
The Parasite Reduction Ratio (PRR) Assay Version 2: Standardized Assessment of Plasmodium falciparum Viability after Antimalarial Treatment In Vitro
 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Results
2.1. Parasite Viability Vs. Standard Readouts
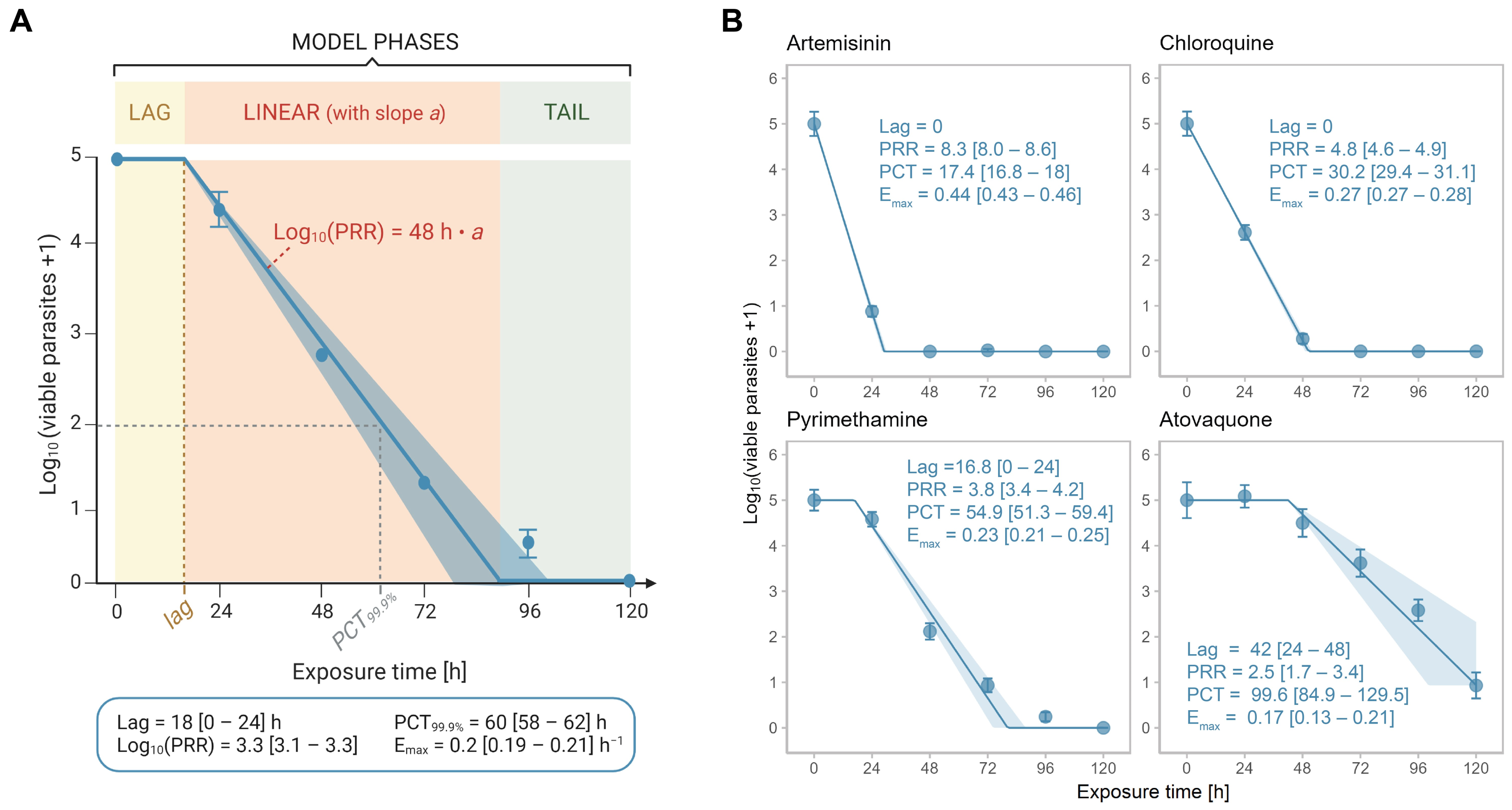
2.2. Optimizing the Protocol: The PRR Assay V2
2.2.1. Reduced Assay Duration
2.2.2. Quality Control 1: Monitoring Undisturbed Parasite Growth
2.2.3. Quality Control 2 and 3: Monitoring Drug Stability and Removal
2.2.4. Automated and Objective Data Analysis
2.2.5. Inclusion of Growth Control Enables Calculation of Emax
3. Discussion
4. Materials and Methods
4.1. Parasite Cultivation
4.2. In Vitro [3H] Hypoxanthine Incorporation Assay
4.3. Parasite Reduction Ratio Assay Version 2
4.4. Quality Controls: Monitoring Drug Stability and -Washout
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Phyo, A.P.; Ashley, E.A.; Anderson, T.J.C.; Bozdech, Z.; Carrara, V.I.; Sriprawat, K.; Nair, S.; White, M.M.; Dziekan, J.; Ling, C.; et al. Declining Efficacy of Artemisinin Combination Therapy Against P. Falciparum Malaria on the Thai–Myanmar Border (2003–2013): The Role of Parasite Genetic Factors. Clin. Infect. Dis. 2016, 63, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Uwimana, A.; Legrand, E.; Stokes, B.H.; Ndikumana, J.-L.M.; Warsame, M.; Umulisa, N.; Ngamije, D.; Munyaneza, T.; Mazarati, J.-B.; Munguti, K.; et al. Emergence and clonal expansion of in vitro artemisinin-resistant Plasmodium falciparum kelch13 R561H mutant parasites in Rwanda. Nat. Med. 2020, 26, 1602–1608. [Google Scholar] [CrossRef] [PubMed]
- Asua, V.; Conrad, M.D.; Aydemir, O.; Duvalsaint, M.; Legac, J.; Duarte, E.; Tumwebaze, P.; Chin, D.M.; Cooper, R.A.; Yeka, A.; et al. Changing Prevalence of Potential Mediators of Aminoquinoline, Antifolate, and Artemisinin Resistance Across Uganda. J. Infect. Dis. 2020, 223, 985–994. [Google Scholar] [CrossRef] [PubMed]
- WHO. World Malaria Report 2022; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- Burrows, J.N.; Duparc, S.; Gutteridge, W.E.; Van Huijsduijnen, R.H.; Kaszubska, W.; MacIntyre, F.; Mazzuri, S.; Möhrle, J.J.; Wells, T.N.C. New developments in anti-malarial target candidate and product profiles. Malar. J. 2017, 16, 26. [Google Scholar] [CrossRef] [PubMed]
- Sanz, L.M.; Crespo, B.; De-Cózar, C.; Ding, X.C.; Llergo, J.L.; Burrows, J.N.; García-Bustos, J.F.; Gamo, F.-J. P. falciparum In Vitro Killing Rates Allow to Discriminate between Different Antimalarial Mode-of-Action. PLoS ONE 2012, 7, e30949. [Google Scholar] [CrossRef]
- Khoury, D.S.; Zaloumis, S.G.; Grigg, M.J.; Haque, A.; Davenport, M.P. Malaria Parasite Clearance: What Are We Really Measuring? Trends Parasitol. 2020, 36, 413–426. [Google Scholar] [CrossRef]
- Rebelo, M.; Pawliw, R.; Gower, J.; Webb, L.; Mitchell, H.; Pava, Z.; Watts, R.E.; Davenport, M.P.; McCarthy, J.S.; Khoury, D.S. Parasite Viability as a Superior Measure of Antimalarial Drug Activity in Humans. J. Infect. Dis. 2020, 223, 2154–2163. [Google Scholar] [CrossRef]
- Radohery, G.F.R.; Walz, A.; Gumpp, C.; Cherkaoui-Rbati, M.H.; Gobeau, N.; Gower, J.; Davenport, M.P.; Rottmann, M.; McCarthy, J.S.; Möhrle, J.J.; et al. Parasite Viability as a Measure of In Vivo Drug Activity in Preclinical and Early Clinical Antimalarial Drug Assessment. Antimicrob. Agents Chemother. 2022, 66, e00114-22. [Google Scholar] [CrossRef]
- Young, R.D.; Rathod, P.K. Clonal viability measurements on Plasmodium falciparum to assess in vitro schizonticidal activity of leupeptin, chloroquine, and 5-fluoroorotate. Antimicrob. Agents Chemother. 1993, 37, 1102–1107. [Google Scholar] [CrossRef]
- Watkins, W.M.; Woodrow, C.; Marsh, K. Falciparum Malaria: Differential Effects of Antimalarial Drugs on Ex Vivo Parasite Viability during the Critical Early Phase of Therapy. Am. J. Trop. Med. Hyg. 1993, 49, 106–112. [Google Scholar] [CrossRef]
- Linares, M.J.P.; Viera, S.; Crespo, B.; Franco, V.; Gómez-Lorenzo, M.G.; Jiménez-Díaz, M.B.; Angulo-Barturen, Í.; Sanz, L.M.; Gamo, F.-J. Identifying rapidly parasiticidal anti-malarial drugs using a simple and reliable in vitro parasite viability fast assay. Malar. J. 2015, 14, 441. [Google Scholar] [CrossRef] [PubMed]
- White, N.J. Assessment of the pharmacodynamic properties of antimalarial drugs in vivo. Antimicrob. Agents Chemother. 1997, 41, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Murray, L.; Stewart, L.B.; Tarr, S.J.; Ahouidi, A.D.; Diakite, M.; Amambua-Ngwa, A.; Conway, D.J. Multiplication rate variation in the human malaria parasite Plasmodium falciparum. Sci. Rep. 2017, 7, 6436. [Google Scholar] [CrossRef] [PubMed]
- Parapini, S.; Olliaro, P.; Navaratnam, V.; Taramelli, D.; Basilico, N. Stability of the Antimalarial Drug Dihydroartemisinin under Physiologically Relevant Conditions: Implications for Clinical Treatment and Pharmacokinetic and In Vitro Assays. Antimicrob. Agents Chemother. 2015, 59, 4046–4052. [Google Scholar] [CrossRef]
- White, N.J. The parasite clearance curve. Malar. J. 2011, 10, 278. [Google Scholar] [CrossRef]
- Flegg, J.A.; Guerin, P.J.; White, N.J.; Stepniewska, K. Standardizing the measurement of parasite clearance in falciparum malaria: The parasite clearance estimator. Malar. J. 2011, 10, 339. [Google Scholar] [CrossRef] [PubMed]
- White, N.J. Pharmacokinetic and Pharmacodynamic Considerations in Antimalarial Dose Optimization. Antimicrob. Agents Chemother. 2013, 57, 5792–5807. [Google Scholar] [CrossRef]
- White, N.J. The assessment of antimalarial drug efficacy. Trends Parasitol. 2002, 18, 458–464. [Google Scholar] [CrossRef]
- Wicha, S.G.; Walz, A.; Cherkaoui-Rbati, M.H.; Bundgaard, N.; Kuritz, K.; Gumpp, C.; Gobeau, N.; Möhrle, J.; Rottmann, M.; Demarta-Gatsi, C. New In Vitro Interaction-Parasite Reduction Ratio Assay for Early Derisk in Clinical Development of Antimalarial Combinations. Antimicrob. Agents Chemother. 2022, 66, e0055622. [Google Scholar] [CrossRef]
- Gassis, S.; Rathod, P.K. Frequency of drug resistance in Plasmodium falciparum: A nonsynergistic combination of 5-fluoroorotate and atovaquone suppresses in vitro resistance. Antimicrob. Agents Chemother. 1996, 40, 914–919. [Google Scholar] [CrossRef]
- Looareesuwan, S.; Viravan, C.; Webster, H.K.; Kyle, D.E.; Hutchinson, D.B.; Canfield, C.J. Clinical Studies of Atovaquone, Alone or in Combination with other Antimalarial Drugs, for Treatment of Acute Uncomplicated Malaria in Thailand. Am. J. Trop. Med. Hyg. 1996, 54, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Giannangelo, C.; Stingelin, L.; Yang, T.; Tilley, L.; Charman, S.A.; Creek, D.J. Parasite-Mediated Degradation of Synthetic Ozonide Antimalarials Impacts In Vitro Antimalarial Activity. Antimicrob. Agents Chemother. 2018, 62, e01566-17. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Xie, S.C.; Cao, P.; Giannangelo, C.; McCaw, J.; Creek, D.J.; Charman, S.A.; Klonis, N.; Tilley, L. Comparison of the Exposure Time Dependence of the Activities of Synthetic Ozonide Antimalarials and Dihydroartemisinin against K13 Wild-Type and Mutant Plasmodium falciparum Strains. Antimicrob. Agents Chemother. 2016, 60, 4501–4510. [Google Scholar] [CrossRef]
- White, N.; Krishna, S. Treatment of malaria: Some considerations and limitations of the current methods of assessment. Trans. R. Soc. Trop. Med. Hyg. 1989, 83, 767–777. [Google Scholar] [CrossRef]
- Lin, W.-H.; Hwang, C.-F.; Chen, L.-W.; Tsen, H.-Y. Viable counts, characteristic evaluation for commercial lactic acid bacteria products. Food Microbiol. 2006, 23, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Koelle, D.; Abbo, H.; Peck, A.; Ziegweid, K.; Corey, L. Direct Recovery of Herpes Simplex Virus (HSV)-Specific T Lymphocyte Clones from Recurrent Genital HSV-2 Lesions. J. Infect. Dis. 1994, 169, 956–961. [Google Scholar] [CrossRef] [PubMed]
- Snyder, C.; Chollet, J.; Santo-Tomas, J.; Scheurer, C.; Wittlin, S. In vitro and in vivo interaction of synthetic peroxide RBx11160 (OZ277) with piperaquine in Plasmodium models. Exp. Parasitol. 2007, 115, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, R.E.; Canfield, C.J.; Haynes, J.D.; Chulay, J.D. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob. Agents Chemother. 1979, 16, 710–718. [Google Scholar] [CrossRef]
- Huber, W.; Koella, J.C. A comparison of three methods of estimating EC50 in studies of drug resistance of malaria parasites. Acta Trop. 1993, 55, 257–261. [Google Scholar] [CrossRef]
- Delves, M.; Plouffe, D.; Scheurer, C.; Meister, S.; Wittlin, S.; Winzeler, E.; Sinden, R.E.; Leroy, D. The Activities of Current Antimalarial Drugs on the Life Cycle Stages of Plasmodium: A Comparative Study with Human and Rodent Parasites. PLoS Med. 2012, 9, e1001169. [Google Scholar] [CrossRef]
- R Core Team R. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Criteria | Example |
|---|---|---|
| Fast without lag phase Fast with lag phase | PRR ≥ 4, lag = 0 h PRR ≥ 4, lag > 0 h | artemisinin, chloroquine |
| Intermediate without lag phase Intermediate with lag phase | 3 ≤ PRR < 4, lag = 0 h 3 ≤ PRR < 4, lag > 0 h | pyrimethamine |
| Slow | PRR < 3 and/or lag ≥ 48 h | atovaquone |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walz, A.; Duffey, M.; Aljayyoussi, G.; Sax, S.; Leroy, D.; Besson, D.; Burrows, J.N.; Cherkaoui-Rbati, M.H.; Gobeau, N.; Westwood, M.-A.; et al. The Parasite Reduction Ratio (PRR) Assay Version 2: Standardized Assessment of Plasmodium falciparum Viability after Antimalarial Treatment In Vitro. Pharmaceuticals 2023, 16, 163. https://doi.org/10.3390/ph16020163
Walz A, Duffey M, Aljayyoussi G, Sax S, Leroy D, Besson D, Burrows JN, Cherkaoui-Rbati MH, Gobeau N, Westwood M-A, et al. The Parasite Reduction Ratio (PRR) Assay Version 2: Standardized Assessment of Plasmodium falciparum Viability after Antimalarial Treatment In Vitro. Pharmaceuticals. 2023; 16(2):163. https://doi.org/10.3390/ph16020163
Chicago/Turabian StyleWalz, Annabelle, Maëlle Duffey, Ghaith Aljayyoussi, Sibylle Sax, Didier Leroy, Dominique Besson, Jeremy N. Burrows, Mohammed H. Cherkaoui-Rbati, Nathalie Gobeau, Marie-Anne Westwood, and et al. 2023. "The Parasite Reduction Ratio (PRR) Assay Version 2: Standardized Assessment of Plasmodium falciparum Viability after Antimalarial Treatment In Vitro" Pharmaceuticals 16, no. 2: 163. https://doi.org/10.3390/ph16020163
APA StyleWalz, A., Duffey, M., Aljayyoussi, G., Sax, S., Leroy, D., Besson, D., Burrows, J. N., Cherkaoui-Rbati, M. H., Gobeau, N., Westwood, M.-A., Siethoff, C., Gamo, F.-J., Mäser, P., & Wittlin, S. (2023). The Parasite Reduction Ratio (PRR) Assay Version 2: Standardized Assessment of Plasmodium falciparum Viability after Antimalarial Treatment In Vitro. Pharmaceuticals, 16(2), 163. https://doi.org/10.3390/ph16020163