016, 115, 78–86. [Google Scholar] [CrossRef]
- Lin, C.-K.; Bai, M.-Y.; Hu, T.-M.; Wang, Y.-C.; Chao, T.-K.; Weng, S.-J.; Huang, R.-L.; Su, P.-H.; Lai, H.-C. Preclinical evaluation of a nanoformulated antihelminthic, niclosamide, in ovarian cancer. Oncotarget 2016, 7, 8993–9006. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y.; Zhang, T.; Zhang, J.; Wu, B. Significantly enhanced bioavailability of niclosamide through submicron lipid emulsions with or without PEG-lipid: A comparative study. J. Microencapsul. 2015, 32, 496–502. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger Release 2022-02: Maestro; Schrödinger, LLC: New York, NY, USA, 2021.
- Schrodinger Release 2022-02: Field-Based QSAR; Schrödinger, LLC: New York, NY, USA, 2021.
- Schrodinger Release 2022-02: QikProp; Schrödinger, LLC: New York, NY, USA, 2021.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Compounds | Structure | Inhibitory Rate | IC50 (μM) a | ||
| R1 | R2 | 22RV1 @1.0 μM | LNCaP95 | 22RV1 | |
| DMSO | −2.10% | Nt b | Nt b | ||
| A1 | 4-Cl | 2′-F | 54.3% | 1.19 ± 0.53 | 0.467 ± 0.131 |
| A2 | 2′-CH3 | 47.1% | 0.620 ± 0.311 | 0.676 ± 0.245 | |
| A3 | 2′-OCH3 | 50.0% | 2.48 ± 0.702 | 0.714 ± 0.303 | |
| A4 | 3′-Cl | 47.5% | 3.43 ± 0.49 | 0.713 ± 0.178 | |
| A5 | 3′-F | 44.2% | 0.937 ± 0.351 | 0.924 ± 0.414 | |
| A6 | 3′-CF3 | 54.4% | 0.214 ± 0.067 | 0.424 ± 0.120 | |
| A7 | 3′-CH3 | 42.5% | 0.789 ± 0.436 | 0.976 ± 0.205 | |
| A8 | 3′-OCH3 | 35.9% | 0.615 ± 0.138 | 2.11 ± 0.52 | |
| A9 | 5-Cl | H | 44.9% | 0.812 ± 0.374 | 0.581 ± 0.289 |
| A10 | 2′-F | 56.0% | 0.373 ± 0.069 | 0.271 ± 0.125 | |
| A11 | 2′-CF3 | −8.30% | nt b | Nt b | |
| A12 | 2′-CH3 | 58.0% | 0.493 ± 0.105 | 0.248 ± 0.063 | |
| A13 | 2′-OCH3 | 57.3% | 0.444 ± 0.119 | 0.158 ± 0.044 | |
| A14 | 3′-Cl | 51.3% | 1.67 ± 0.25 | 0.386 ± 0.171 | |
| A15 | 3′-F | 55.4% | 0.755 ± 0.221 | 0.280 ± 0.092 | |
| A16 | 3′-CF3 | 47.5% | 0.693 ± 0.314 | 0.556 ± 0.112 | |
| A17 | 3′-CH3 | 50.1% | 0.867 ± 0.298 | 0.324 ± 0.058 | |
| A18 | 3′-OCH3 | 55.4% | 2.11 ± 0.46 | 0.483 ± 0.145 | |
| A19 | 5-Br | 2′-Cl | 13.40% | Nt b | nt b |
| A20 | 2′-OCH3 | 60.2% | 0.304 ± 0.053 | 0.242 ± 0.044 | |
| B1 | 5-Cl | 3′, 4′-diCF3 | 59.2% | 0.239 ± 0.072 | 0.258 ± 0.097 |
| B2 | 2′-Cl, 4′-NH2 | 19.60% | nt b | nt b | |
| B3 | 3′-CF3, 4′-CN | 34.9% | 0.354 ± 0.066 | 0.695 ± 0.246 | |
| B4 | 5-Br | 3′, 4′-diCF3 | 53.1% | 0.213 ± 0.042 | 0.307 ± 0.059 |
| B5 | 5-F | 2′-Cl, 4′-CF3 | 59.2% | 0.384 ± 0.088 | 0.375 ± 0.063 |
| B6 | 2′-F, 4′-CF3 | 62.5% | 0.185 ± 0.041 | 0.196 ± 0.032 | |
| B7 | 2′-CN, 4′-CF3 | 42.3% | 4.38 ± 1.33 | 7.92 ± 2.56 | |
| B8 | 3′-CN, 4′-CF3 | 60.4% | 0.396 ± 0.065 | 0.240 ± 0.048 | |
| B9 | 3′, 4′-diCF3 | 84.3% | 0.130 ± 0.033 | 0.0997 ± 0.0221 | |
| B10 |  | 42.3% | 0.627 ± 0.225 | 0.547 ± 0.153 | |
| B11 |  | 47.7% | 0.392 ± 0.136 | 0.581 ± 0.205 | |
| B12 | 2′-Cl, 4′-CN | 44.3% | 14.0 ± 5.6 | 3.23 ± 1.84 | |
| B13 | 2′-F, 4′-CN | 65.6% | 0.231 ± 0.072 | 0.184 ± 0.061 | |
| B14 | 4,5-diF | 3′, 4′-diCF3 | 47.3% | 0.199 ± 0.055 | 0.382 ± 0.078 |
| B15 | 5-CN | 44.8% | 2.35 ± 0.53 | 1.04 ± 0.26 | |
| B16 | 5-CF3 | 64.6% | 0.113 ± 0.044 | 0.111 ± 0.023 | |
| C1 | 5-Cl | 3′, 4′-diCF3 | 53.1% | 0.299± | 0.320 ± 0.115 |
| C2 C3 | 2′-Cl, 4′-NO2 | 19.0% | nt b | nt b | |
| 3′-CF3, 4′-NO2 | 43.7% | 0.330 ± 0.081 | 0.557 ± 0.127 | ||
| C4 | 5-Br | 2′-Cl, 4′-NO2 | 31.2% | nt b | nt b |
| C5 | 3′-CF3, 4′-NO2 | 58.3% | 0.536 ± 0.201 | 0.306 ± 0.058 | |
| C6 | 2-TfO, 4-Cl | 3′-CH3, 4′-NO2 | 59.4% | 0.485 ± 0.147 | 0.268 ± 0.066 |
| C7 | 2-AcO, 5-Cl | 2′-Cl, 4′-NO2 | 42.9% | 0.386 ± 0.098 | 0.481 ± 0.113 |
| C8 | 2-TfO, 5-Cl | 59.2% | 0.196 ± 0.064 | 0.252 ± 0.092 | |
| Niclosamide | 55.8% | 0.372 ± 0.074 | 0.284 ± 0.059 | ||
| Enzalutamide | 24.1% | >10,000 | 695 | ||
| Compound | LNCaP95 | 22RV1 | ||
|---|---|---|---|---|
| AR Downregulation IC50 (µM) a | AR-V7 Downregulation IC50 (µM) a | AR Downregulation IC50 (µM) a | AR-V7 Downregulation IC50 (µM) a | |
| Niclosamide | 0.633 ± 0.252 | 0.644 ± 0.315 | 0.707 ± 0.248 | 6.98 ± 0.401 |
| B1 | 0.463 ± 0.224 | 0.280 ± 0.117 | 0.158 ± 0.087 | 4.28 ± 1.36 |
| B3 | 0.781 ± 0.512 | 0.850 ± 0.246 | 9.93 ± 2.41 | 0.354 ± 0.134 |
| B9 | 0.551 ± 0.173 | 0.467 ± 0.204 | 0.722 ± 0.158 | 2.86 ± 0.95 |
| B14 | 0.498 ± 0.185 | 0.491 ± 0.156 | 2.864 ± 0.793 | >10 |
| No | Compd | Obs pIC50 | Pred pIC50 | δ | No | Compd | Obs pIC50 | Pred pIC50 | δ |
|---|---|---|---|---|---|---|---|---|---|
| 1 | A1 | 6.331 | 6.204 | 0.127 | 21 | B4 | 6.513 | 6.617 | −0.104 |
| 2 | A2 | 6.170 | 6.011 | 0.159 | 22 | B5 | 6.426 | 6.460 | −0.034 |
| 3 | A3 | 6.146 | 6.472 | −0.326 | 23 | B6 | 6.708 | 6.867 | −0.159 |
| 4 | A4 * | 6.147 | 6.115 | 0.032 | 24 | B7 | 5.101 | 5.124 | −0.023 |
| 5 | A5 | 6.034 | 6.117 | −0.083 | 25 | B8 * | 6.620 | 6.652 | −0.032 |
| 6 | A6 | 6.373 | 6.090 | 0.283 | 26 | B9 | 7.004 | 6.645 | 0.359 |
| 7 | A7 | 6.011 | 6.120 | −0.109 | 27 | B10 | 6.262 | 6.272 | −0.010 |
| 8 | A8 * | 5.676 | 5.905 | −0.229 | 28 | B11 * | 6.236 | 6.128 | 0.108 |
| 9 | A9 | 6.236 | 6.581 | −0.345 | 29 | B12 | 5.491 | 5.908 | −0.417 |
| 10 | A10 | 6.567 | 6.518 | 0.049 | 30 | B13 | 6.735 | 6.329 | 0.406 |
| 11 | A12 | 6.606 | 6.327 | 0.279 | 31 | B14 | 6.418 | 6.591 | −0.173 |
| 12 | A13 | 6.801 | 6.771 | 0.030 | 32 | B15 | 5.983 | 6.092 | −0.109 |
| 13 | A14 | 6.413 | 6.442 | −0.009 | 33 | B16 | 6.955 | 6.895 | 0.060 |
| 14 | A15 | 6.553 | 6.412 | 0.141 | 34 | C1 | 6.495 | 6.524 | −0.034 |
| 15 | A16 | 6.255 | 6.390 | −0.135 | 35 | C3 | 6.254 | 6.331 | −0.077 |
| 16 | A17 | 6.489 | 6.416 | 0.073 | 36 | C5 * | 6.514 | 6.365 | 0.149 |
| 17 | A18 | 6.316 | 6.219 | 0.097 | 37 | C6 * | 6.572 | 6.420 | 0.152 |
| 18 | A20 * | 6.616 | 6.807 | −0.191 | 38 | C7 * | 6.318 | 6.442 | −0.124 |
| 19 | B1 | 6.588 | 6.586 | 0.002 | 39 | C8 | 6.599 | 6.615 | −0.016 |
| 20 | B3 | 6.158 | 6.262 | −0.104 | 40 | Niclosamide | 6.547 | 6.350 | 0.197 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, B.; Mottamal, M.; Zhong, Q.; Bratton, M.; Zhang, C.; Guo, S.; Hossain, A.; Ma, P.; Zhang, Q.; Wang, G.; et al. Design, Synthesis, and Evaluation of Niclosamide Analogs as Therapeutic Agents for Enzalutamide-Resistant Prostate Cancer. Pharmaceuticals 2023, 16, 735. https://doi.org/10.3390/ph16050735
Kang B, Mottamal M, Zhong Q, Bratton M, Zhang C, Guo S, Hossain A, Ma P, Zhang Q, Wang G, et al. Design, Synthesis, and Evaluation of Niclosamide Analogs as Therapeutic Agents for Enzalutamide-Resistant Prostate Cancer. Pharmaceuticals. 2023; 16(5):735. https://doi.org/10.3390/ph16050735
Chicago/Turabian StyleKang, Borui, Madhusoodanan Mottamal, Qiu Zhong, Melyssa Bratton, Changde Zhang, Shanchun Guo, Ahamed Hossain, Peng Ma, Qiang Zhang, Guangdi Wang, and et al. 2023. "Design, Synthesis, and Evaluation of Niclosamide Analogs as Therapeutic Agents for Enzalutamide-Resistant Prostate Cancer" Pharmaceuticals 16, no. 5: 735. https://doi.org/10.3390/ph16050735
APA StyleKang, B., Mottamal, M., Zhong, Q., Bratton, M., Zhang, C., Guo, S., Hossain, A., Ma, P., Zhang, Q., Wang, G., & Payton-Stewart, F. (2023). Design, Synthesis, and Evaluation of Niclosamide Analogs as Therapeutic Agents for Enzalutamide-Resistant Prostate Cancer. Pharmaceuticals, 16(5), 735. https://doi.org/10.3390/ph16050735