

Trypanosoma cruzi Sirtuin 2 as a Relevant Druggable Target: New Inhibitors Developed by Computer-Aided Drug Design

 , , ,
, , ,
Abstract
1. Introduction
2. Results and Discussion
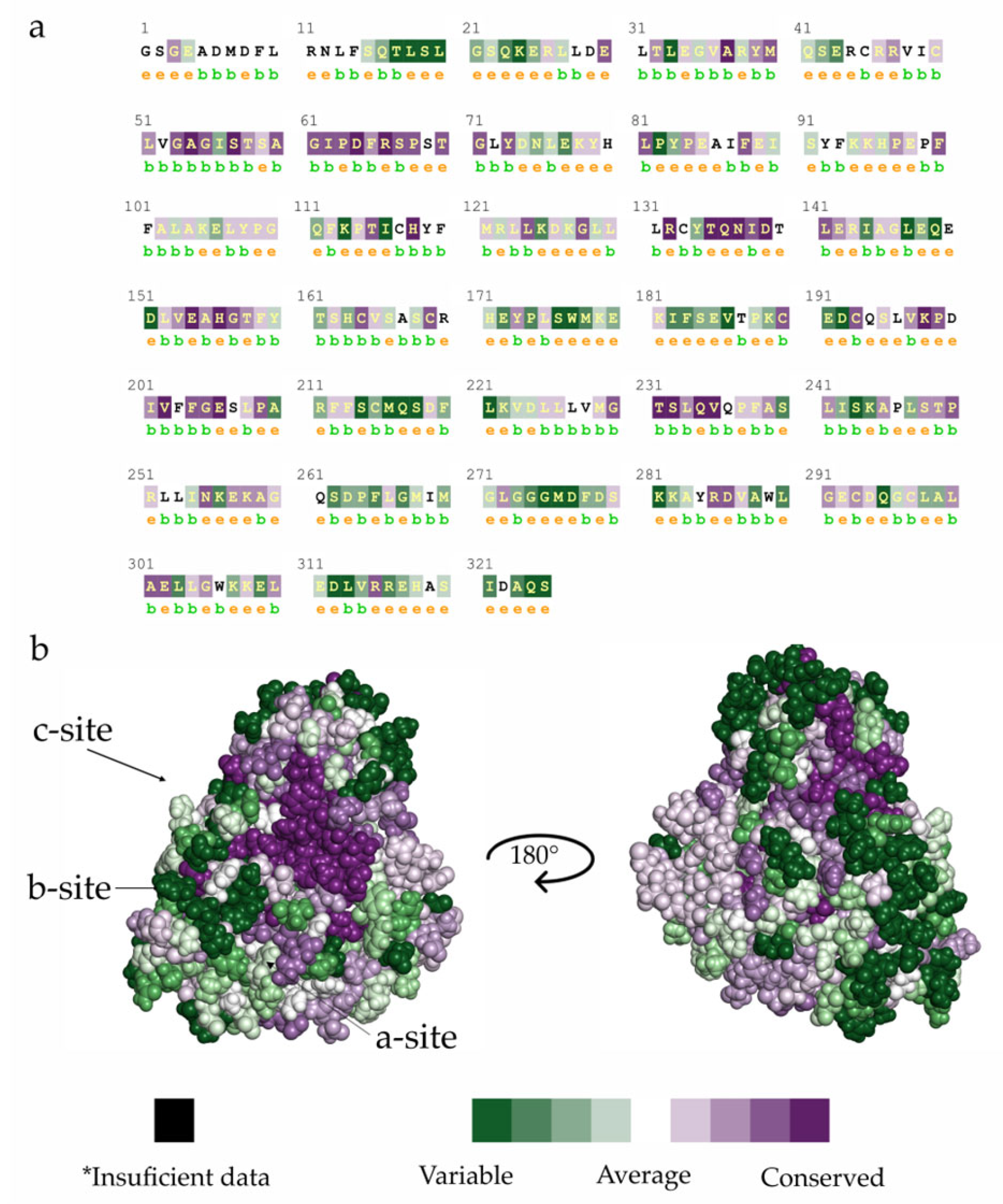
2.1. TcSir2 Structure Harbours a Conserved NAD+ Binding Site
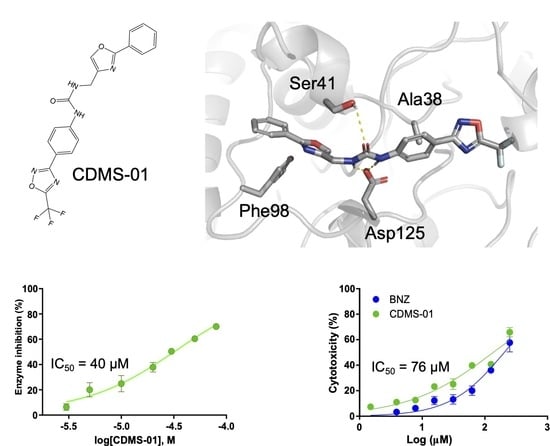
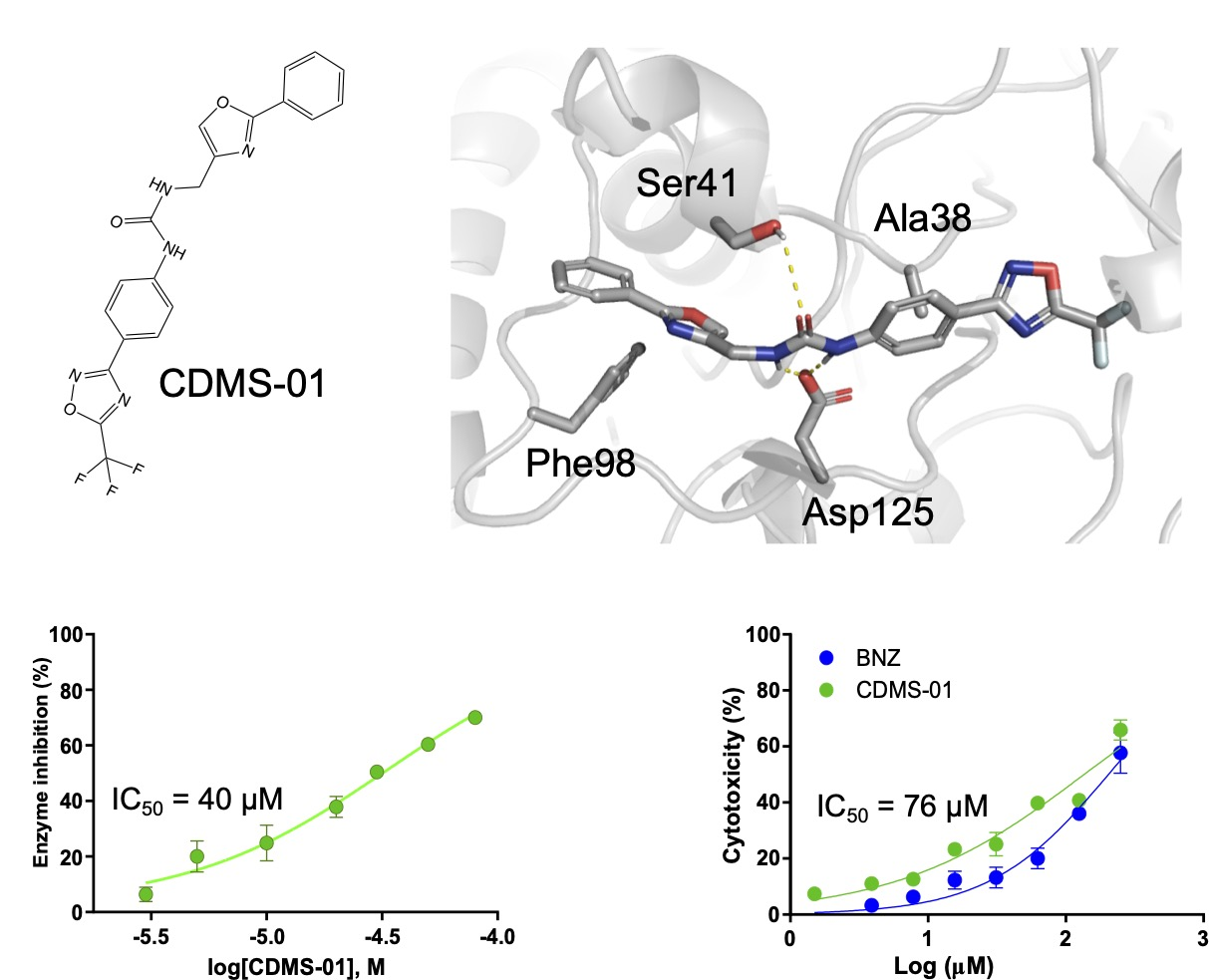
2.2. TcSir2 Inhibitor Virtual Screening and Experimental Validation
3. Materials and Methods
3.1. Computer-Aided Drug Discovery
3.1.1. Comparative Modelling and Conservation Analysis
3.1.2. Molecular Dynamics Simulation
3.1.3. Molecular Interactions Fields, Pharmacophore and Virtual Screening
3.2. Experimental Validation of Compound Inhibition
3.2.1. Chemicals and Biological Reagents
3.2.2. TcSir2 Recombinant Expression
3.2.3. Recombinant TcSir2 Activity Assay
3.2.4. Evaluation of In Vitro Trypanocidal Activity with Amastigote Forms
3.2.5. Mammalian Cell Viability Assay
3.2.6. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ioset, J.-R.; Chatelain, E. Drug discovery and development for neglected diseases: The DNDi model. Drug Des. Dev. Ther. 2011, 5, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Torrico, M.C.; Solano, M.; Guzmán, J.M.; Parrado, R.; Suarez, E.; Alonzo-Vega, C.; Truyens, C.; Carlier, Y.; Torrico, F. Estimation of the parasitemia in Trypanosoma cruzi human infection: High parasitemias are associated with severe and fatal congenital Chagas disease. Rev. Soc. Bras. Med. Trop. 2005, 38, 58–61. [Google Scholar] [PubMed]
- Alberca, L.N.; Sbaraglini, M.L.; Balcazar, D.; Fraccaroli, L.; Carrillo, C.; Medeiros, A.; Benitez, D.; Comini, M.; Talevi, A. Discovery of novel polyamine analogs with anti-protozoal activity by computer guided drug repositioning. J. Comput. Mol. Des. 2016, 30, 305–321. [Google Scholar] [CrossRef] [PubMed]
- DNDi. Drugs for Neglected Diseases Initiative. 2015. Available online: https://dndi.org/ (accessed on 18 May 2015).
- Clayton, J. Chagas disease: Pushing through the pipeline. Nature 2010, 465, S12–S15. [Google Scholar] [CrossRef]
- Moretti, N.S.; Augusto, L.D.S.; Clemente, T.M.; Antunes, R.P.P.; Yoshida, N.; Torrecilhas, A.C.; Cano, M.I.N.; Schenkman, S. Characterization of Trypanosoma cruzi Sirtuins as Possible Drug Targets for Chagas Disease. Antimicrob. Agents Chemother. 2015, 59, 4669–4679. [Google Scholar] [CrossRef]
- Ritagliati, C.; Alonso, V.L.; Manarin, R.; Cribb, P.; Serra, E.C. Overexpression of Cytoplasmic TcSIR2RP1 and Mitochondrial TcSIR2RP3 Impacts on Trypanosoma cruzi Growth and Cell Invasion. PLoS Neglected Trop. Dis. 2015, 9, e0003725. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Weinert, B.T.; Nishida, Y.; Verdin, E.; Mann, M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Callinan, P.A.; Feinberg, A. The emerging science of epigenomics. Hum. Mol. Genet. 2006, 15, R95–R101. [Google Scholar] [CrossRef] [PubMed]
- Menzies, K.J.; Zhang, H.; Katsyuba, E.; Auwerx, J. Protein acetylation in metabolism—Metabolites and cofactors. Nat. Rev. Endocrinol. 2016, 12, 43–60. [Google Scholar] [CrossRef]
- Van de Ven, R.A.; Santos, D.; Haigis, M.C. Mitochondrial Sirtuins and Molecular Mechanisms of Aging. Trends Mol. Med. 2017, 23, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Alsford, S.; Kawahara, T.; Isamah, C.; Horn, D. A sirtuin in the African trypanosome is involved in both DNA repair and telomeric gene silencing but is not required for antigenic variation. Mol. Microbiol. 2007, 63, 724–736. [Google Scholar] [CrossRef] [PubMed]
- Avalos, J.L.; Celic, I.; Muhammad, S.; Cosgrove, M.S.; Boeke, J.D.; Wolberger, C. Structure of a Sir2 Enzyme Bound to an Acetylated p53 Peptide. Mol. Cell 2002, 10, 523–535. [Google Scholar] [CrossRef]
- Gaspar, L.; Coron, R.P.; Lin, P.K.; Costa, D.M.; Perez-Cabezas, B.; Tavares, J.; Roura-Ferrer, M.; Ramos, I.; Ronin, C.; Major, L.L.; et al. Inhibitors of Trypanosoma cruzi Sir2 related protein 1 as potential drugs against Chagas disease. PLoS Negl. Trop. Dis. 2018, 12, e0006180. [Google Scholar] [CrossRef] [PubMed]
- Tavares, J.; Ouaissi, A.; Kongâthooâlin, P.; Loureiro, I.; Kaur, S.; Roy, N.; Cordeiro-Da-Silva, A. Bisnaphthalimidopropyl derivatives as inhibitors of Leishmania SIR2 related protein 1. ChemMedChem 2010, 5, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Veiga-Santos, P.; Reignault, L.C.; Huber, K.; Bracher, F.; DE Souza, W.; DE Carvalho, T.M.U. Inhibition of NAD+-dependent histone deacetylases (sirtuins) causes growth arrest and activates both apoptosis and autophagy in the pathogenic protozoan Trypanosoma cruzi. Parasitology 2014, 141, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Ronin, C.; Costa, D.M.; Tavares, J.; Faria, J.; Ciesielski, F.; Ciapetti, P.; Smith, T.K.; MacDougall, J.; Cordeiro-Da-Silva, A.; Pemberton, I.K. The crystal structure of the Leishmania infantum Silent Information Regulator 2 related protein 1: Implications to protein function and drug design. PLoS ONE 2018, 13, e0193602. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.Y.; Fernandez, I.; Anmangandla, A.; Lu, X.; Bai, J.J.; Lin, H. Pharmacological Advantage of SIRT2-Selective versus pan-SIRT1–3 Inhibitors. ACS Chem. Biol. 2021, 16, 1266–1275. [Google Scholar] [CrossRef] [PubMed]
- Sanders, B.D.; Jackson, B.; Marmorstein, R. Structural basis for sirtuin function: What we know and what we don’t. Biochim. et Biophys. Acta Proteins Proteom. 2010, 1804, 1604–1616. [Google Scholar] [CrossRef] [PubMed]
- Parenti, M.D.; Bruzzone, S.; Nencioni, A.; Del Rio, A. Selectivity hot-spots of sirtuin catalytic cores. Mol. Biosyst. 2015, 11, 2263–2272. [Google Scholar] [CrossRef] [PubMed]
- Celniker, G.; Nimrod, G.; Ashkenazy, H.; Glaser, F.; Martz, E.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf: Using Evolutionary Data to Raise Testable Hypotheses about Protein Function. Isr. J. Chem. 2013, 53, 199–206. [Google Scholar] [CrossRef]
- Berezin, C.; Glaser, F.; Rosenberg, J.; Paz, I.; Pupko, T.; Fariselli, P.; Casadio, R.; Ben-Tal, N. ConSeq: The identification of functionally and structurally important residues in protein sequences. Bioinformatics 2004, 20, 1322–1324. [Google Scholar] [CrossRef] [PubMed]
- Landau, M.; Mayrose, I.; Rosenberg, Y.; Glaser, F.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2005: The projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005, 33, W299–W302. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, G.M.; De Magalhães, J.G.; Maltarollo, V.G.; Kronenberger, T.; Ganesan, A.; Emery, F.D.S.; Trossini, G.H.G. QSAR studies on the human sirtuin 2 inhibition by non-covalent 7,5,2-anilinobenzamide derivatives. J. Biomol. Struct. Dyn. 2019, 38, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.; Sobrinho-Junior, E.; Brito, L.; Nicolau, L.; Carvalho, T.; Moura, A.; Rodrigues, K.; Carneiro, S.; Arcanjo, D.; Citó, A.; et al. Anti-Leishmania activity of essential oil of Myracrodruon urundeuva (Engl.) Fr. All.: Composition, cytotoxity and possible mechanisms of action. Exp. Parasitol. 2017, 175, 59–67. [Google Scholar] [CrossRef]
- Yang, L.; Ma, X.; Yuan, C.; He, Y.; Li, L.; Fang, S.; Xia, W.; He, T.; Qian, S.; Xu, Z.; et al. Discovery of 2-((4,6-dimethylpyrimidin-2-yl)thio)- N -phenylacetamide derivatives as new potent and selective human sirtuin 2 inhibitors. Eur. J. Med. Chem. 2017, 134, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Chiba, J.; Iimura, S.; Yoneda, Y.; Watanabe, T.; Muro, F.; Tsubokawa, M.; Iigou, Y.; Satoh, A.; Takayama, G.; Yokoyama, M.; et al. Synthesis and biological evaluation of benzoic acid derivatives as potent, orally active VLA-4 antagonists. Bioorganic Med. Chem. 2007, 15, 1679–1693. [Google Scholar] [CrossRef]
- Eren, G.; Bruno, A.; Guntekin-Ergun, S.; Cetin-Atalay, R.; Ozgencil, F.; Ozkan, Y.; Gozelle, M.; Kaya, S.G.; Costantino, G. Pharmacophore modeling and virtual screening studies to identify novel selective SIRT2 inhibitors. J. Mol. Graph. Model. 2019, 89, 60–73. [Google Scholar] [CrossRef]
- Söding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.S.; Eramian, D.; Shen, M.-Y.; Pieper, U.; Sali, A. Comparative Protein Structure Modeling Using Modeller. Curr. Protoc. Bioinform. 2006, 15, 5–6. [Google Scholar] [CrossRef]
- Ferreira, G.M.; Kronenberger, T.; de Almeida, C.; Sampaio, J.; Terra, C.F.; Pinto, E.; Trossini, G.H.G. Inhibition of Porcine Aminopeptidase M (pAMP) by the Pentapeptide Microginins. Molecules 2019, 24, 4369. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sac-erdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, New York, NY, USA, 11 November 2006; p. 84. [Google Scholar]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Cruciani, G. Molecular Interaction Fields: Applications in Drug Discovery and ADME Prediction; Wiley-VCH: Hoboken, NJ, USA, 2006. [Google Scholar]
- Rocha, J.R.; Freitas, R.F.; A Montanari, C. The GRID/CPCA approach in drug discovery. Expert Opin. Drug Discov. 2010, 5, 333–346. [Google Scholar] [CrossRef]
- Lipinski, C.A. Rule of five in 2015 and beyond: Target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions. Adv. Drug Deliv. Rev. 2016, 101, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Tripos. SYBYL® Expert Molecular Modeling Environment; Tripos: St. Louis, MO, USA, 2017. [Google Scholar]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins Struct. Funct. Bioinform. 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; MacKerell, A.D. Computer-Aided Drug Design Methods. Methods Mol. Biol. 2017, 1520, 85–106. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.C.; Von Hippel, P.H. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989, 182, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Springer: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Adan, A.; Kiraz, Y.; Baran, Y. Cell Proliferation and Cytotoxicity Assays. Curr. Pharm. Biotechnol. 2016, 17, 1213–1221. [Google Scholar] [CrossRef] [PubMed]
- Copeland, R.A. Evaluation of Enzyme Inhibitors in Drug Discovery; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (µM) | IC50 T. cruzi ama (µM) | EC50 Cell Toxicity (µM) | SIama |
|---|---|---|---|---|
| CDMS-01 | 39.98 ± 0.05 | 76.68 ± 0.02 | 135.91 ± 0.50 | 1.77 |
| CDMS-02 | 240.21 ± 1.25 | 183.91 ± 0.01 | 100.13 ± 0.10 | 0.54 |
| CDMS-03 | 482.21 ± 1.35 | 163.17 ± 0.02 | 245.91 ± 0.13 | 1.5 |
| CDMS-04 | ND | 15.16 ± 0.03 | 62.35 ± 0.15 | 4.11 |
| CDMS-05 | ND | 177.17 ± 0.03 | 239.97 ± 0.31 | 1.35 |
| CDMS-06 | 332.13 ± 0.55 | 169.40 ± 0.02 | 226.83 ± 0.56 | 1.33 |
| Nicotinamide | 987.4 ± 0.64 | ND* | ND* | ND* |
| BZN | 9.33 ± 0.01 | 4.55 ± 0.03 | 204.05 ± 0.81 | 44.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, G.M.; Kronenberger, T.; Maltarollo, V.G.; Poso, A.; de Moura Gatti, F.; Almeida, V.M.; Marana, S.R.; Lopes, C.D.; Tezuka, D.Y.; de Albuquerque, S.; et al. Trypanosoma cruzi Sirtuin 2 as a Relevant Druggable Target: New Inhibitors Developed by Computer-Aided Drug Design. Pharmaceuticals 2023, 16, 428. https://doi.org/10.3390/ph16030428
Ferreira GM, Kronenberger T, Maltarollo VG, Poso A, de Moura Gatti F, Almeida VM, Marana SR, Lopes CD, Tezuka DY, de Albuquerque S, et al. Trypanosoma cruzi Sirtuin 2 as a Relevant Druggable Target: New Inhibitors Developed by Computer-Aided Drug Design. Pharmaceuticals. 2023; 16(3):428. https://doi.org/10.3390/ph16030428
Chicago/Turabian StyleFerreira, Glaucio Monteiro, Thales Kronenberger, Vinicius Gonçalves Maltarollo, Antti Poso, Fernando de Moura Gatti, Vitor Medeiros Almeida, Sandro Roberto Marana, Carla Duque Lopes, Daiane Yukie Tezuka, Sérgio de Albuquerque, and et al. 2023. "Trypanosoma cruzi Sirtuin 2 as a Relevant Druggable Target: New Inhibitors Developed by Computer-Aided Drug Design" Pharmaceuticals 16, no. 3: 428. https://doi.org/10.3390/ph16030428
APA StyleFerreira, G. M., Kronenberger, T., Maltarollo, V. G., Poso, A., de Moura Gatti, F., Almeida, V. M., Marana, S. R., Lopes, C. D., Tezuka, D. Y., de Albuquerque, S., da Silva Emery, F., & Trossini, G. H. G. (2023). Trypanosoma cruzi Sirtuin 2 as a Relevant Druggable Target: New Inhibitors Developed by Computer-Aided Drug Design. Pharmaceuticals, 16(3), 428. https://doi.org/10.3390/ph16030428