
Effects of Apocynin, a NADPH Oxidase Inhibitor, in the Protection of the Heart from Ischemia/Reperfusion Injury


Abstract

{kind=link}
{kind=link}
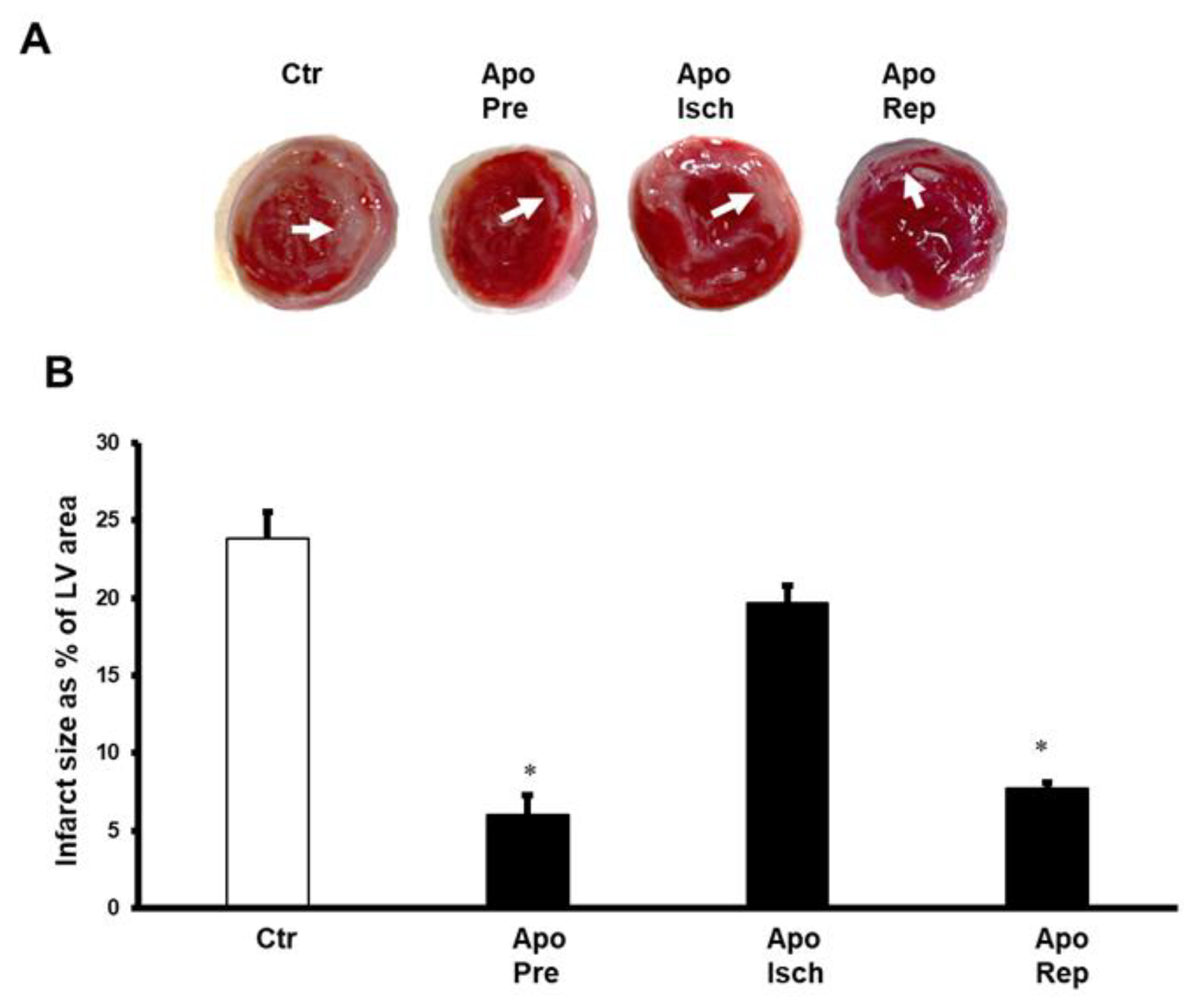{kind=link}
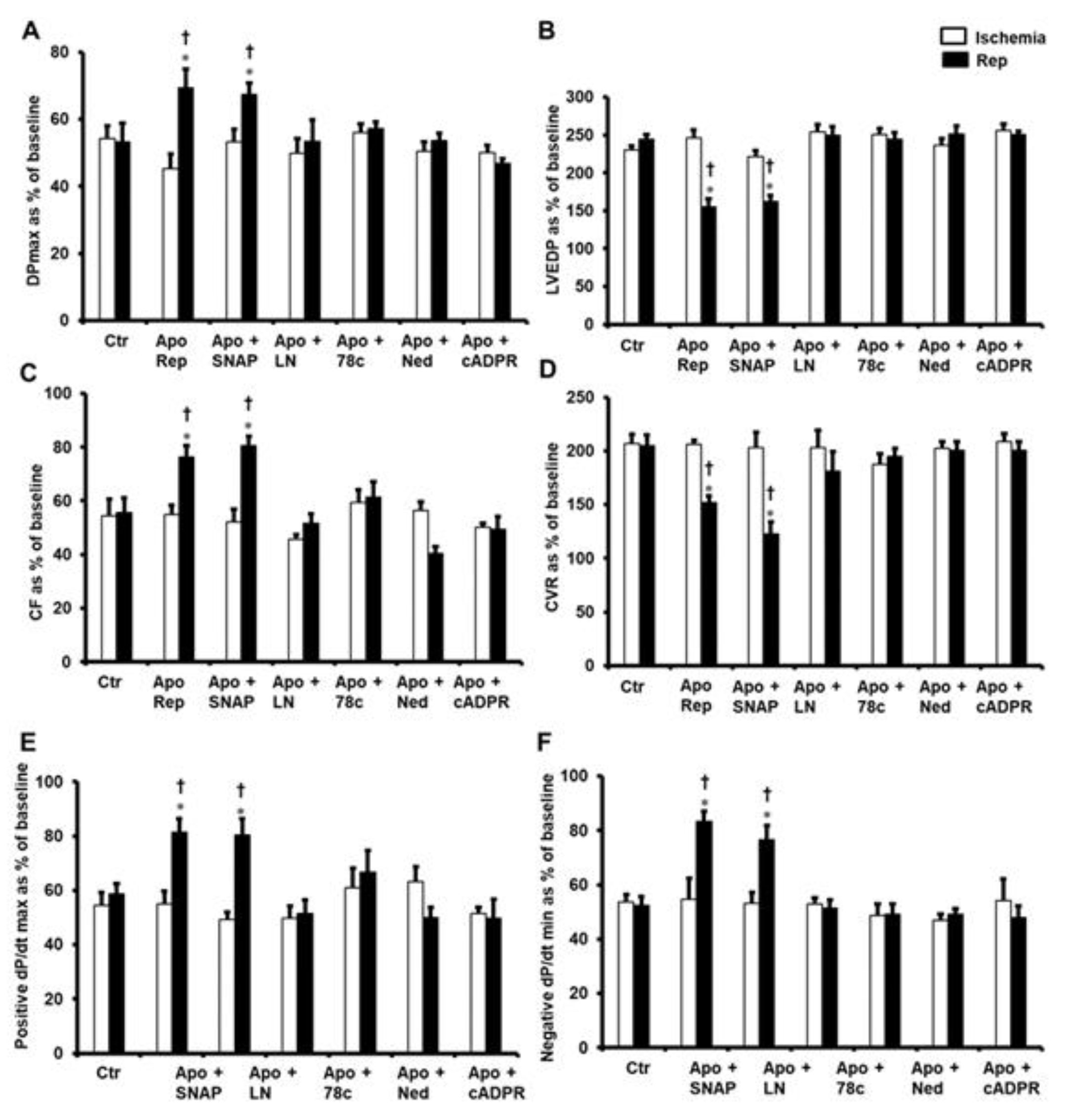{kind=link}
{kind=link}
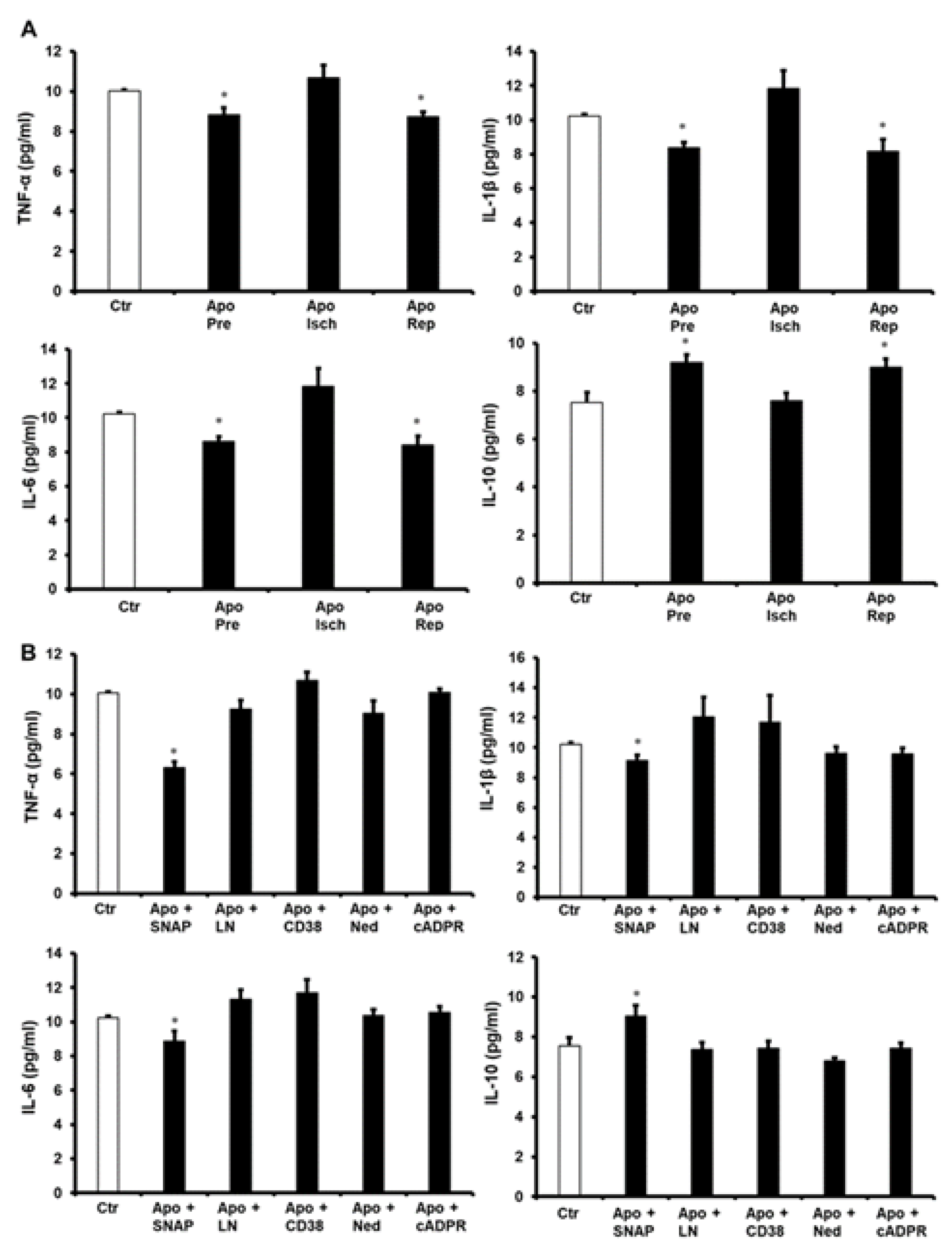{kind=link}
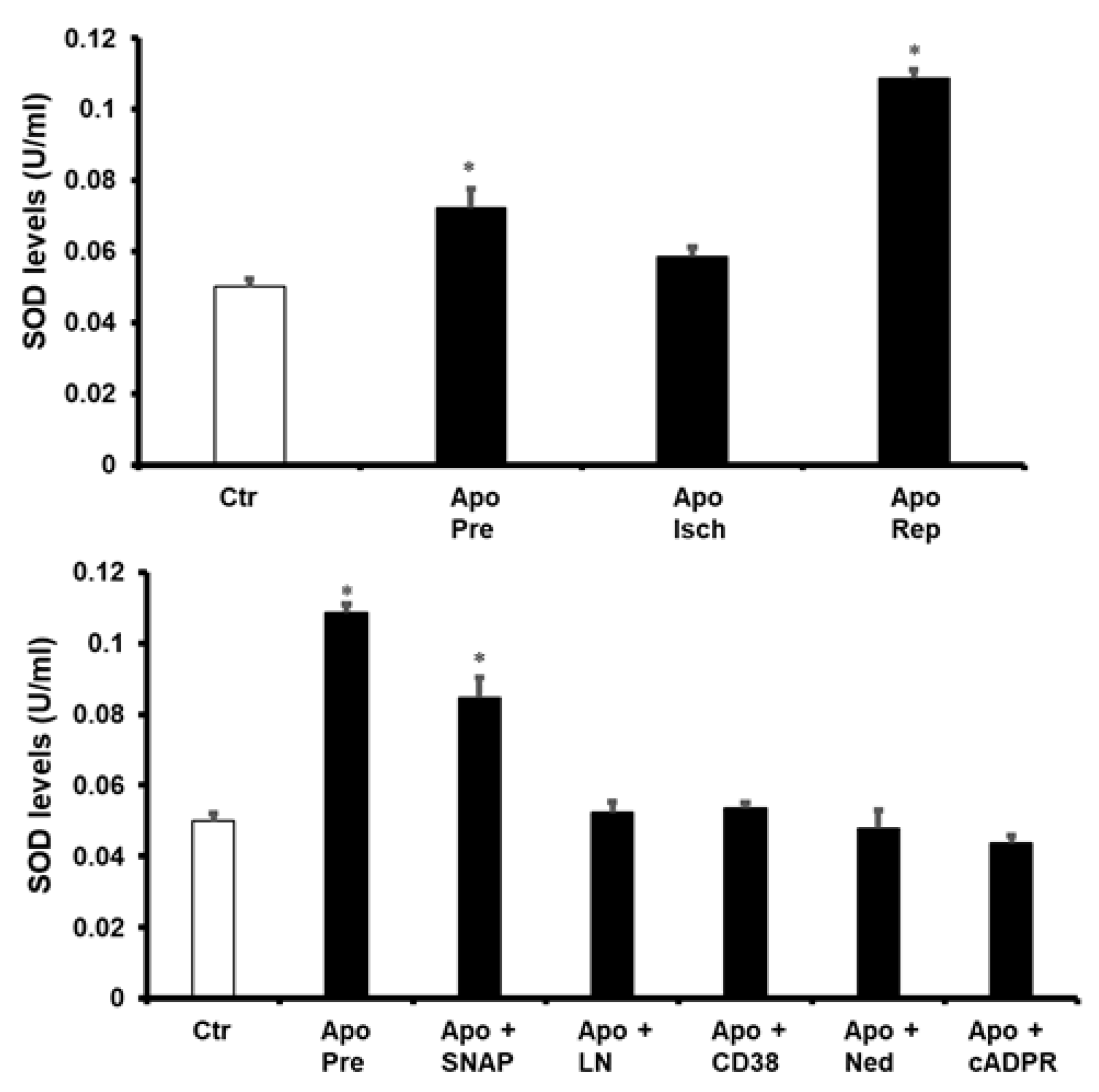{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
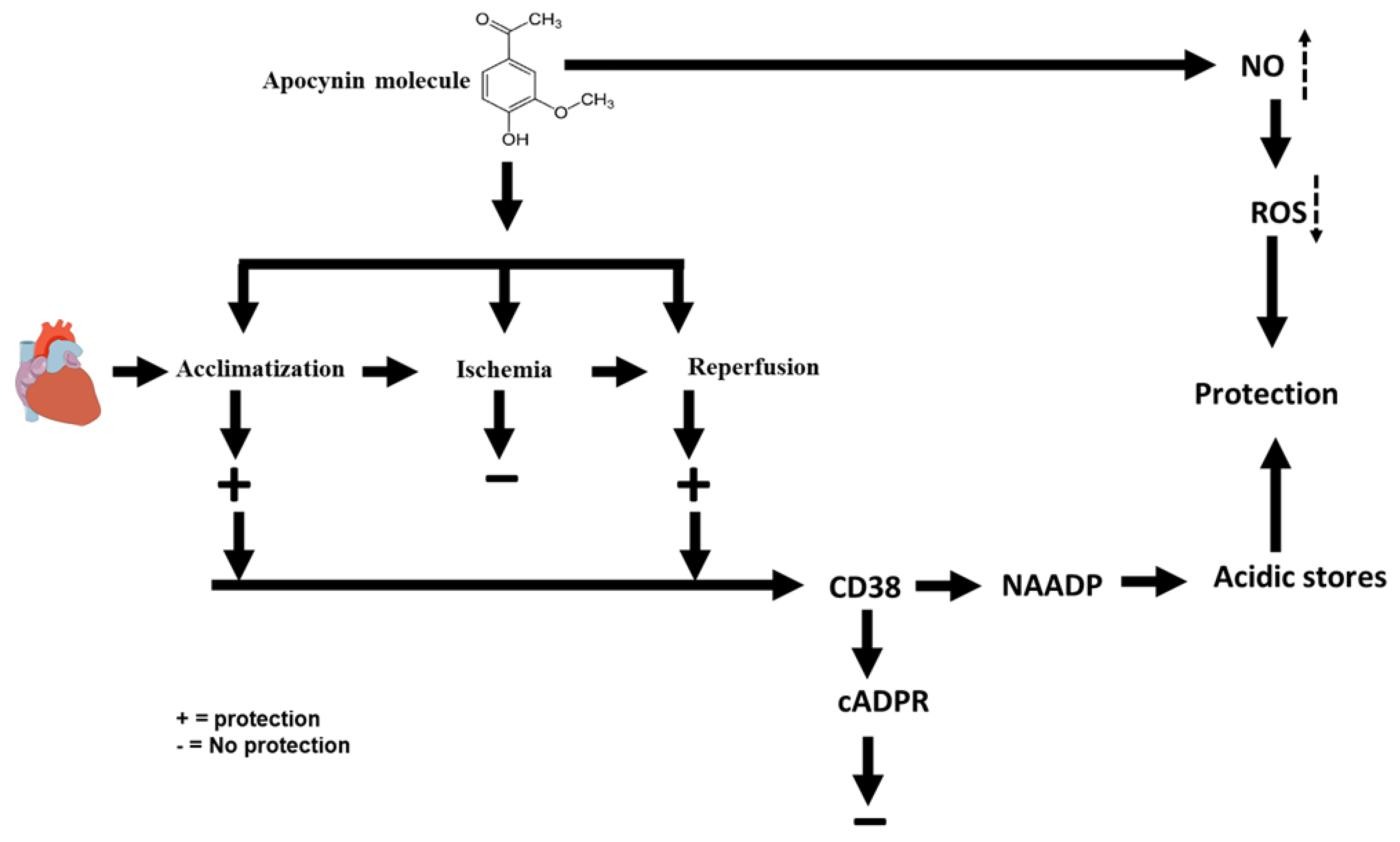
2. Results
3. Discussion
4. Materials and Methods
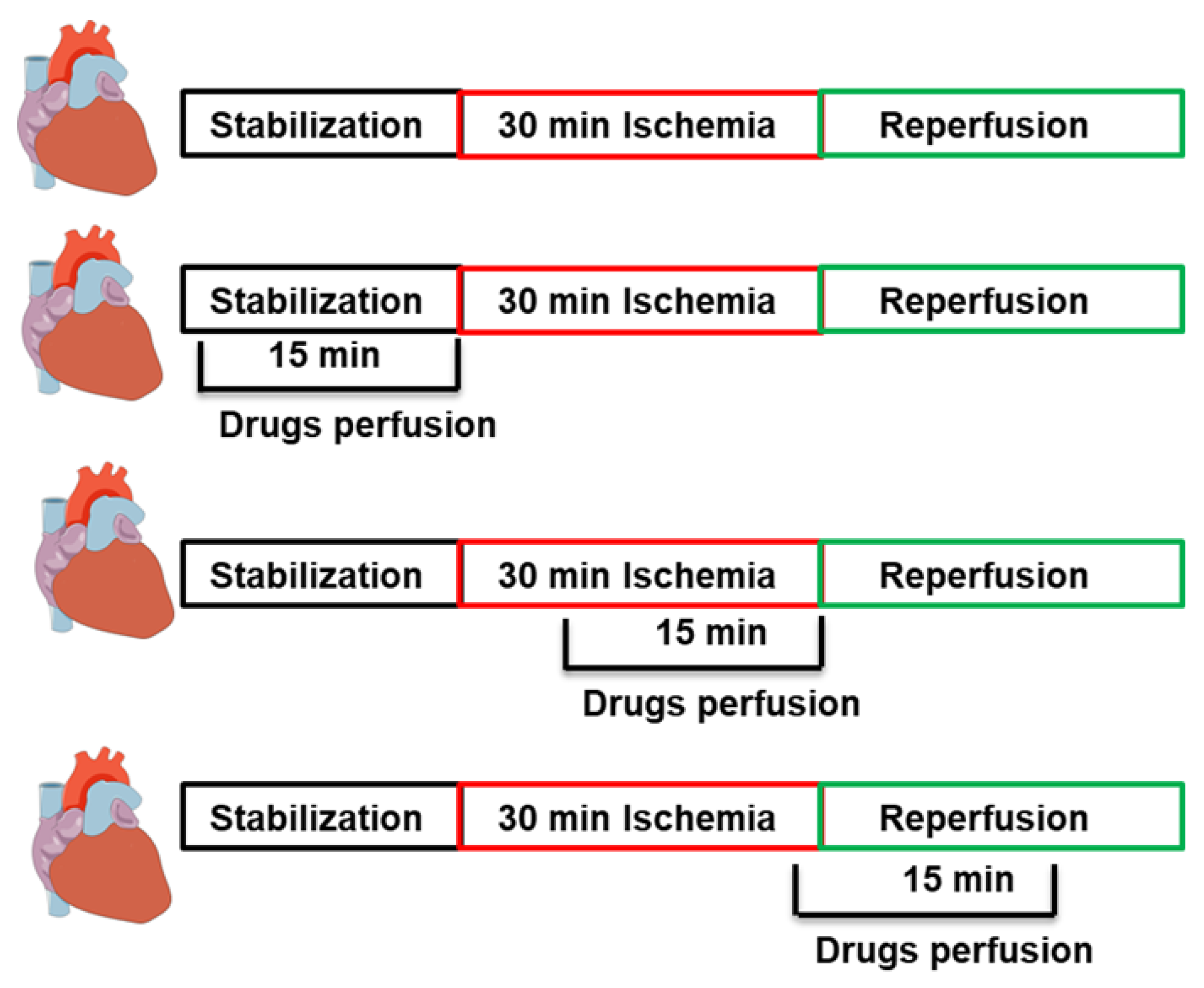
4.1. Experimental Procedure
4.2. Study Protocol
4.3. Cardiac Hemodynamics Assessment
4.4. Sample Storage
4.5. Evaluation of Cardiac Injury by Measurements of Infarct Size and Cardiac Enzyme Levels
4.6. Protein Extraction
4.7. Enzyme-Linked Immunosorbent Assay (ELISA)
4.8. Antioxidant Evaluation
4.9. Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2020 Update: A Report from the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart disease and stroke statistics—2019 update: A report from the American heart association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef] [PubMed]
- Mensah, G.A.; Wei, G.S.; Sorlie, P.D.; Fine, L.J.; Rosenberg, Y.; Kaufmann, P.G.; Mussolino, M.E.; Hsu, L.; Addou, E.; Engelgau, M.M.; et al. Decline in Cardiovascular Mortality. Circ. Res. 2017, 120, 366–380. [Google Scholar] [CrossRef]
- Cung, T.-T.; Morel, O.; Cayla, G.; Rioufol, G.; Garcia-Dorado, D.; Angoulvant, D.; Bonnefoy-Cudraz, E.; Guérin, P.; Elbaz, M.; Delarche, N.; et al. Cyclosporine before PCI in Patients with Acute Myocardial Infarction. New Engl. J. Med. 2015, 373, 1021–1031. [Google Scholar] [CrossRef]
- Hart, B.A.T.; Copray, S.; Philippens, I. Apocynin, a Low Molecular Oral Treatment for Neurodegenerative Disease. BioMed. Res. Int. 2014, 2014, 298020. [Google Scholar] [CrossRef]
- Petrônio, M.S.; Zeraik, M.L.; Da Fonseca, L.M.; Ximenes, V.F. Apocynin: Chemical and Biophysical Properties of a NADPH Oxidase Inhibitor. Molecules 2013, 18, 2821–2839. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M. Apocynin, NADPH Oxidase, and Vascular Cells: A complex matter. Hypertension 2008, 51, 172–174. [Google Scholar] [CrossRef]
- Muijsers, R.B.; van Den Worm, E.; Folkerts, G.; Beukelman, C.J.; Koster, A.S.; Postma, D.S.; Nijkamp, F.P. Apocynin inhibits peroxynitrite formation by murine macrophages. Br. J. Pharmacol. 2000, 130, 932–936. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Stefanska, J.; Pawliczak, R. Apocynin: Molecular Aptitudes. Mediat. Inflamm. 2008, 2008, 106507. [Google Scholar] [CrossRef]
- Uysal, A.; Sahna, E.; Özgüler, I.M.; Burma, O.; Ilhan, N. Effects of apocynin, an NADPH oxidase inhibitor, on levels of ADMA, MPO, iNOS and TLR4 induced by myocardial ischemia reperfusion. Perfusion 2015, 30, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Parker, W.; Devine, I.; Ondrasik, R.; Habtamu, T.; Bartol, K.D.; Casey, B.; Patel, H.; Chau, W.; Kuhn, T.; et al. Apocynin Exerts Dose-Dependent Cardioprotective Effects by Attenuating Reactive Oxygen Species in Ischemia/Reperfusion. Cardiovasc. Pharmacol. 2016, 5, 1–7. [Google Scholar] [CrossRef]
- Chen, S.; Meng, X.-F.; Zhang, C. Role of NADPH Oxidase-Mediated Reactive Oxygen Species in Podocyte Injury. BioMed. Res. Int. 2013, 2013, 839761. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, S.; Tsutsui, H.; Sadoshima, J. Physiological and pathological functions of NADPH oxidases during myocardial ischemia–reperfusion. Trends Cardiovasc. Med. 2014, 24, 202–205. [Google Scholar] [CrossRef]
- Ago, T.; Kuroda, J.; Pain, J.; Fu, C.; Li, H.; Sadoshima, J. Upregulation of Nox4 by Hypertrophic Stimuli Promotes Apoptosis and Mitochondrial Dysfunction in Cardiac Myocytes. Circ. Res. 2010, 106, 1253–1264. [Google Scholar] [CrossRef]
- Krijnen, P.A.; Meischl, C.; Hack, C.E.; Meijer, C.J.; Visser, C.A.; Roos, D.; Niessen, H.W. Increased Nox2 expression in human cardiomyocytes after acute myocardial infarction. J. Clin. Pathol. 2003, 56, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, S.; Kuroda, J.; Ago, T.; Zhai, P.; Ikeda, Y.; Oka, S.; Fong, G.H.; Tian, R.; Sadoshima, J. Broad Suppression of NADPH Oxidase Activity Exacerbates Ischemia/Reperfusion Injury Through Inadvertent Downregulation of Hypoxia-inducible Factor-1α and Upregulation of Peroxisome Proliferator–activated Receptor-α. Circ. Res. 2013, 112, 1135–1149. [Google Scholar] [CrossRef]
- Qin, F.; Simeone, M.; Patel, R. Inhibition of NADPH oxidase reduces myocardial oxidative stress and apoptosis and improves cardiac function in heart failure after myocardial infarction. Free. Radic. Biol. Med. 2007, 43, 271–281. [Google Scholar] [CrossRef]
- Boslett, J.; Hemann, C.; Zhao, Y.J.; Lee, H.-C.; Zweier, J.L. Luteolinidin Protects the Postischemic Heart through CD38 Inhibition with Preservation of NAD(P)(H). J. Pharmacol. Exp. Ther. 2017, 361, 99–108. [Google Scholar] [CrossRef]
- Davidson, S.M.; Foote, K.; Kunuthur, S.; Gosain, R.; Tan, N.; Tyser, R.; Zhao, Y.J.; Graeff, R.; Ganesan, A.; Duchen, M.R.; et al. Inhibition of NAADP signalling on reperfusion protects the heart by preventing lethal calcium oscillations via two-pore channel 1 and opening of the mitochondrial permeability transition pore. Cardiovasc. Res. 2015, 108, 357–366. [Google Scholar] [CrossRef]
- Zhan, K.-Y.; Kai-Yu, Z.; Liu, C.-H.; Luo, J.-H.; Yang, W. Detrimental or beneficial: The role of TRPM2 in ischemia/reperfusion injury. Acta Pharmacol. Sin. 2016, 37, 4–12. [Google Scholar] [CrossRef]
- Lee, H.C. Structure and Enzymatic Functions of Human CD38. Mol. Med. 2006, 12, 317–323. [Google Scholar] [CrossRef]
- Boslett, J.; Reddy, N.; Alzarie, Y.A.; Zweier, J.L. Inhibition of CD38 with the Thiazoloquin(az)olin(on)e 78c Protects the Heart against Postischemic Injury. J. Pharmacol. Exp. Ther. 2019, 369, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Boslett, J.; Helal, M.; Chini, E.; Zweier, J.L. Genetic deletion of CD38 confers post-ischemic myocardial protection through preserved pyridine nucleotides. J. Mol. Cell. Cardiol. 2018, 118, 81–94. [Google Scholar] [CrossRef]
- Boslett, J.; Hemann, C.; Christofi, F.L.; Zweier, J.L. Characterization of CD38 in the major cell types of the heart: Endothelial cells highly express CD38 with activation by hypoxia-reoxygenation triggering NAD(P)H depletion. Am. J. Physiol. Cell Physiol. 2018, 314, C297–C309. [Google Scholar] [CrossRef] [PubMed]
- Reyes, L.A.; Boslett, J.; Varadharaj, S.; De Pascali, F.; Hemann, C.; Druhan, L.J.; Ambrosio, G.; El-Mahdy, M.; Zweier, J.L. Depletion of NADP(H) due to CD38 activation triggers endothelial dysfunction in the postischemic heart. Proc. Natl. Acad. Sci. USA 2015, 112, 11648–11653. [Google Scholar] [CrossRef]
- Guse, A.H. Second messenger function and the structure-activity relationship of cyclic adenosine diphosphoribose (cADPR). FEBS J. 2005, 272, 4590–4597. [Google Scholar] [CrossRef]
- Xie, G.-H.; Rah, S.-Y.; Yi, K.S.; Han, M.-K.; Chae, S.-W.; Im, M.-J.; Kim, U.-H. Increase of intracellular Ca2+ during ischemia/reperfusion injury of heart is mediated by cyclic ADP-ribose. Biochem. Biophys. Res. Commun. 2003, 307, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Eraslan, E.; Tanyeli, A.; Polat, E. 8-Br-cADPR, a TRPM2 ion channel antagonist, inhibits renal ischemia–reperfusion injury. J. Cell. Physiol. 2019, 234, 4572–4581. [Google Scholar] [CrossRef] [PubMed]
- Altamirano, F.; Wang, Z.V.; Hill, J.A. Cardioprotection in ischaemia-reperfusion injury: Novel mechanisms and clinical translation. J. Physiol. 2015, 593, 3773–3788. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C. Cyclic ADP-ribose and Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) as Messengers for Calcium Mobilization. J. Biol. Chem. 2012, 287, 31633–31640. [Google Scholar] [CrossRef]
- Calcraft, P.J.; Ruas, M.; Pan, Z.; Cheng, X.; Arredouani, A.; Hao, X.; Tang, J.; Rietdorf, K.; Teboul, L.; Chuang, K.-T.; et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 2009, 459, 596–600. [Google Scholar] [CrossRef]
- Khalaf, A.; Babiker, F. Discrepancy in calcium release from the sarcoplasmic reticulum and intracellular acidic stores for the protection of the heart against ischemia/reperfusion injury. J. Physiol. Biochem. 2016, 72, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Riganti, C.; Costamagna, C.; Doublier, S.; Miraglia, E.; Polimeni, M.; Bosia, A.; Ghigo, D. The NADPH oxidase inhibitor apocynin induces nitric oxide synthesis via oxidative stress. Toxicol. Appl. Pharmacol. 2008, 228, 277–285. [Google Scholar] [CrossRef]
- Liu, X.; Huang, Y.; Pokreisz, P.; Vermeersch, P.; Marsboom, G.; Swinnen, M.; Verbeken, E.; Santos, J.; Pellens, M.; Gillijns, H.; et al. Nitric Oxide Inhalation Improves Microvascular Flow and Decreases Infarction Size After Myocardial Ischemia and Reperfusion. J. Am. Coll. Cardiol. 2007, 50, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.-J.; Ji, S.-K.; Liu, B.; Zhang, H.-F.; Yang, Z.-B.; Ma, Q.-L. NADPH oxidase inhibitor apocynin attenuates ischemia/reperfusion induced myocardial injury in rats. Zhonghua Xin Xue Guan Bing Za Zhi 2012, 40, 991–996. [Google Scholar]
- Lee, S.; Paudel, O.; Jiang, Y.; Yang, X.-R.; Sham, J.S.K. CD38 Mediates Angiotensin II–Induced Intracellular Ca2+ Release in Rat Pulmonary Arterial Smooth Muscle Cells. Am. J. Respir. Cell Mol. Biol. 2015, 52, 332–341. [Google Scholar] [CrossRef]
- Cheng, H.-P.; Wei, S.; Wei, L.-P.; Verkhratsky, A. Calcium signaling in physiology and pathophysiology. Acta Pharmacol. Sin. 2006, 27, 767–772. [Google Scholar] [CrossRef]
- Arredouani, A.; Evans, A.M.; Ma, J.; Parrington, J.; Zhu, M.X.; Galione, A. An emerging role for NAADP-mediated Ca2+ signaling in the pancreatic β-cell. Islets 2010, 2, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Walseth, T.F.; Guse, A.H. NAADP: From Discovery to Mechanism. Front. Immunol. 2021, 12, 703326. [Google Scholar] [CrossRef] [PubMed]
- Djerada, Z.; Peyret, H.; Dukic, S.; Millart, H. Extracellular NAADP affords cardioprotection against ischemia and reperfusion injury and involves the P2Y11-like receptor. Biochem. Biophys. Res. Commun. 2013, 434, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Schulz, R.; Kelm, M.; Heusch, G. Nitric oxide in myocardial ischemia/reperfusion injury. Cardiovasc. Res. 2004, 61, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Kawano, T.; Zoga, V.; Kimura, M.; Liang, M.-Y.; Wu, H.-E.; Gemes, G.; McCallum, J.B.; Kwok, W.-M.; Hogan, Q.H.; Sarantopoulos, C.D. Nitric Oxide Activates ATP-Sensitive Potassium Channels in Mammalian Sensory Neurons: Action by Direct S-Nitrosylation. Mol. Pain 2009, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, N.; Sato, T.; Ohler, A.; O'Rourke, B.; Marbán, E. Activation of Mitochondrial ATP-Dependent Potassium Channels by Nitric Oxide. Circulation 2000, 101, 439–445. [Google Scholar] [CrossRef]
- Kobara, M.; Tatsumi, T.; Takeda, M.; Mano, A.; Yamanaka, S.; Shiraishi, J.; Keira, N.; Matoba, S.; Asayama, J.; Nakagawa, M. The dual effects of nitric oxide synthase inhibitors on ischemia-reperfusion injury in rat hearts. Basic Res. Cardiol. 2003, 98, 319–328. [Google Scholar] [CrossRef]
- Zhang, Y.; Bissing, J.W.; Xu, L.; Ryan, A.J.; Martin, S.M.; Miller, F.J., Jr.; Kregel, K.C.; Buettner, G.R.; Kerber, R.E. Nitric oxide synthase inhibitors decrease coronary sinus-free radical concentration and ameliorate myocardial stunning in an ischemia-reperfusion model. J. Am. Coll. Cardiol. 2001, 38, 546–554. [Google Scholar] [CrossRef]
- Hamilton, C.A.; Brosnan, M.J.; Al-Benna, S.; Berg, G.; Dominiczak, A. NAD(P)H Oxidase Inhibition Improves Endothelial Function in Rat and Human Blood Vessels. Hypertension 2002, 40, 755–762. [Google Scholar] [CrossRef]
- Gouda, E.; Babiker, F. Gum Arabic protects the rat heart from ischemia/reperfusion injury through anti-inflammatory and antioxidant pathways. Sci. Rep. 2022, 12, 17235. [Google Scholar] [CrossRef]
- Ismaeil, A.; Babiker, F.; Al-Sabah, S. Discrepancy between the Actions of Glucagon-like Peptide-1 Receptor Ligands in the Protection of the Heart against Ischemia Reperfusion Injury. Pharmaceuticals 2022, 15, 720. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, S.; Babiker, F.; Akhtar, U.A.; Benter, I.F. Mitigating Cardiotoxicity of Dendrimers: Angiotensin-(1-7) via Its Mas Receptor Ameliorates PAMAM-Induced Cardiac Dysfunction in the Isolated Mammalian Heart. Pharmaceutics 2022, 14, 2673. [Google Scholar] [CrossRef]
- Al-Kouh, A.; Babiker, F.; Al-Bader, M. Renin–Angiotensin System Antagonism Protects the Diabetic Heart from Ischemia/Reperfusion Injury in Variable Hyperglycemia Duration Settings by a Glucose Transporter Type 4-Mediated Pathway. Pharmaceuticals 2023, 16, 238. [Google Scholar] [CrossRef]
- Babiker, F.A.; Al-Jarallah, A.; Joseph, S. Understanding pacing postconditioning-mediated cardiac protection: A role of oxidative stress and a synergistic effect of adenosine. J. Physiol. Biochem. 2017, 73, 175–185. [Google Scholar] [CrossRef]
- Babiker, F.A.; Van Golde, J.; Vanagt, W.Y.; Prinzen, F.W. Pacing Postconditioning: Impact of Pacing Algorithm, Gender, and Diabetes on Its Myocardial Protective Effects. J. Cardiovasc. Transl. Res. 2012, 5, 727–734. [Google Scholar] [CrossRef]
- Babiker, F.; Al-Kouh, A.; Kilarkaje, N. Lead exposure induces oxidative stress, apoptosis, and attenuates protection of cardiac myocytes against ischemia–reperfusion injury. Drug Chem. Toxicol. 2019, 42, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Gouda, A.; Babiker, F. Micronized flavonoid fraction Daflon 500 protects heart against ischemia–reperfusion injury: An old medicine for a new target. Front. Life Sci. 2020, 13, 556–568. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohammad, A.; Babiker, F.; Al-Bader, M. Effects of Apocynin, a NADPH Oxidase Inhibitor, in the Protection of the Heart from Ischemia/Reperfusion Injury. Pharmaceuticals 2023, 16, 492. https://doi.org/10.3390/ph16040492
Mohammad A, Babiker F, Al-Bader M. Effects of Apocynin, a NADPH Oxidase Inhibitor, in the Protection of the Heart from Ischemia/Reperfusion Injury. Pharmaceuticals. 2023; 16(4):492. https://doi.org/10.3390/ph16040492
Chicago/Turabian StyleMohammad, Ali, Fawzi Babiker, and Maie Al-Bader. 2023. "Effects of Apocynin, a NADPH Oxidase Inhibitor, in the Protection of the Heart from Ischemia/Reperfusion Injury" Pharmaceuticals 16, no. 4: 492. https://doi.org/10.3390/ph16040492
APA StyleMohammad, A., Babiker, F., & Al-Bader, M. (2023). Effects of Apocynin, a NADPH Oxidase Inhibitor, in the Protection of the Heart from Ischemia/Reperfusion Injury. Pharmaceuticals, 16(4), 492. https://doi.org/10.3390/ph16040492