
Advances in Protozoan Epigenetic Targets and Their Inhibitors for the Development of New Potential Drugs

 , , , , and
, , , , and 
Abstract
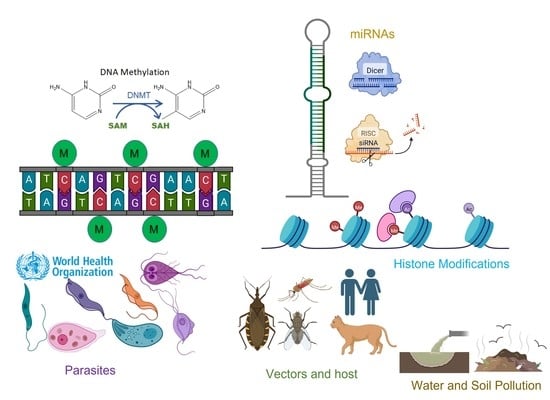
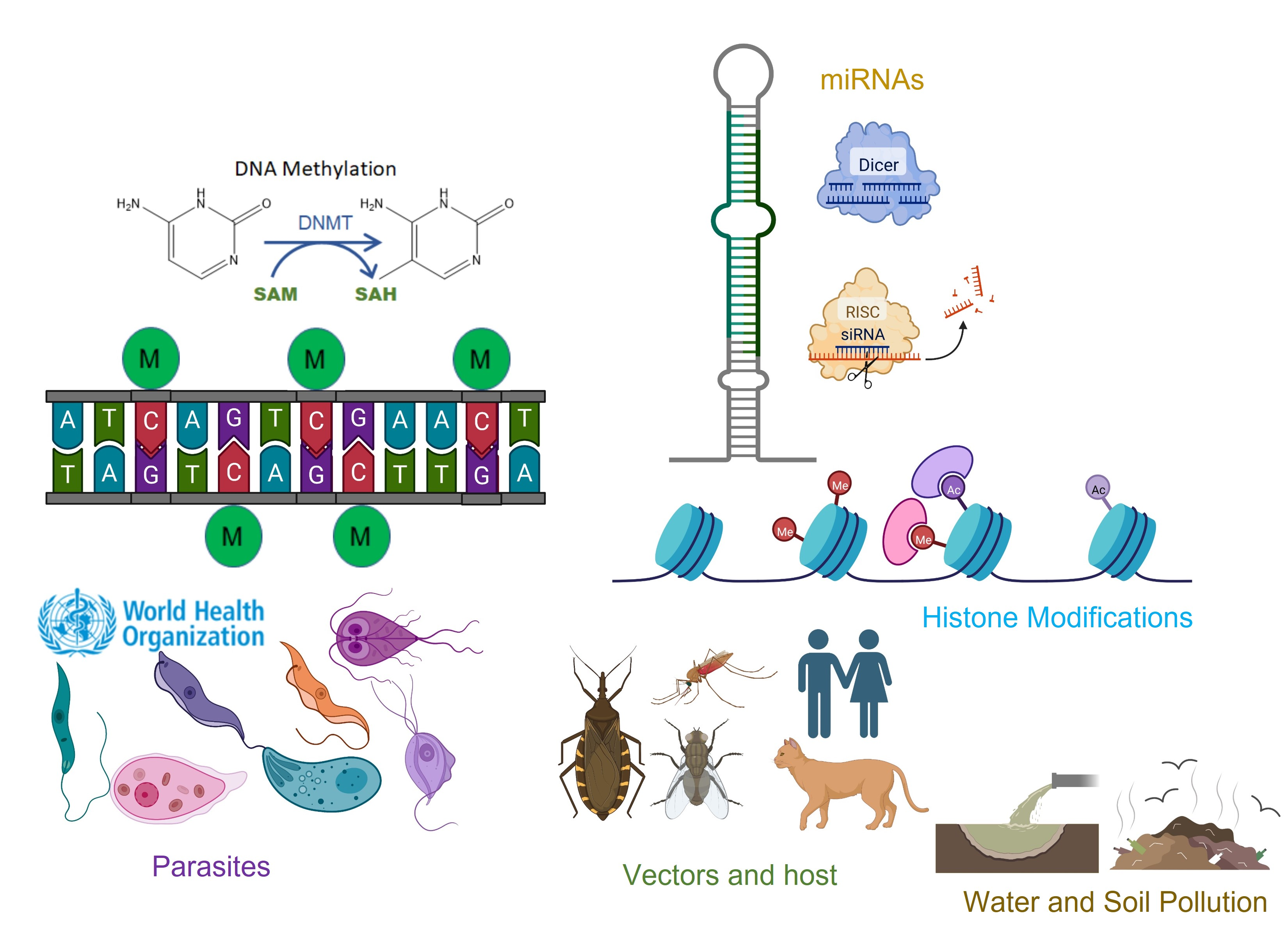
1. Introduction
2. Trypanosoma cruzi
Histone Post-Translational Modifications (HPTMs)
3. Trypanosoma brucei
3.1. Variable Surface Glycoprotein (VSG)
3.2. Histone Post-Translational Modifications (HPTMs)
3.3. Histones Variants
3.4. RNA Interference
3.5. Base J
4. Leishmania spp.
4.1. Histone Post-Translational Modifications (HPTMs)
4.2. Non-Coding RNAs (ncRNA)
5. Entamoeba histolytica
5.1. Histone Post-Translational Modifications (HPTMs)
5.2. RNA Interference
5.3. DNA Methylation
6. Giardia lamblia
6.1. Variant-Specific Surface Protein
6.2. Histone Post-Translational Modifications (HPTMs)
6.3. RNA Interference
7. Toxoplasma gondii
Histone Post-Translational Modifications (HPTMs)
8. Trichomonas vaginalis
8.1. Histone Post-Translational Modifications (HPTMs)
8.2. RNA Interference
8.3. Histone Variants
8.4. DNA Methylation
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Molyneux, D. Neglected tropical diseases. Commun. Eye Health 2013, 26, 21–24. [Google Scholar]
- Sbaraglini, M.L.; Vanrell, M.C.; Bellera, C.L.; Benaim, G.; Carrillo, C.; Talevi, A.; Romano, P.S. Neglected Tropical Protozoan Diseases: Drug Repositioning as a Rational Option. Curr. Top. Med. Chem. 2016, 16, 2201–2222. [Google Scholar] [CrossRef] [PubMed]
- Herrick, J.A.; Nordstrom, M.; Maloney, P.; Rodriguez, M.; Naceanceno, K.; Gallo, G.; Mejia, R.; Hershow, R. Parasitic infections represent a significant health threat among recent immigrants in Chicago. Parasitol. Res. 2020, 119, 1139–1148. [Google Scholar] [CrossRef] [PubMed]
- Dalmasso, M.C.; Sullivan, W.J., Jr.; Angel, S.O. Canonical and variant histones of protozoan parasites. Front Biosci. 2011, 16, 2086–2105. [Google Scholar] [CrossRef] [PubMed]
- Croken, M.M.; Nardelli, S.C.; Kim, K. Chromatin modifications; epigenetics; and how protozoan parasites regulate their lives. Trends Parasitol. 2012, 28, 202–213. [Google Scholar] [CrossRef]
- Mittal, N.; Subramanian, G.; Bütikofer, P.; Madhubala, R. Unique posttranslational modifications in eukaryotic translation factors and their roles in protozoan parasite viability and pathogenesis. Mol. Biochem. Parasitol. 2013, 187, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Nardelli, S.C.; Ting, L.M.; Kim, K. Techniques to study epigenetic control and the epigenome in parasites. Methods Mol. Biol. 2015, 1201, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes. Dev. 2009, 1, 781–783. [Google Scholar] [CrossRef]
- Dymock, B.W. The rise of epigenetic drug discovery. Future Med. Chem. 2016, 8, 1523–1524. [Google Scholar] [CrossRef] [PubMed]
- Miranda Furtado, C.L.; Dos Santos Luciano, M.C.; Silva Santos, R.D.; Furtado, G.P.; Moraes, M.O.; Pessoa, C. Epidrugs: Targeting epigenetic marks in cancer treatment. Epigenetics 2019, 14, 1164–1176. [Google Scholar] [CrossRef]
- Vitiello, M.; Tuccoli, A.; Poliseno, L. Long non-coding RNAs in cancer: Implications for personalized therapy. Cell Oncol. 2015, 38, 17–28. [Google Scholar] [CrossRef]
- Heerboth, S.; Lapinska, K.; Snyder, N.; Leary, M.; Rollinson, S.; Sarkar, S. Use of epigenetic drugs in disease: An overview. Genet. Epigenet. 2014, 27, 9–19. [Google Scholar] [CrossRef]
- Lundstrom, K. Epigenetics: New possibilities for drug discovery. Future Med. Chem. 2017, 9, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Ghorbaninejad, M.; Khademi-Shirvan, M.; Hosseini, S.; Baghaban Eslaminejad, M. Epidrugs: Novel epigenetic regulators that open a new window for targeting osteoblast differentiation. Stem Cell Res. Ther. 2020, 28, 456. [Google Scholar] [CrossRef] [PubMed]
- Deschamps, P.; Lara, E.; Marande, W.; López-García, P.; Ekelund, F.; Moreira, D. Phylogenomic analysis of kinetoplastids supports that trypanosomatids arose from within bodonids. Mol. Biol. Evol. 2011, 28, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Lukeš, J.; Skalický, T.; Týč, J.; Votýpka, J.; Yurchenko, V. Evolution of parasitism in kinetoplastid flagellates. Mol. Biochem. Parasitol. 2014, 195, 115–122. [Google Scholar] [CrossRef]
- Guhl, F.; Ramírez, J.D. Trypanosoma cruzi I diversity: Towards the need of genetic subdivision? Acta Trop. 2011, 119, 1–4. [Google Scholar] [CrossRef]
- Zingales, B. Trypanosoma cruzi genetic diversity: Something new for something known about Chagas disease manifestations; serodiagnosis and drug sensitivity. Acta Trop. 2018, 184, 38–52. [Google Scholar] [CrossRef]
- Moretti, N.S.; Mortara, R.A.; Schenkman, S. Trypanosoma cruzi. Trends Parasitol. 2020, 36, 404–405. [Google Scholar] [CrossRef]
- WHO Expert Committee on the Control of Chagas Disease (2000: Brasilia; Brazil) & World Health Organization. Control of Chagas Disease: Second Report of the WHO Expert Committee; World Health Organization: Rome, Italy, 2002. [Google Scholar]
- Lidani, K.C.F.; Andrade, F.A.; Bavia, L.; Damasceno, F.S.; Beltrame, M.H.; Messias-Reason, I.J.; Sandri, T.L. Chagas Disease: From Discovery to a Worldwide Health Problem. Front Public Health 2019, 7, 166. [Google Scholar] [CrossRef]
- Ribeiro, A.L.; Nunes, M.P.; Teixeira, M.M.; Rocha, M.O. Diagnosis and management of Chagas disease and cardiomyopathy. Nat. Rev. Cardiol. 2012, 9, 576–589. [Google Scholar] [CrossRef] [PubMed]
- Urbina, J.A. Specific chemotherapy of Chagas disease: Relevance; current limitations and new approaches. Acta Trop. 2010, 115, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Leiby, D.A.; Herron, R.M., Jr.; Read, E.J.; Lenes, B.A.; Stumpf, R.J. Trypanosoma cruzi in Los Angeles and Miami blood donors: Impact of evolving donor demographics on seroprevalence and implications for transfusion transmission. Transfusion 2002, 42, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Steverding, D. The history of Chagas disease. Parasit. Vectors 2014, 7, 317. [Google Scholar] [CrossRef] [PubMed]
- Norman, F.F.; López-Vélez, R. Chagas disease and breast-feeding. Emerg. Infect. Dis. 2013, 19, 1561–1566. [Google Scholar] [CrossRef] [PubMed]
- Holloway, G.A.; Charman, W.N.; Fairlamb, A.H.; Brun, R.; Kaiser, M.; Kostewicz, E.; Novello, P.M.; Parisot, J.P.; Richardson, J.; Street, I.P.; et al. Trypanothione reductase high-throughput screening campaign identifies novel classes of inhibitors with antiparasitic activity. Antimicrob. Agents Chemother. 2009, 53, 2824–2833. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, J.D.S.; Costa, D.S.; Cunha-Júnior, E.F.; Andrade-Neto, V.V.; Fairlamb, A.H.; Wyllie, S.; Goulart, M.O.F.; Santos, D.C.; Silva, T.L.; Alves, M.A.; et al. Monocyclic Nitro-heteroaryl Nitrones with Dual Mechanism of Activation: Synthesis and Antileishmanial Activity. ACS Med. Chem. Lett. 2021, 12, 1405–1412. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucl. Acids Res. 2021, 8, D1388–D1395. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Page Last Reviewed: 14 June 2021. Available online: https://www.cdc.gov/parasites/chagas/health_professionals/tx.html (accessed on 15 January 2023).
- Mayo Clinic Health System. 2018. Available online: https://www.mayoclinichealthsystem.org/ (accessed on 29 October 2022).
- National Library of Medicine. 2004. Available online: https://pubchem.ncbi.nlm.nih.gov/ (accessed on 5 January 2023).
- Felsenfeld, G.; Groudine, M. Controlling the double helix. Nature 2003, 421, 448–453. [Google Scholar] [CrossRef]
- Huang, B.; Li, G.; Jiang, X.H. Fate determination in mesenchymal stem cells: A perspective from histone-modifying enzymes. Stem Cell Res. Ther. 2015, 6, 35. [Google Scholar] [CrossRef]
- Saha, S. Histone Modifications and Other Facets of Epigenetic Regulation in Trypanosomatids: Leaving Their Mark. Mbio 2020, 11, e01079-20. [Google Scholar] [CrossRef]
- Campo, V.A. Comparative effects of histone deacetylases inhibitors and resveratrol on Trypanosoma cruzi replication, differentiation, infectivity, and gene expression. Int. J. Parasitol. Drugs Drug. Resist. 2017, 7, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.E.; Tekiel, V.; Campo, V.A. In vitro evaluation of Resveratrol as a potential pre-exposure prophylactic drug against Trypanosoma cruzi infection. Int. J. Parasitol. Drugs Drug. Resist. 2022, 20, 54–64. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Zeng, L.; Zhou, M.M. Bromodomain: An acetyl-lysine binding domain. FEBS Lett. 2002, 513, 124–128. [Google Scholar] [CrossRef]
- Sanchez, R.; Meslamani, J.; Zhou, M.M. The bromodomain: From epigenome reader to druggable target. Biochim. Biophys. Acta 2014, 1839, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Garnier, J.M.; Sharp, P.P.; Burns, C.J. BET bromodomain inhibitors: A patent review. Expert. Opin. Pat. 2014, 24, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Jeffers, V.; Yang, C.; Huang, S.; Sullivan, W.J., Jr. Bromodomains in Protozoan Parasites: Evolution, Function, and Opportunities for Drug Development. Microbiol. Mol. Biol. Rev. 2017, 81, e00047-16. [Google Scholar] [CrossRef]
- Ritagliati, C.; Villanova, G.V.; Alonso, V.L.; Zuma, A.A.; Cribb, P.; Motta, M.C.; Serra, E.C. Glycosomal bromodomain factor 1 from Trypanosoma cruzi enhances trypomastigote cell infection and intracellular amastigote growth. Biochem. J. 2016, 473, 73–85. [Google Scholar] [CrossRef]
- Villanova, G.V.; Nardelli, S.C.; Cribb, P.; Magdaleno, A.; Silber, A.M.; Motta, M.C.; Schenkman, S.; Serra, E. Trypanosoma cruzi bromodomain factor 2 (BDF2) binds to acetylated histones and is accumulated after UV irradiation. Int. J. Parasitol. 2009, 39, 665–673. [Google Scholar] [CrossRef]
- Alonso, V.L.; Villanova, G.V.; Ritagliati, C.; Machado Motta, M.C.; Cribb, P.; Serra, E.C. Trypanosoma cruzi bromodomain factor 3 binds acetylated α-tubulin and concentrates in the flagellum during metacyclogenesis. Eukaryot. Cell 2014, 13, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Pezza, A.; Tavernelli, L.E.; Alonso, V.L.; Perdomo, V.; Gabarro, R.; Prinjha, R.; Rodríguez Araya, E.; Rioja, I.; Docampo, R.; Calderón, F.; et al. Essential Bromodomain TcBDF2 as a Drug Target against Chagas Disease. ACS Infect. Dis. 2022, 8, 1062–1074. [Google Scholar] [CrossRef] [PubMed]
- Büscher, P.; Cecchi, G.; Jamonneau, V.; Priotto, G. Human African trypanosomiasis. Lancet 2017, 390, 2397–2409. [Google Scholar] [CrossRef] [PubMed]
- Fèvre, E.M.; Picozzi, K.; Jannin, J.; Welburn, S.C.; Maudlin, I. Human African trypanosomiasis: Epidemiology and control. Adv. Parasitol. 2006, 61, 167–221. [Google Scholar] [CrossRef] [PubMed]
- Maurice, J. New WHO plan targets the demise of sleeping sickness. Lancet 2013, 381, 13–14. [Google Scholar] [CrossRef]
- Francisco, K.R.; Monti, L.; Yang, W.; Park, H.; Liu, L.J.; Watkins, K.; Amarasinghe, D.K.; Nalli, M.; Roberto Polaquini, C.; Regasini, L.O.; et al. Structure-activity relationship of dibenzylideneacetone analogs against the neglected disease pathogen, Trypanosoma brucei. Bioorg. Med. Chem. Lett. 2023, 81, 129123. [Google Scholar] [CrossRef]
- Fairlamb, A.H. Chemotherapy of human African trypanosomiasis: Current and future prospects. Trends Parasitol. 2003, 19, 488–494. [Google Scholar] [CrossRef]
- Yun, O.; Priotto, G.; Tong, J.; Flevaud, L.; Chappuis, F. NECT is next: Implementing the new drug combination therapy for Trypanosoma brucei gambiense sleeping sickness. PLoS Negl. Trop. Dis. 2010, 4, e720. [Google Scholar] [CrossRef]
- Barrett, M.P.; Vincent, I.M.; Burchmore, R.J.; Kazibwe, A.J.; Matovu, E. Drug resistance in human African trypanosomiasis. Future Microbiol. 2011, 6, 1037–1047. [Google Scholar] [CrossRef]
- Baker, N.; de Koning, H.P.; Mäser, P.; Horn, D. Drug resistance in African trypanosomiasis: The melarsoprol and pentamidine story. Trends Parasitol. 2013, 29, 110–118. [Google Scholar] [CrossRef]
- Kennedy, P.G.E. Update on human African trypanosomiasis (sleeping sickness). J. Neurol. 2019, 266, 2334–2337. [Google Scholar] [CrossRef]
- Kennedy, P.G. Clinical features; diagnosis; and treatment of human African trypanosomiasis (sleeping sickness). Lancet Neurol. 2013, 12, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Franco, J.R.; Simarro, P.P.; Diarra, A.; Jannin, J.G. Epidemiology of human African trypanosomiasis. Clin. Epidemiol. 2014, 6, 257–275. [Google Scholar] [CrossRef]
- Brun, R.; Blum, J.; Chappuis, F.; Burri, C. Human African trypanosomiasis. Lancet 2010, 375, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Donkor, I.O.; Huang, T.L.; Tao, B.; Rattendi, D.; Lane, S.; Vargas, M.; Goldberg, B.; Bacchi, C. Trypanocidal activity of conformationally restricted pentamidine congeners. J. Med. Chem. 2003, 46, 1041–1048. [Google Scholar] [CrossRef]
- Ding, D.; Meng, Q.; Gao, G.; Zhao, Y.; Wang, Q.; Nare, B.; Jacobs, R.; Rock, F.; Alley, M.R.; Plattner, J.J.; et al. Design; synthesis; and structure-activity relationship of Trypanosoma brucei leucyl-tRNA synthetase inhibitors as antitrypanosomal agents. J. Med. Chem. 2011, 54, 1276–1287. [Google Scholar] [CrossRef] [PubMed]
- Vicik, R.; Hoerr, V.; Glaser, M.; Schultheis, M.; Hansell, E.; McKerrow, J.H.; Holzgrabe, U.; Caffrey, C.R.; Ponte-Sucre, A.; Moll, H.; et al. Aziridine-2;3-dicarboxylate inhibitors targeting the major cysteine protease of Trypanosoma brucei as lead trypanocidal agents. Bioorg. Med. Chem. Lett. 2006, 16, 2753–2757. [Google Scholar] [CrossRef]
- Cross, G.A.; Kim, H.S.; Wickstead, B. Capturing the variant surface glycoprotein repertoire (the VSGnome) of Trypanosoma brucei Lister 427. Mol. Biochem. Parasitol. 2014, 195, 59–73. [Google Scholar] [CrossRef]
- Stanne, T.M.; Rudenko, G. Active VSG expression sites in Trypanosoma brucei are depleted of nucleosomes. Eukaryot. Cell 2010, 9, 136–147. [Google Scholar] [CrossRef]
- Figueiredo, L.M.; Cross, G.A. Nucleosomes are depleted at the VSG expression site transcribed by RNA polymerase I in African trypanosomes. Eukaryot. Cell 2010, 9, 148–154. [Google Scholar] [CrossRef]
- Aresta-Branco, F.; Pimenta, S.; Figueiredo, L.M. A transcription-independent epigenetic mechanism is associated with antigenic switching in Trypanosoma brucei. Nucl. Acids Res. 2016, 44, 3131–3146. [Google Scholar] [CrossRef]
- Alsford, S.; Horn, D. Cell-cycle-regulated control of VSG expression site silencing by histones and histone chaperones ASF1A and CAF-1b in Trypanosoma brucei. Nucl. Acids Res. 2012, 40, 10150–10160. [Google Scholar] [CrossRef] [PubMed]
- Günzl, A.; Kirkham, J.K.; Nguyen, T.N.; Badjatia, N.; Park, S.H. Mono-allelic VSG expression by RNA polymerase I in Trypanosoma brucei: Expression site control from both ends? Gene 2015, 556, 68–73. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Narayanan, M.S.; Rudenko, G. TDP1 is an HMG chromatin protein facilitating RNA polymerase I transcription in African trypanosomes. Nucl. Acids Res. 2013, 41, 2981–2992. [Google Scholar] [CrossRef]
- Janzen, C.J.; Hake, S.B.; Lowell, J.E.; Cross, G.A. Selective di- or trimethylation of histone H3 lysine 76 by two DOT1 homologs is important for cell cycle regulation in Trypanosoma brucei. Mol. Cell 2006, 23, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, L.M.; Janzen, C.J.; Cross, G.A. A histone methyltransferase modulates antigenic variation in African trypanosomes. PLoS Biol. 2008, 6, e161. [Google Scholar] [CrossRef] [PubMed]
- Landeira, D.; Navarro, M. Nuclear repositioning of the VSG promoter during developmental silencing in Trypanosoma brucei. J. Cell Biol. 2007, 176, 133–139. [Google Scholar] [CrossRef]
- Alsford, S.; Kawahara, T.; Isamah, C.; Horn, D. A sirtuin in the African trypanosome is involved in both DNA repair and telomeric gene silencing but is not required for antigenic variation. Mol. MicroBiol. 2007, 63, 724–736. [Google Scholar] [CrossRef]
- Mandava, V.; Fernandez, J.P.; Deng, H.; Janzen, C.J.; Hake, S.B.; Cross, G.A. Histone modifications in Trypanosoma brucei. Mol. Biochem. Parasitol. 2007, 156, 41–50. [Google Scholar] [CrossRef]
- Ingram, A.K.; Horn, D. Histone deacetylases in Trypanosoma brucei: Two are essential and another is required for normal cell cycle progression. Mol. MicroBiol. 2002, 45, 89–97. [Google Scholar] [CrossRef]
- Wang, Q.P.; Kawahara, T.; Horn, D. Histone deacetylases play distinct roles in telomeric VSG expression site silencing in African trypanosomes. Mol. MicroBiol. 2010, 77, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Carrillo, A.K.; Guiguemde, W.A.; Guy, R.K. Evaluation of histone deacetylase inhibitors (HDACi) as therapeutic leads for human African trypanosomiasis (HAT). Bioorg. Med. Chem. 2015, 23, 5151–5155. [Google Scholar] [CrossRef] [PubMed]
- Debler, E.W.; Jain, K.; Warmack, R.A.; Feng, Y.; Clarke, S.G.; Blobel, G.; Stavropoulos, P. A glutamate/aspartate switch controls product specificity in a protein arginine methyltransferase. Proc. Natl. Acad. Sci. USA 2016, 113, 2068–2073. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Litt, M.; Felsenfeld, G. Methylation of histone H4 by arginine methyltransferase PRMT1 is essential in vivo for many subsequent histone modifications. Genes. Dev. 2005, 19, 1885–1893. [Google Scholar] [CrossRef]
- Wang, Q.; Rosa, B.A.; Nare, B.; Powell, K.; Valente, S.; Rotili, D.; Mai, A.; Marshall, G.R.; Mitreva, M. Targeting Lysine Deacetylases (KDACs) in Parasites. PLoS Negl. Trop. Dis. 2015, 9, e0004026. [Google Scholar] [CrossRef]
- Maree, J.P.; Patterton, H.G. The epigenome of Trypanosoma brucei: A regulatory interface to an unconventional transcriptional machine. Biochim. Biophys. Acta 2014, 1839, 743–750. [Google Scholar] [CrossRef]
- Ngô, H.; Tschudi, C.; Gull, K.; Ullu, E. Double-stranded RNA induces mRNA degradation in Trypanosoma brucei. Proc. Natl. Acad. Sci. USA 1998, 95, 4687–4692. [Google Scholar] [CrossRef]
- Kolev, N.G.; Tschudi, C.; Ullu, E. RNA interference in protozoan parasites: Achievements and challenges. Eukaryot. Cell 2011, 10, 1156–1163. [Google Scholar] [CrossRef]
- Shi, H.; Djikeng, A.; Tschudi, C.; Ullu, E. Argonaute protein in the early divergent eukaryote Trypanosoma brucei: Control of small interfering RNA accumulation and retroposon transcript abundance. Mol. Cell Biol. 2004, 24, 420–427. [Google Scholar] [CrossRef]
- Patrick, K.L.; Shi, H.; Kolev, N.G.; Ersfeld, K.; Tschudi, C.; Ullu, E. Distinct and overlapping roles for two Dicer-like proteins in the RNA interference pathways of the ancient eukaryote Trypanosoma brucei. Proc. Natl. Acad. Sci. USA 2009, 106, 17933–17938. [Google Scholar] [CrossRef]
- Subramaniam, C.; Veazey, P.; Redmond, S.; Hayes-Sinclair, J.; Chambers, E.; Carrington, M.; Gull, K.; Matthews, K.; Horn, D.; Field, M.C. Chromosome-wide analysis of gene function by RNA interference in the african trypanosome. Eukaryot. Cell 2006, 5, 1539–1549. [Google Scholar] [CrossRef]
- El-Sayed, N.M.; Myler, P.J.; Blandin, G.; Berriman, M.; Crabtree, J.; Aggarwal, G.; Caler, E.; Renauld, H.; Worthey, E.A.; Hertz-Fowler, C.; et al. Comparative genomics of trypanosomatid parasitic protozoa. Science 2005, 309, 404–409. [Google Scholar] [CrossRef]
- Cross, M.; Kieft, R.; Sabatini, R.; Wilm, M.; de Kort, M.; van der Marel, G.A.; van Boom, J.H.; van Leeuwen, F.; Borst, P. The modified base J is the target for a novel DNA-binding protein in kinetoplastid protozoans. EMBO J. 1999, 18, 6573–6581. [Google Scholar] [CrossRef]
- van Leeuwen, F.; Wijsman, E.R.; Kieft, R.; van der Marel, G.A.; van Boom, J.H.; Borst, P. Localization of the modified base J in telomeric VSG gene expression sites of Trypanosoma brucei. Genes. Dev. 1997, 11, 3232–3241. [Google Scholar] [CrossRef][Green Version]
- van Leeuwen, F.; Wijsman, E.R.; Kuyl-Yeheskiely, E.; van der Marel, G.A.; van Boom, J.H.; Borst, P. The telomeric GGGTTA repeats of Trypanosoma brucei contain the hypermodified base J in both strands. Nucl. Acids Res. 1996, 24, 2476–2482. [Google Scholar] [CrossRef]
- van Leeuwen, F.; Kieft, R.; Cross, M.; Borst, P. Biosynthesis and function of the modified DNA base beta-D-glucosyl-hydroxymethyluracil in Trypanosoma brucei. Mol. Cell Biol. 1998, 18, 5643–5651. [Google Scholar] [CrossRef]
- van Leeuwen, F.; Kieft, R.; Cross, M.; Borst, P. Tandemly repeated DNA is a target for the partial replacement of thymine by beta-D-glucosyl-hydroxymethyluracil in Trypanosoma brucei. Mol. Biochem. Parasitol. 2000, 109, 133–145. [Google Scholar] [CrossRef]
- Bernards, A.; van Harten-Loosbroek, N.; Borst, P. Modification of telomeric DNA in Trypanosoma brucei; a role in antigenic variation? Nucl. Acids Res. 1984, 12, 4153–4170. [Google Scholar] [CrossRef]
- de Vries, H.J.; Reedijk, S.H.; Schallig, H.D. Cutaneous leishmaniasis: Recent developments in diagnosis and management. Am. J. Clin. Dermatol. 2015, 16, 99–109. [Google Scholar] [CrossRef]
- Pearson, R.D.; Sousa, A.Q. Clinical spectrum of Leishmaniasis. Clin. Infect. Dis. 1996, 22, 1–13. [Google Scholar] [CrossRef]
- Savoia, D. Recent updates and perspectives on leishmaniasis. J. Infect. Dev. Ctries. 2015, 9, 588–596. [Google Scholar] [CrossRef]
- Desjeux, P. Leishmaniasis. Nat. Rev. Microbiol. 2004, 2, 692–693. [Google Scholar] [CrossRef]
- World Health Organization (WHO). January 2023. Available online: https://www.who.int/es/news-room/fact-sheets/detail/leishmaniasis (accessed on 5 February 2023).
- Mokni, M. Leishmanioses cutanées [Cutaneous leishmaniasis]. Ann. Derm. Venereol. 2019, 146, 232–246. [Google Scholar] [CrossRef]
- Ghorbani, M.; Farhoudi, R. Leishmaniasis in humans: Drug or vaccine therapy? Drug. Des. Devel. Ther. 2017, 12, 25–40. [Google Scholar] [CrossRef]
- Freitas-Junior, L.H.; Chatelain, E.; Kim, H.A.; Siqueira-Neto, J.L. Visceral leishmaniasis treatment: What do we have; what do we need and how to deliver it? Int. J. Parasitol. Drugs Drug. Resist. 2012, 2, 11–19. [Google Scholar] [CrossRef]
- van Griensven, J.; Diro, E. Visceral leishmaniasis. Infect. Dis. Clin. North. Am. 2012, 26, 309–322. [Google Scholar] [CrossRef]
- Goto, H.; Lauletta-Lindoso, J.A. Cutaneous and mucocutaneous leishmaniasis. Infect. Dis. Clin. North. Am. 2012, 26, 293–307. [Google Scholar] [CrossRef]
- Bates, P.A. Revising Leishmania’s life cycle. Nat. Microbiol. 2018, 3, 529–530. [Google Scholar] [CrossRef]
- Molaie, S.; Ghaffarifar, F.; Hasan, Z.M.; Dalimi, A. Enhancement Effect of Shark Cartilage Extract on Treatment of Leishmania infantum with Artemisinin and Glucantime and Evaluation of killing Factors and Apoptosis in-vitro Condition. Iran. J. Pharm. Res. 2019, 18, 887–902. [Google Scholar] [CrossRef]
- Masmoudi, A.; Maalej, N.; Mseddi, M.; Souissi, A.; Turki, H.; Boudaya, S.; Bouassida, S.; Zahaf, A. Glucantime par voie parentérale: Bénéfice versus toxicité [Glucantime injection: Benefit versus toxicity]. Med. Mal. Infect. 2005, 35, 42–45. [Google Scholar] [CrossRef]
- Ephros, M.; Waldman, E.; Zilberstein, D. Pentostam induces resistance to antimony and the preservative chlorocresol in Leishmania donovani promastigotes and axenically grown amastigotes. Antimicrob. Agents Chemother. 1997, 41, 1064–1068. [Google Scholar] [CrossRef]
- Vermeersch, M.; da Luz, R.I.; Toté, K.; Timmermans, J.P.; Cos, P.; Maes, L. In vitro susceptibilities of Leishmania donovani promastigote and amastigote stages to antileishmanial reference drugs: Practical relevance of stage-specific differences. Antimicrob. Agents Chemother. 2009, 53, 3855–3859. [Google Scholar] [CrossRef]
- Muhammad, M.T.; Ghouri, N.; Khan, K.M.; Arshia-Choudhary, M.I.; Perveen, S. Synthesis of Thiocarbohydrazones and Evaluation of their in vitro Antileishmanial Activity. Med. Chem. 2018, 14, 725–732. [Google Scholar] [CrossRef]
- Gadelha, E.P.; Talhari, S.; Guerra, J.A.; Neves, L.O.; Talhari, C.; Gontijo, B.; Silva-Junior, R.M.; Talhari, A.C. Efficacy and safety of a single dose pentamidine (7mg/kg) for patients with cutaneous leishmaniasis caused by L. guyanensis: A pilot study. An. Bras. Dermatol. 2015, 90, 807–813. [Google Scholar] [CrossRef]
- Andrade-Neto, V.V.; Matos-Guedes, H.L.; Gomes, D.C.; Canto-Cavalheiro, M.M.; Rossi-Bergmann, B.; Torres-Santos, E.C. The stepwise selection for ketoconazole resistance induces upregulation of C14-demethylase (CYP51) in Leishmania amazonensis. Mem. Inst. Oswaldo Cruz 2012, 107, 416–419. [Google Scholar] [CrossRef]
- Saenz, R.E.; Paz, H.; Berman, J.D. Efficacy of ketoconazole against Leishmania braziliensis panamensis cutaneous leishmaniasis. Am. J. Med. 1990, 89, 147–155. [Google Scholar] [CrossRef]
- de Morais-Teixeira, E.; Gallupo, M.K.; Rodrigues, L.F.; Romanha, A.J.; Rabello, A. In vitro interaction between paromomycin sulphate and four drugs with leishmanicidal activity against three New World Leishmania species. J. Antimicrob. Chemother. 2014, 69, 150–154. [Google Scholar] [CrossRef][Green Version]
- Anderson, B.A.; Wong, I.L.; Baugh, L.; Ramasamy, G.; Myler, P.J.; Beverley, S.M. Kinetoplastid-specific histone variant functions are conserved in Leishmania major. Mol. Biochem. Parasitol. 2013, 191, 53–57. [Google Scholar] [CrossRef]
- Thomas, S.; Green, A.; Sturm, N.R.; Campbell, D.A.; Myler, P.J. Histone acetylations mark origins of polycistronic transcription in Leishmania major. BMC Genomics 2009, 10, 1–15. [Google Scholar] [CrossRef]
- Chandra, U.; Yadav, A.; Kumar, D.; Saha, S. Cell cycle stage-specific transcriptional activation of cyclins mediated by HAT2-dependent H4K10 acetylation of promoters in Leishmania donovani. PLoS Pathog. 2017, 13, e1006615. [Google Scholar] [CrossRef]
- Kumar, D.; Saha, S. HAT3-mediated acetylation of PCNA precedes PCNA monoubiquitination following exposure to UV radiation in Leishmania donovani. Nucl. Acids Res. 2015, 43, 5423–5441. [Google Scholar] [CrossRef]
- Vizuet-de-Rueda, J.C.; Florencio-Martínez, L.E.; Padilla-Mejía, N.E.; Manning-Cela, R.; Hernández-Rivas, R.; Martínez-Calvillo, S. Ribosomal RNA Genes in the Protozoan Parasite Leishmania major possess a nucleosomal structure. Protist 2016, 167, 121–135. [Google Scholar] [CrossRef]
- Fioravanti, R.; Mautone, N.; Rovere, A.; Rotili, D.; Mai, A. Targeting histone acetylation/deacetylation in parasites: An update (2017-2020). Curr. Opin. Chem. Biol. 2020, 57, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Scholte, L.L.S.; Mourão, M.M.; Pais, F.S.-M.; Melesina, J.; Robaa, D.; Volpini, A.C.; Sippl, W.; Pierce, R.J.; Oliveira, G.; Nahum, L.A. Evolutionary Relationships among Protein Lysine Deacetylases of Parasites Causing Neglected Diseases. Infect. Genet. Evol. 2017, 53, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Ângelo de Souza, L.; Silva, E.; Bastos, M.; de Melo Agripino, J.; Souza-Onofre, T.; Apaza-Calla, L.F.; Heimburg, T.; Ghazy, E.; Bayer, T.; Ferraz da Silva, V.H.; et al. Histone deacetylases inhibitors as new potential drugs against Leishmania braziliensis; the main causative agent of new world tegumentary leishmaniasis. Biochem. Pharmacol. 2020, 180, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Loeuillet, C.; Touquet, B.; Oury, B.; Eddaikra, N.; Pons, J.L.; Guichou, J.F.; Labesse, G.; Sereno, D. Synthesis of aminophenylhydroxamate and aminobenzylhydroxamate derivatives and in vitro screening for antiparasitic and histone deacetylase inhibitory activity. Int. J. Parasitol. Drugs Drug. Resist. 2018, 8, 59–66. [Google Scholar] [CrossRef]
- Melesina, J.; Robaa, D.; Pierce, R.J.; Romier, C.; Sippl, W. Homology modeling of parasite histone deacetylases to guide the structure-based design of selective inhibitors. J. Mol. Graph. Model. 2015, 62, 342–361. [Google Scholar] [CrossRef]
- Di Bello, E.; Noce, B.; Fioravanti, R.; Zwergel, C.; Valente, S.; Rotili, D.; Fianco, G.; Trisciuoglio, D.; Mourão, M.M.; Sales, P., Jr.; et al. Effects of Structurally Different HDAC Inhibitors against Trypanosoma cruzi; Leishmania; and Schistosoma mansoni. ACS Infect. Dis. 2022, 8, 1356–1366. [Google Scholar] [CrossRef]
- Chua, M.J.; Arnold, M.S.; Xu, W.; Lancelot, J.; Lamotte, S.; Späth, G.F.; Prina, E.; Pierce, R.J.; Fairlie, D.P.; Skinner-Adams, T.S.; et al. Effect of clinically approved HDAC inhibitors on Plasmodium; Leishmania and Schistosoma parasite growth. Int. J. Parasitol. Drugs Drug. Resist. 2017, 7, 42–50. [Google Scholar] [CrossRef]
- Bayer-Santos, E.; Marini, M.M.; da Silveira, J.F. Non-coding RNAs in Host-Pathogen Interactions: Subversion of Mammalian Cell Functions by Protozoan Parasites. Front Microbiol. 2017, 8, 1–36. [Google Scholar] [CrossRef]
- Ruy, P.C.; Monteiro-Teles, N.M.; Miserani-Magalhães, R.D.; Freitas-Castro, F.; Dias, L.; Aquino-Defina, T.P.; Rosas De Vasconcelos, E.J.; Myler, P.J.; Kaysel-Cruz, A. Comparative transcriptomics in Leishmania braziliensis: Disclosing differential gene expression of coding and putative noncoding RNAs across developmental stages. RNA Biol. 2019, 16, 639–660. [Google Scholar] [CrossRef]
- Dumas, C.; Chow, C.; Müller, M.; Papadopoulou, B. A novel class of developmentally regulated noncoding RNAs in Leishmania. Eukaryot. Cell 2006, 5, 2033–2046. [Google Scholar] [CrossRef]
- Madej, M.J.; Alfonzo, J.D.; Hüttenhofer, A. Small ncRNA transcriptome analysis from kinetoplast mitochondria of Leishmania tarentolae. Nucl. Acids Res. 2007, 35, 1544–1554. [Google Scholar] [CrossRef]
- Blum, B.; Bakalara, N.; Simpson, L. A model for RNA editing in kinetoplastid mitochondria: “guide” RNA molecules transcribed from maxicircle DNA provide the edited information. Cell 1990, 60, 189–198. [Google Scholar] [CrossRef]
- Blum, B.; Simpson, L. Guide RNAs in kinetoplastid mitochondria have a nonencoded 3’ oligo(U) tail involved in recognition of the preedited region. Cell 1990, 62, 391–397. [Google Scholar] [CrossRef]
- Sturm, N.R.; Simpson, L. Kinetoplast DNA minicircles encode guide RNAs for editing of cytochrome oxidase subunit III mRNA. Cell 1990, 61, 879–884. [Google Scholar] [CrossRef]
- Bercu, T.E.; Petri, W.A.; Behm, J.W. Amebic colitis: New insights into pathogenesis and treatment. Curr. Gastroenterol. Rep. 2007, 9, 429–433. [Google Scholar] [CrossRef]
- Ximénez, C.; Cerritos, R.; Rojas, L.; Dolabella, S.; Morán, P.; Shibayama, M.; González, E.; Valadez, A.; Hernández, E.; Valenzuela, O.; et al. Human amebiasis: Breaking the paradigm? Int. J. Env. Res. Public Health. 2010, 7, 1105–1120. [Google Scholar] [CrossRef]
- Ralston, K.S.; Petri, W.A., Jr. Tissue destruction and invasion by Entamoeba histolytica. Trends Parasitol. 2011, 27, 254–263. [Google Scholar] [CrossRef]
- Al Saqur, I.M.; Al-Warid, H.S.; Albahadely, H.S. The prevalence of Giardia lamblia and Entamoeba histolytica/dispar among Iraqi provinces. Karbala Int. J. Mod. Sci. 2017, 3, 93–96. [Google Scholar] [CrossRef]
- Haque, R.; Huston, C.D.; Hughes, M.; Houpt, E.; Petri, W.A., Jr. Amebiasis. N. Engl. J. Med. 2003, 348, 1565–1573. [Google Scholar] [CrossRef]
- Wuerz, T.; Kane, J.B.; Boggild, A.K.; Krajden, S.; Keystone, J.S.; Fuksa, M.; Kain, K.C.; Warren, R.; Kempston, J.; Anderson, J. A review of amoebic liver abscess for clinicians in a nonendemic setting. Can. J. Gastroenterol. 2012, 26, 729–733. [Google Scholar] [CrossRef]
- Cheepsattayakorn, A.; Cheepsattayakorn, R. Parasitic pneumonia and lung involvement. Biomed. Res. Int. 2014, 2014, 1–18. [Google Scholar] [CrossRef]
- Carrero, J.C.; Reyes-López, M.; Serrano-Luna, J.; Shibayama, M.; Unzueta, J.; León-Sicairos, N.; de la Garza, M. Intestinal amoebiasis: 160 years of its first detection and still remains as a health problem in developing countries. Int. J. Med. Microbiol. 2020, 310, 1–15. [Google Scholar] [CrossRef]
- Kantor, M.; Abrantes, A.; Estevez, A.; Schiller, A.; Torrent, J.; Gascon, J.; Hernandez, R.; Ochner, C. Entamoeba Histolytica: Updates in Clinical Manifestation; Pathogenesis; and Vaccine Development. Can. J. Gastroenterol. Hepatol. 2018, 2018, 4601420. [Google Scholar] [CrossRef]
- Bustos-Brito, C.; Joseph-Nathan, P.; Burgueño-Tapia, E.; Martínez-Otero, D.; Nieto-Camacho, A.; Calzada, F.; Yépez-Mulia, L.; Esquivel, B.; Quijano, L. Structure and Absolute Configuration of Abietane Diterpenoids from Salvia clinopodioides: Antioxidant; Antiprotozoal; and Antipropulsive Activities. J. Nat. Prod. 2019, 82, 1207–1216. [Google Scholar] [CrossRef]
- González-Garza, M.T.; Said-Fernández, S. Confiabilidad del medio de cultivo pehps para la evaluación in vitro de drogas antiamibianas. Reliability of the PEHPS culture medium for the in vitro evaluation of antiamebic drugs. Arch. Invest. Med. 1989, 20, 29–32. [Google Scholar]
- Ramakrishnan, G.; Gilchrist, C.A.; Musa, H.; Torok, M.S.; Grant, P.A.; Mann, B.J.; Petri, W.A., Jr. Histone acetyltransferases and deacetylase in Entamoeba histolytica. Mol. Biochem. Parasitol. 2004, 138, 205–216. [Google Scholar] [CrossRef]
- Torres-Guerrero, H.; Peattie, D.A.; Meza, I. Chromatin organization in Entamoeba histolytica. Mol. Biochem. Parasitol. 1991, 45, 121–130. [Google Scholar] [CrossRef]
- Huguenin, M.; Bracha, R.; Chookajorn, T.; Mirelman, D. Epigenetic transcriptional gene silencing in Entamoeba histolytica: Insight into histone and chromatin modifications. Parasitology 2010, 137, 619–627. [Google Scholar] [CrossRef]
- Lozano-Amado, D.; Herrera-Solorio, A.M.; Valdés, J.; Alemán-Lazarini, L.; de Almaraz-Barrera, M.J.; Luna-Rivera, E.; Vargas, M.; Hernández-Rivas, R. Identification of repressive and active epigenetic marks and nuclear bodies in Entamoeba histolytica. Parasit. Vectors 2016, 14, 19. [Google Scholar] [CrossRef]
- Byers, J.; Faigle, W.; Eichinger, D. Colonic short-chain fatty acids inhibit encystation of Entamoeba invadens. Cell Microbiol. 2005, 7, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Lozano-Amado, D.; Ávila-López, P.A.; Hernández-Montes, G.; Briseño-Díaz, P.; Vargas, M.; Lopez-Rubio, J.J.; Carrero, J.C.; Hernández-Rivas, R. A class I histone deacetylase is implicated in the encystation of Entamoeba invadens. Int. J. Parasitol. 2020, 50, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Ehrenkaufer, G.M.; Eichinger, D.J.; Singh, U. Trichostatin A effects on gene expression in the protozoan parasite Entamoeba histolytica. BMC Genom. 2007, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wood, A.; Shilatifard, A. Posttranslational modifications of histones by methylation. Adv. Prot. Chem. 2004, 67, 201–222. [Google Scholar] [CrossRef]
- Borbolla-Vázquez, J.; Orozco, E.; Betanzos, A.; Rodríguez, M.A. Entamoeba histolytica: Protein arginine transferase 1a methylates arginine residues and potentially modify the H4 histone. Parasit. Vectors 2015, 8, 1–13. [Google Scholar] [CrossRef]
- Borbolla-Vázquez, J.; Orozco, E.; Medina-Gómez, C.; Martínez-Higuera, A.; Javier-Reyna, R.; Chávez, B.; Betanzos, A.; Rodríguez, M.A. Identification and functional characterization of lysine methyltransferases of Entamoeba histolytica. Mol. Microbiol. 2016, 101, 351–365. [Google Scholar] [CrossRef]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing; erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef]
- Anbar, M.; Bracha, R.; Nuchamowitz, Y.; Li, Y.; Florentin, A.; Mirelman, D. Involvement of a short interspersed element in epigenetic transcriptional silencing of the amoebapore gene in Entamoeba histolytica. Eukaryot. Cell 2005, 4, 1775–1784. [Google Scholar] [CrossRef]
- Dam, S.; Lohia, A. Entamoeba histolytica sirtuin EhSir2a deacetylates tubulin and regulates the number of microtubular assemblies during the cell cycle. Cell Microbiol. 2010, 12, 1002–1014. [Google Scholar] [CrossRef][Green Version]
- Zhang, H.; Ehrenkaufer, G.M.; Hall, N.; Singh, U. Small RNA pyrosequencing in the protozoan parasite Entamoeba histolytica reveals strain-specific small RNAs that target virulence genes. BMC Genomics 2013, 25, 1–19. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lavi, T.; Siman-Tov, R.; Ankri, S. EhMLBP is an essential constituent of the Entamoeba histolytica epigenetic machinery and a potential drug target. Mol. Microbiol. 2008, 69, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Ankri, S.; Padilla-Vaca, F.; Stolarsky, T.; Koole, L.; Katz, U.; Mirelman, D. Antisense inhibition of expression of the light subunit (35 kDa) of the Gal/GalNac lectin complex inhibits Entamoeba histolytica virulence. Mol. Microbiol. 1999, 33, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Mirelman, D.; Anbar, M.; Bracha, R. Trophozoites of Entamoeba histolytica epigenetically silenced in several genes are virulence-attenuated. Parasite 2008, 15, 266–274. [Google Scholar] [CrossRef][Green Version]
- Nurkanto, A.; Jeelani, G.; Yamamoto, T.; Naito, Y.; Hishiki, T.; Mori, M.; Suematsu, M.; Shiomi, K.; Hashimoto, T.; Nozaki, T. Characterization and validation of Entamoeba histolytica pantothenate kinase as a novel anti-amebic drug target. Int. J. Parasitol. Drugs Drug. Resist. 2018, 8, 125–136. [Google Scholar] [CrossRef]
- Nurkanto, A.; Jeelani, G.; Yamamoto, T.; Hishiki, T.; Naito, Y.; Suematsu, M.; Hashimoto, T.; Nozaki, T. Biochemical, Metabolomic, and Genetic Analyses of Dephospho Coenzyme A Kinase Involved in Coenzyme A Biosynthesis in the Human Enteric Parasite Entamoeba histolytica. Front Microbiol. 2018, 30, 2902. [Google Scholar] [CrossRef]
- Foda, B.M.; Singh, U. Dimethylated H3K27 Is a Repressive Epigenetic Histone Mark in the Protist Entamoeba histolytica and Is Significantly Enriched in Genes Silenced via the RNAi Pathway. J. Biol. Chem. 2015, 290, 1–17. [Google Scholar] [CrossRef]
- Tovy, A.; Ankri, S. Epigenetics in the unicellular parasite Entamoeba histolytica. Future Microbiol. 2010, 5, 1875–1884. [Google Scholar] [CrossRef]
- Fisher, O.; Siman-Tov, R.; Ankri, S. Characterization of cytosine methylated regions and 5-cytosine DNA methyltransferase (Ehmeth) in the protozoan parasite Entamoeba histolytica. Nucl. Acids Res. 2004, 32, 287–297. [Google Scholar] [CrossRef]
- Schulz, E.C.; Roth, H.M.; Ankri, S.; Ficner, R. Structure analysis of Entamoeba histolytica DNMT2 (EhMeth). PLoS ONE 2012, 7, e38728. [Google Scholar] [CrossRef]
- Tovy, A.; Siman, T.R.; Gaentzsch, R.; Helm, M.; Ankri, S. A new nuclear function of the Entamoeba histolytica glycolytic enzyme enolase: The metabolic regulation of cytosine-5 methyltransferase 2 (Dnmt2) activity. PLoS Pathog. 2010, 6, e1000775. [Google Scholar] [CrossRef] [PubMed]
- Bernes, S.; Siman-Tov, R.; Ankri, S. Epigenetic and classical activation of Entamoeba histolytica heat shock protein 100 (EHsp100) expression. FEBS Lett. 2005, 579, 6395–6402. [Google Scholar] [CrossRef] [PubMed]
- Ortega, Y.R.; Adam, R.D. Giardia: Overview and update. Clin. Infect. Dis. 1997, 25, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Adam, R.D. Giardia duodenalis: Biology and Pathogenesis. Clin. Microbiol. Rev. 2021, 34, e0002419. [Google Scholar] [CrossRef]
- Huang, D.B.; White, A.C. An updated review on Cryptosporidium and Giardia. Gastroenterol. Clin. North. Am. 2006, 35, 291–314. [Google Scholar] [CrossRef]
- Hill, D.R. Giardiasis. Issues in diagnosis and management. Infect. Dis. Clin. North. Am. 1993, 7, 503–525. [Google Scholar] [CrossRef]
- Lanata, C.F.; Fischer-Walker, C.L.; Olascoaga, A.C.; Torres, C.X.; Aryee, M.J.; Black, R.E.; Child Health Epidemiology Reference Group of the World Health Organization and UNICEF. Global causes of diarrheal disease mortality in children <5 years of age: A systematic review. PLoS ONE 2013, 8, e72788. [Google Scholar] [CrossRef]
- Feng, Y.; Xiao, L. Zoonotic potential and molecular epidemiology of Giardia species and giardiasis. Clin. Microbiol. Rev. 2011, 24, 110–140. [Google Scholar] [CrossRef]
- Riches, A.; Hart, C.J.S.; Trenholme, K.R.; Skinner-Adams, T.S. Anti-Giardia Drug Discovery: Current Status and Gut Feelings. J. Med. Chem. 2020, 63, 13330–13354. [Google Scholar] [CrossRef]
- Pipiková, J.; Papajová, I.; Majláthová, V.; Šoltys, J.; Bystrianska, J.; Schusterová, I.; Vargová, V. First report on Giardia duodenalis assemblage F in Slovakian children living in poor environmental conditions. J. Microbiol. Immunol. Infect. 2020, 53, 148–156. [Google Scholar] [CrossRef]
- Hernández-Ceruelos, A.; Romero-Quezada, L.C.; Ruvalcaba-Ledezma, J.C.; López-Contreras, L. Therapeutic uses of metronidazole and its side effects: An update. Eur. Rev. Med. Pharm. Sci. 2019, 23, 397–401. [Google Scholar] [CrossRef]
- Cedillo-Rivera, R.; Muñoz, O. In-vitro susceptibility of Giardia lamblia to albendazole; mebendazole and other chemotherapeutic agents. J. Med. Microbiol. 1992, 37, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.K.C.; Leung, A.A.M.; Wong, A.H.C.; Sergi, C.M.; Kam, J.K.M. Giardiasis: An Overview. Recent. Pat. Inflamm. Allergy Drug. Discov. 2019, 13, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Gargantini, P.R.; Serradell, M.D.C.; Ríos, D.N.; Tenaglia, A.H.; Luján, H.D. Antigenic variation in the intestinal parasite Giardia lamblia. Curr. Opin. Microbiol. 2016, 32, 52–58. [Google Scholar] [CrossRef]
- Kulakova, L.; Singer, S.M.; Conrad, J.; Nash, T.E. Epigenetic mechanisms are involved in the control of Giardia lamblia antigenic variation. Mol. Microbiol. 2006, 61, 1533–1542. [Google Scholar] [CrossRef]
- Prucca, C.G.; Slavin, I.; Quiroga, R.; Elías, E.V.; Rivero, F.D.; Saura, A.; Carranza, P.G.; Luján, H.D. Antigenic variation in Giardia lamblia is regulated by RNA interference. Nature 2008, 11, 750–754. [Google Scholar] [CrossRef]
- Salusso, A.; Zlocowski, N.; Mayol, G.F.; Zamponi, N.; Rópolo, A.S. Histone methyltransferase 1 regulates the encystation process in the parasite Giardia lamblia. FEBS J. 2017, 284, 2396–2409. [Google Scholar] [CrossRef]
- Peng, L.; Seto, E. Deacetylation of nonhistone proteins by HDACs and the implications in cancer. Handb. Exp. Pharmacol. 2011, 206, 39–56. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Sonda, S.; Morf, L.; Bottova, I.; Baetschmann, H.; Rehrauer, H.; Caflisch, A.; Hakimi, M.A.; Hehl, A.B. Epigenetic mechanisms regulate stage differentiation in the minimized protozoan Giardia lamblia. Mol. Microbiol. 2010, 76, 48–67. [Google Scholar] [CrossRef]
- Carranza, P.G.; Gargantini, P.R.; Prucca, C.G.; Torri, A.; Saura, A.; Svärd, S.; Lujan, H.D. Specific histone modifications play critical roles in the control of encystation and antigenic variation in the early-branching eukaryote Giardia lamblia. Int. J. Biochem. Cell Biol. 2016, 81, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Saraiya, A.A.; Li, W.; Wang, C.C. Transition of a microRNA from repressing to activating translation depending on the extent of base pairing with the target. PLoS ONE 2013, 8, e55672. [Google Scholar] [CrossRef]
- Liao, J.Y.; Guo, Y.H.; Zheng, L.L.; Li, Y.; Xu, W.L.; Zhang, Y.C.; Zhou, H.; Lun, Z.R.; Ayala, F.J.; Qu, L.H. Both endo-siRNAs and tRNA-derived small RNAs are involved in the differentiation of primitive eukaryote Giardia lamblia. Proc. Natl. Acad. Sci. USA 2014, 111, 14159–14164. [Google Scholar] [CrossRef] [PubMed]
- Dubey, J.P.; Lindsay, D.S.; Speer, C.A. Structures of Toxoplasma gondii tachyzoites, bradyzoites, and sporozoites and biology and development of tissue cysts. Clin. Microbiol. Rev. 1998, 11, 267–299. [Google Scholar] [CrossRef] [PubMed]
- Bartošová-Sojková, P.; Oppenheim, R.D.; Soldati-Favre, D.; Lukeš, J. Epicellular Apicomplexans: Parasites “On the Way In”. PLoS Pathog. 2015, 24, e1005080. [Google Scholar] [CrossRef]
- Zhao, X.Y.; Ewald, S.E. The molecular biology and immune control of chronic Toxoplasma gondii infection. J. Clin. Investig. 2020, 130, 3370–3380. [Google Scholar] [CrossRef]
- Flegr, J.; Klapilová, K.; Kaňková, S. Toxoplasmosis can be a sexually transmitted infection with serious clinical consequences. Not all routes of infection are created equal. Med. Hypoth. 2014, 83, 286–289. [Google Scholar] [CrossRef]
- Zhang, Y.; Lai, B.S.; Juhas, M.; Zhang, Y. Toxoplasma gondii secretory proteins and their role in invasion and pathogenesis. Microbiol. Res. 2019, 227, 126293. [Google Scholar] [CrossRef]
- Fallahi, S.; Rostami, A.; Nourollahpour-Shiadeh, M.; Behniafar, H.; Paktinat, S. An updated literature review on maternal-fetal and reproductive disorders of Toxoplasma gondii infection. J. Gynecol. Obstet. Hum. Reprod. 2018, 47, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Weiss, L.M.; Dubey, J.P. Toxoplasmosis: A history of clinical observations. Int. J. Parasitol. 2009, 39, 895–901. [Google Scholar] [CrossRef]
- Chen, X.; Chen, B.; Hou, X.; Zheng, C.; Yang, X.; Ke, J.; Hu, X.; Tan, F. Association between Toxoplasma gondii infection and psychiatric disorders in Zhejiang; Southeastern China. Acta Trop. 2019, 192, 82–86. [Google Scholar] [CrossRef]
- Fuglewicz, A.J.; Piotrowski, P.; Stodolak, A. Relationship between toxoplasmosis and schizophrenia: A review. Adv. Clin. Exp. Med. 2017, 26, 1031–1036. [Google Scholar] [CrossRef]
- Robert-Gangneux, F.; Dardé, M.L. Epidemiology of and diagnostic strategies for toxoplasmosis. Clin. Microbiol. Rev. 2012, 25, 264–296. [Google Scholar] [CrossRef]
- Neville, A.J.; Zach, S.J.; Wang, X.; Larson, J.J.; Judge, A.K.; Davis, L.A.; Vennerstrom, J.L.; Davis, P.H. Clinically Available Medicines Demonstrating Anti-Toxoplasma Activity. Antimicrob. Agents Chemother. 2015, 59, 7161–7169. [Google Scholar] [CrossRef]
- Deng, Y.; Wu, T.; Zhai, S.Q.; Li, C.H. Recent progress on anti-Toxoplasma drugs discovery: Design; synthesis and screening. Eur. J. Med. Chem. 2019, 183, 111711. [Google Scholar] [CrossRef]
- Gangjee, A.; Jain, H.D.; Queener, S.F.; Kisliuk, R.L. The effect of 5-alkyl modification on the biological activity of pyrrolo[2;3-d]pyrimidine containing classical and nonclassical antifolates as inhibitors of dihydrofolate reductase and as antitumor and/or antiopportunistic infection agents. J. Med. Chem. 2008, 51, 4589–4600. [Google Scholar] [CrossRef]
- Gangjee, A.; Vidwans, A.P.; Vasudevan, A.; Queener, S.F.; Kisliuk, R.L.; Cody, V.; Li, R.; Galitsky, N.; Luft, J.R.; Pangborn, W. Structure-based design and synthesis of lipophilic 2;4-diamino-6-substituted quinazolines and their evaluation as inhibitors of dihydrofolate reductases and potential antitumor agents. J. Med. Chem. 1998, 41, 3426–3434. [Google Scholar] [CrossRef] [PubMed]
- Tenório, R.P.; Carvalho, C.S.; Pessanha, C.S.; de Lima, J.G.; de Faria, A.R.; Alves, A.J.; de Melo, E.J.; Góes, A.J. Synthesis of thiosemicarbazone and 4-thiazolidinone derivatives and their in vitro anti-Toxoplasma gondii activity. Bioorg. Med. Chem. Lett. 2005, 15, 2575–2578. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, W.J., Jr.; Hakimi, M.A. Histone mediated gene activation in Toxoplasma gondii. Mol. Biochem. Parasitol. 2006, 148, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Gissot, M.; Kelly, K.A.; Ajioka, J.W.; Greally, J.M.; Kim, K. Epigenomic modifications predict active promoters and gene structure in Toxoplasma gondii. PLoS Pathog. 2007, 3, e77. [Google Scholar] [CrossRef] [PubMed]
- Meissner, M.; Soldati, D. The transcription machinery and the molecular toolbox to control gene expression in Toxoplasma gondii and other protozoan parasites. Microb. Infect. 2005, 7, 1376–1384. [Google Scholar] [CrossRef] [PubMed]
- Saksouk, N.; Bhatti, M.M.; Kieffer, S.; Smith, A.T.; Musset, K.; Garin, J.; Sullivan, W.J., Jr.; Cesbron-Delauw, M.F.; Hakimi, M.A. Histone-modifying complexes regulate gene expression pertinent to the differentiation of the protozoan parasite Toxoplasma gondii. Mol. Cell Biol. 2005, 25, 10301–10314. [Google Scholar] [CrossRef] [PubMed]
- Lau, Y.L.; Lee, W.C.; Gudimella, R.; Zhang, G.; Ching, X.T.; Razali, R.; Aziz, F.; Anwar, A.; Fong, M.Y. Deciphering the Draft Genome of Toxoplasma gondii RH Strain. PLoS ONE 2016, 11, e0157901. [Google Scholar] [CrossRef]
- Bougdour, A.; Maubon, D.; Baldacci, P.; Ortet, P.; Bastien, O.; Bouillon, A.; Barale, J.C.; Pelloux, H.; Ménard, R.; Hakimi, M.A. Drug inhibition of HDAC3 and epigenetic control of differentiation in Apicomplexa parasites. J. Exp. Med. 2009, 206, 953–966. [Google Scholar] [CrossRef]
- Hanquier, J.; Gimeno, T.; Jeffers, V.; Sullivan, W.J., Jr. Evaluating the GCN5b bromodomain as a novel therapeutic target against the parasite Toxoplasma gondii. Exp. Parasitol. 2020, 211, 107868. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, M.M.; Livingston, M.; Mullapudi, N.; Sullivan, W.J., Jr. Pair of unusual GCN5 histone acetyltransferases and ADA2 homologues in the protozoan parasite Toxoplasma gondii. Eukaryot. Cell 2006, 5, 62–76. [Google Scholar] [CrossRef]
- Fleck, K.; Nitz, M.; Jeffers, V. “Reading” a new chapter in protozoan parasite transcriptional regulation. PLoS Pathog. 2021, 17, e1010056. [Google Scholar] [CrossRef]
- Mouveaux, T.; Rotili, D.; Boissavy, T.; Roger, E.; Pierrot, C.; Mai, A.; Gissot, M. A potent HDAC inhibitor blocks Toxoplasma gondii tachyzoite growth and profoundly disrupts parasite gene expression. Int. J. Antimicrob. Agents 2022, 59, 106526. [Google Scholar] [CrossRef]
- Araujo-Silva, C.A.; De Souza, W.; Martins-Duarte, E.S.; Vommaro, R.C. HDAC inhibitors Tubastatin A and SAHA affect parasite cell division and are potential anti-Toxoplasma gondii chemotherapeutics. Int. J. Parasitol. Drugs Drug. Resist. 2021, 15, 25–35. [Google Scholar] [CrossRef]
- Jublot, D.; Cavaillès, P.; Kamche, S.; Francisco, D.; Fontinha, D.; Prudêncio, M.; Guichou, J.F.; Labesse, G.; Sereno, D.; Loeuillet, C. A Histone Deacetylase (HDAC) Inhibitor with Pleiotropic In Vitro Anti-Toxoplasma and Anti-Plasmodium Activities Controls Acute and Chronic Toxoplasma Infection in Mice. Int. J. Mol. Sci. 2022, 23, 3254. [Google Scholar] [CrossRef]
- Van der Pol, B. Trichomonas vaginalis infection: The most prevalent nonviral sexually transmitted infection receives the least public health attention. Clin. Infect. Dis. 2007, 44, 23–25. [Google Scholar] [CrossRef]
- Silver, B.J.; Guy, R.J.; Kaldor, J.M.; Jamil, M.S.; Rumbold, A.R. Trichomonas vaginalis as a cause of perinatal morbidity: A systematic review and meta-analysis. Sex. Transm. Dis. 2014, 41, 369–376. [Google Scholar] [CrossRef]
- Seña, A.C.; Miller, W.C.; Hobbs, M.M.; Schwebke, J.R.; Leone, P.A.; Swygard, H.; Atashili, J.; Cohen, M.S. Trichomonas vaginalis infection in male sexual partners: Implications for diagnosis; treatment; and prevention. Clin. Infect. Dis. 2007, 44, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Swygard, H.; Miller, W.C.; Kaydos-Daniels, S.C.; Cohen, M.S.; Leone, P.A.; Hobbs, M.M.; Seña, A.C. Targeted screening for Trichomonas vaginalis with culture using a two-step method in women presenting for STD evaluation. Sex. Transm. Dis. 2004, 31, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Cudmore, S.L.; Delgaty, K.L.; Hayward-McClelland, S.F.; Petrin, D.P.; Garber, G.E. Treatment of infections caused by metronidazole-resistant Trichomonas vaginalis. Clin. Microbiol. Rev. 2004, 17, 783–793. [Google Scholar] [CrossRef]
- Edwards, T.; Burke, P.; Smalley, H.; Hobbs, G. Trichomonas vaginalis: Clinical relevance; pathogenicity and diagnosis. Crit. Rev. Microbiol. 2016, 42, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Patel, O.P.S.; Jesumoroti, O.J.; Legoabe, L.J.; Beteck, R.M. Metronidazole-conjugates: A comprehensive review of recent developments towards synthesis and medicinal perspective. Eur. J. Med. Chem. 2021, 210, 112994. [Google Scholar] [CrossRef]
- Ribeiro, K.C.; Monteiro-Leal, L.H.; Benchimol, M. Contributions of the axostyle and flagella to closed mitosis in the protists Tritrichomonas foetus and Trichomonas vaginalis. J. Eukaryot. Microbiol. 2000, 47, 481–492. [Google Scholar] [CrossRef]
- Voleman, L.; Doležal, P. Mitochondrial dynamics in parasitic protists. PLoS Pathog. 2019, 15, e1008008. [Google Scholar] [CrossRef]
- Kulda, J. Trichomonads, hydrogenosomes and drug resistance. Int. J. Parasitol. 1999, 29, 199–212. [Google Scholar] [CrossRef]
- Gunderson, J.; Hinkle, G.; Leipe, D.; Morrison, H.G.; Stickel, S.K.; Odelson, D.A.; Breznak, J.A.; Nerad, T.A.; Müller, M.; Sogin, M.L. Phylogeny of trichomonads inferred from small-subunit rRNA sequences. J. Eukaryot. Microbiol. 1995, 42, 411–415. [Google Scholar] [CrossRef]
- Roger, A.J.; Clark, C.G.; Doolittle, W.F. A possible mitochondrial gene in the early-branching amitochondriate protist Trichomonas vaginalis. Proc. Natl. Acad. Sci. USA 1996, 93, 14618–14622. [Google Scholar] [CrossRef]
- Lizarraga, A.; Muñoz, D.; Strobl-Mazzulla, P.H.; de Miguel, N. Toward incorporating epigenetics into regulation of gene expression in the parasite Trichomonas vaginalis. Mol. Microbiol. 2021, 115, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Song, M.J.; Kim, M.; Choi, Y.; Yi, M.H.; Kim, J.; Park, S.J.; Yong, T.S.; Kim, H.P. Epigenome mapping highlights chromatin-mediated gene regulation in the protozoan parasite Trichomonas vaginalis. Sci. Rep. 2017, 7, 45365. [Google Scholar] [CrossRef] [PubMed]
- Pachano, T.; Nievas, Y.R.; Lizarraga, A.; Johnson, P.J.; Strobl-Mazzulla, P.H.; de Miguel, N. Epigenetics regulates transcription and pathogenesis in the parasite Trichomonas vaginalis. Cell Microbiol. 2017, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Carlton, J.M.; Hirt, R.P.; Silva, J.C.; Delcher, A.L.; Schatz, M.; Zhao, Q.; Wortman, J.R.; Bidwell, S.L.; Alsmark, U.C.; Besteiro, S.; et al. Draft genome sequence of the sexually transmitted pathogen Trichomonas vaginalis. Science 2007, 315, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Warring, S.D.; Blow, F.; Avecilla, G.; Orosco, J.C.; Sullivan, S.A.; Carlton, J.M. Small RNAs Are Implicated in Regulation of Gene and Transposable Element Expression in the Protist Trichomonas vaginalis. mSphere 2021, 6, e01061-20. [Google Scholar] [CrossRef]
- Cong, P.; Luo, Y.; Bao, W.; Hu, S. Genomic organization and promoter analysis of the Trichomonas vaginalis core histone gene families. Parasitol. Int. 2010, 59, 29–34. [Google Scholar] [CrossRef]
- Smith, A.J.; Chudnovsky, L.; Simoes-Barbosa, A.; Delgadillo-Correa, M.G.; Jonsson, Z.O.; Wohlschlegel, J.A.; Johnson, P.J. Novel core promoter elements and a cognate transcription factor in the divergent unicellular eukaryote Trichomonas vaginalis. Mol. Cell Biol. 2011, 31, 1444–1458. [Google Scholar] [CrossRef]
- Zubácová, Z.; Hostomská, J.; Tachezy, J. Histone H3 Variants in Trichomonas vaginalis. Eukaryot. Cell 2012, 11, 654–661. [Google Scholar] [CrossRef][Green Version]
- Chen, K.; Zhao, B.S.; He, C. Nucleic Acid Modifications in Regulation of Gene Expression. Cell Chem. Biol. 2016, 23, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.Z.; Blanco, M.A.; Greer, E.L.; He, C.; Shi, Y. DNA N(6)-methyladenine: A new epigenetic mark in eukaryotes? Nat. Rev. Mol. Cell Biol. 2015, 16, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Lizarraga, A.; O’Brown, Z.K.; Boulias, K.; Roach, L.; Greer, E.L.; Johnson, P.J.; Strobl-Mazzulla, P.H.; de Miguel, N. Adenine DNA methylation; 3D genome organization; and gene expression in the parasite Trichomonas vaginalis. Proc. Natl. Acad. Sci. USA 2020, 117, 13033–13043. [Google Scholar] [CrossRef] [PubMed]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drugs | IC50 (µM) | Doses | Side Effects |
|---|---|---|---|
| Benznidazole | 0.0018 [27] | Oral-Solid: 100 mg tablet; 50 mg tablet (Adults) [30]; 12.5 mg tablets by two months. | Chills, chest pain, fever, severe rash, unusual bleeding or bruising, unusual tiredness or weakness, and painful or difficult urination [31]. |
| Nifurtimox | 0.11 [28] | Oral-Solid: 30 mg tablet; 120 mg tablet; 250 mg tablet by two months [32]. | Decreased appetite, diarrhea, dizziness, fever, headache, nausea, stomach pain, vomiting, and weight loss [31]. |
| Drugs | IC50 (µM) | Doses | Side Effects |
|---|---|---|---|
| Pentamidine | 0.0008 [59] | Parenteral-General injections-IM: 200 mg (as isethionate) powder for injection by one week [32]. | Pain and swelling at the injection site, hypotension, vomiting, blood dyscrasias, and renal damage [31]. |
| Suramin | 0.005 [60] | Doses are based on body weight and must be determined by a physician within approximately twenty-one days [32]. | Nausea, vomiting, diarrhea, headache, skin tingling, and weakness [31]. |
| Melarsoprol | 0.0026 [61] | Parenteral-General injections-IV: 3.6% in 5 mL ampoule solution (180 mg of the active compound by ten days) [32]. | Convulsions, fever, loss of consciousness, rashes, bloody stools, nausea, and vomiting [31]. |
| Eflornithine | 0.026 | Parenteral-General injections-IV: 200 mg per mL in 100 mL bottle (hydrochloride) by one week [32]. | Sore throat and fever, unusual bleeding or bruising, unusual tiredness or weakness, convulsions (seizures), and loss of hearing [31]. |
| Nifurtimox | 0.005 | Oral-Solid: 120 mg by ten days [32]. | Sore throat and fever, unusual bleeding or bruising, unusual tiredness or weakness, convulsions (seizures), and loss of hearing [31]. |
| Drugs | IC50 | Doses | Side Effects |
|---|---|---|---|
| Glucantime | 100 µg/mL [104]. | Adults and children, 20 mg per kg of body weight per day injected into a muscle for twenty to twenty-eight 28 days [105]. | Fever, arthralgia, myalgia, nausea and vomiting, erythema nodosum, and skin rash [31]. |
| Pentostam | 24 µg/mL [106]. | Adults 100 mg 3 times a day for three months. Children younger than 16 years of age. Use and dose must be determined by a physician [32]. | Black, tarry stools; bleeding of the gums; blindness; blood in the urine or stools; blurred, decreased, or other vision changes; bruising; burning, dry, or itching eyes; chills; and cough [31]. |
| Amphotericin B | 0.6 to 0.7 μM [107]. | Adults and children: 3 to 6 mg per kilogram of body weight once daily for seven days, injected slowly into a vein [31]. | Chills, fever, irregular heartbeat, muscle cramps or pain, unusual tiredness, or weakness [31]. |
| Pentamidine | 5.09 µM [108]. | A single dose of 7 mg/kg [109]. | Decrease in urination, sore throat, fever, unusual bleeding, or bruising [31]. |
| Ketoconazole | 2 µM [111]. | Oral 600 mg/day for twenty-eight days [110]. | Bleeding gums, blood in the urine or stools, blurred vision, burning, itching. Ketoconazole can cause serious harm to the liver that may result in a liver transplant or cause death [31]. |
| Miltefosine | 3.8 μM [112]. | Oral-Solid: 10 mg; 50 mg for twenty days cutaneous form and twenty-eight to forty days visceral form [32]. | Abdominal or stomach pain; bloating or swelling of the face, arms, hands, lower legs, or feet; and possible teratogenicity [31]. |
| Paromomycin | 133.8 μM [112]. | Parenteral-General injections-IM: 750 mg paromomycin base (as sulfate) for twenty-eight days [32]. | Abdominal or stomach cramps, diarrhea, and nausea [31]. |
| Drugs | IC50 (µM) | Doses | Side Effects |
|---|---|---|---|
| Paromomycin | Not available | Oral-Liquid: 125 mg per 5 mL as sulfate. Oral-Solid: 250 mg as sulfate for five to six days [32]. | Abdominal or stomach cramps, diarrhea, and nausea [31]. |
| Metronidazole | 0.23 [141] | Oral-Liquid: 200 mg per 5 mL (as benzoate) Oral-Solid: 200 to 500 mg tablet Parenteral-General injections-unspecified: 500 mg in 100 mL vial for five to ten days [32]. | Agitation, back pain, blindness, confusion, decreased vision, depression, dizziness, fever, headache, irritability, nausea, seizures, vomiting, and weakness in the arms, hands, legs, or feet [31]. |
| Diiodohydroxyquin | 0.082 [142] | Tab 210 mg three times Tab 650 mg three times for ten days [32]. | Vomiting, nausea, diarrhea, headaches, skin rashes, and anal pruritis [31]. |
| Diloxanide furoate | Not available | Adults, oral, 500 mg every 8 h for ten days Children > 25 kg, 20 mg/kg per day orally in three separate doses for ten days [32]. | Feeling sick (nausea) or being sick (vomiting), loss of appetite, diarrhea, stomach (abdominal) cramps, and wind (flatulence) [31]. |
| Drugs | IC50 (mg/L) | Doses | Side Effects |
|---|---|---|---|
| Metronidazole | 0.21 [177] | 15 mg/kg/day (maximum 750 mg/day) orally in three doses for five to ten days [178]. | Nausea, abdominal pain, and diarrhea. Rare cases of neurotoxicity, optic neuropathy, peripheral neuropathy, and encephalopathy. Genotoxic effects in animal models are controversial in humans [176]. |
| Tinidazole | 0.14 [177] | 50 mg/kg (maximum 2 g) orally, single dose [178]. | Chest tightness, change in consciousness, cough, loss of consciousness, and trouble breathing [31]. |
| Nitazoxanide | 15 nM [177] | 7.5 mg/kg orally twice a day for three days [178]. | Abdominal or stomach pain, headache, nausea, and urine changes [31]. |
| Albendazole | 0.01 [177] | 10 to 15 mg/kg (maximum 400 mg) orally once daily for five days [178]. | Stomach pain; black, tarry stools; bleeding gums; and blood in the urine or stools [31]. |
| Mebendazole | 0.06 [177] | 100 mg orally twice a day for three days [178]. | Black, tarry stools; chills, convulsions, cough, or hoarseness; dark urine; fever with or without chills; and a general feeling of tiredness or weakness [31]. |
| Furazolidone | 0.62 [177] | 2 mg/kg (maximum 100 mg) orally, four times daily for seven days [178]. | Abdominal or stomach pain, diarrhea, headache, nausea, or vomiting [31]. |
| Secnidazole | 0.62 [177] | 30 mg/kg (maximum 2 g) orally, single dose [178]. | Change in taste, Diarrhea, headache, loss of taste, nausea, stomach pain, and vomiting [31]. |
| Ornidazole | 0.12 [177] | 20 to 40 mg/kg (maximum 2 g) orally, single dose [178]. | Abdominal or stomach pain; anxiety; black, tarry stools; bleeding gums; blood in the urine or stools; blurred vision; body aches; or chest pain [31]. |
| Drugs | IC50 (µM) | Doses | Side Effects |
|---|---|---|---|
| Spiramycin | Not available | Adults and teenagers—1 to 2 g, two times a day, or 500 mg to 1 g at least four to six weeks after the absence of clinical symptoms [32]. | Skin rash and itching, unusual bleeding or bruising [31]. |
| Pyrimethamine | 0.08 [201] | Oral-Solid: 25 mg for one to three weeks [32]. | Chest pain, dry cough, fever, rapid or trouble breathing, skin rash, unusual tiredness or weakness [31]. |
| Sulfadiazine | 100 [202] | 100 mg/kg daily, orally, divided twice a day for three to four weeks [32]. | Hives; difficulty breathing; swelling of the face, lips, tongue, or throat [31]. |
| Trimethoprim | 0.0072 [203] | 10 mg/kg/day for four weeks [32]. | Skin rash or itching; black tarry stools; blood in urine or stools; bluish fingernails, lips, or skin; changes in facial skin color; and chills [31]. |
| Sulfamethoxazole | Not available | One 800 mg tablet of sulfamethoxazole and 160 mg of trimethoprim or four teaspoonfuls or 20 mL of oral liquid every 12 h for ten to fourteen days [32]. | Black, tarry stools; blistering, peeling, or loosening of the skin; chest pain or tightness [31]. |
| Drugs | IC50 (µM) | Doses | Side Effects |
|---|---|---|---|
| Metronidazole | 0.068 [222] | Adults: A tablet of 2 g, in a single dose of 1 g twice a day for one day. The capsule 375 mg twice a day for seven days. Children: Use and dose must be determined by a physician [29]. | Vomiting, blindness, back pain, burning, numbness, tingling or painful sensations in the hands or feet, dizziness, and drowsiness [31]. |
| Tinidazole | Not available | Adults 2 g given once as a single dose. Children: Use and dose must be determined by a physician [29]. | Chest tightness, change in consciousness, loss of consciousness, cough, and trouble breathing [31]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaona-López, C.; Vazquez-Jimenez, L.K.; Gonzalez-Gonzalez, A.; Delgado-Maldonado, T.; Ortiz-Pérez, E.; Nogueda-Torres, B.; Moreno-Rodríguez, A.; Vázquez, K.; Saavedra, E.; Rivera, G. Advances in Protozoan Epigenetic Targets and Their Inhibitors for the Development of New Potential Drugs. Pharmaceuticals 2023, 16, 543. https://doi.org/10.3390/ph16040543
Gaona-López C, Vazquez-Jimenez LK, Gonzalez-Gonzalez A, Delgado-Maldonado T, Ortiz-Pérez E, Nogueda-Torres B, Moreno-Rodríguez A, Vázquez K, Saavedra E, Rivera G. Advances in Protozoan Epigenetic Targets and Their Inhibitors for the Development of New Potential Drugs. Pharmaceuticals. 2023; 16(4):543. https://doi.org/10.3390/ph16040543
Chicago/Turabian StyleGaona-López, Carlos, Lenci K. Vazquez-Jimenez, Alonzo Gonzalez-Gonzalez, Timoteo Delgado-Maldonado, Eyrá Ortiz-Pérez, Benjamín Nogueda-Torres, Adriana Moreno-Rodríguez, Karina Vázquez, Emma Saavedra, and Gildardo Rivera. 2023. "Advances in Protozoan Epigenetic Targets and Their Inhibitors for the Development of New Potential Drugs" Pharmaceuticals 16, no. 4: 543. https://doi.org/10.3390/ph16040543
APA StyleGaona-López, C., Vazquez-Jimenez, L. K., Gonzalez-Gonzalez, A., Delgado-Maldonado, T., Ortiz-Pérez, E., Nogueda-Torres, B., Moreno-Rodríguez, A., Vázquez, K., Saavedra, E., & Rivera, G. (2023). Advances in Protozoan Epigenetic Targets and Their Inhibitors for the Development of New Potential Drugs. Pharmaceuticals, 16(4), 543. https://doi.org/10.3390/ph16040543