

Template Entrance Channel as Possible Allosteric Inhibition and Resistance Site for Quinolines Tricyclic Derivatives in RNA Dependent RNA Polymerase of Bovine Viral Diarrhea Virus

 , ,
, ,  and
and
Abstract
1. Introduction
2. Results
2.1. Identification of the Most Likely Poses of Compounds
2.2. Molecular Dynamics Simulations
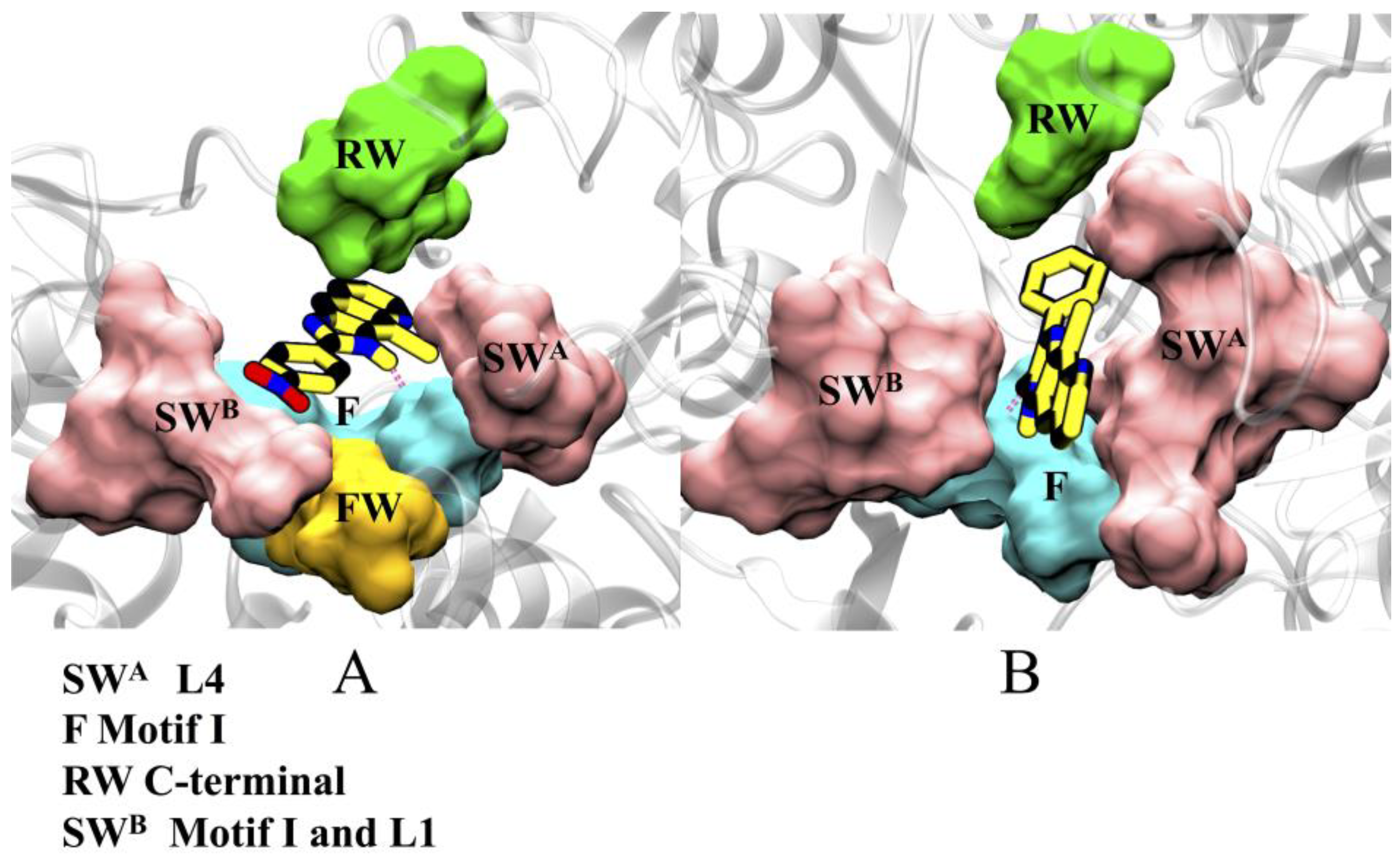
2.3. Characterization of Binding Site
2.4. Protein Complexed to Ligand 2h (COM1)
2.5. Unbinding Mechanism and Escape of Ligand 2h
2.6. Protein Complexed to Ligand 5m (COM2)
2.7. Unbinding Mechanism and Escape of Ligand 5m
2.8. Possible Mechanism of Inhibition
2.9. Mutation in Hot-Spot Residues Most Likely Drives the Drug Resistance
- The identification of critical residues contributing to the stability of the most stable state (mini-1);
- Mutational scanning of key residues;
- Already reported resistant mutations localized in the binding site.
- All of the reported resistant mutants for different classes of compounds are localized in the vicinity of each other, making the zone a hot spot;
- These mutations are common, even for different classes of drugs, such as F224S/Y, the resistant mutant for indole, imidazopyridine and pyrimidine-amine (Table S8), indicating the criticality of these residues.
- We assume that by mutating these residues, the RdRp can resume its function (possibly with different rates to WT). However, the selection of the respective residue depends on the location of drug binding, and how the particular mutation changes the dynamics of the RdRp to render it resistant against the respective drug. In the case of 2h, the order of possible mutants is A392 > I261 > F224 (Figure 8). The mutational changes suggest that the favorable mutation for 2h could be, in the case of 392 aromatic/acidic/Gln/Asn, for residue 261 Gln/Glu/Arg, and for residue 224 Gln/Asn/Asp/Arg/Tyr. For 2m, the order of preference is I261 > A392 > F224.
3. Discussion
4. Methods
4.1. Structural Retreival and Preparation of RdRp and Ligands
4.2. Molecular Docking for Site Identification of Compounds 2h and 5m
4.2.1. AutoLigand (AL)
4.2.2. Blind Docking (BD)
4.3. Guided Docking (GD)
4.4. Conventional Molecular Dynamics (MD) Simulations
4.5. Analysis of MD Simulation Trajectories
4.6. Metadynamics
Choice of Reaction Coordinates
4.7. MM-PBSA Calculations
4.8. Bioluminate: Scanning of Possible Substitution to Predict the Resistant Mutations
5. Conclusions
- The detailed insights obtained from our multi-step computational studies revealed two significant outcomes: (i) A common binding site for compounds 2h and 5m, as they share the key residues and are localized at the template entrance site. The binding of compounds at the template entry site occludes the entrance passage, and their significant interactions with loop L2 (motif G), loop L4 (beta-hairpin motif), and motif I (motif F), make crucial motifs essential for biological function unavailable, therefore, causing inhibition. (ii) Three amino acids, I261, P262, and N264, belonging to motif I of the fingertip region, played a significant role in establishing 2h and 5m potency. This supports the development of a pharmacophore model for establishing more potent leads.
- In summary, identifying a new binding site at the template entry channel appears as a “hot spot” for developing broad-spectrum antiviral drugs against infectious RdRps families, which are known to have a common structural and functional skeleton. Furthermore, identifying the most probable resistant mutation against compound 2h, using residue scanning methods, indicates that A392 is the most likely residue to mutate, possibly rendering RdRp resistant against 2h. Based on the reported mutation, there is the possibility of A392E being resistant against 2h, as it makes the mutant protein more stable, and does not allow 2h to enter the binding site. The computationally-derived mechanistic (inhibition and resistance) studies provide precious insights for improving the reported leads’ specificities and inhibitory potencies, and rationalizing the design of effective bioactive inhibitors with low susceptibility to resistance.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baginski, S.G.; Pevear, D.C.; Seipel, M.; Sun, S.C.C.; Benetatos, C.A.; Chunduru, S.K.; Rice, C.M.; Collett, M.S. Mechanism of action of a pestivirus antiviral compound. Proc. Natl. Acad. Sci. USA 2000, 97, 7981–7986. [Google Scholar] [CrossRef]
- Castro, E.F.; Casal, J.J.; de Marco, M.J.E.; Battini, L.; Fabiani, M.; Fernández, G.A.; Bruno, A.M.; Cavallaro, L.V.; Bollini, M. Identification of potent bovine viral diarrhea virus inhibitors by a structure-based virtual screening approach. Bioorg. Med. Chem. Lett. 2019, 29, 262–266. [Google Scholar] [CrossRef]
- Fulton, R.W.; Ridpath, J.F.; Saliki, J.T.; Briggs, R.E.; Confer, A.W.; Burge, L.J.; Purdy, C.W.; Loan, R.W.; Duff, G.C.; Payton, M.E. Bovine viral diarrhea virus (BVDV) 1b: Predominant BVDV subtype in calves with respiratory disease. Can. J. Vet. Res. 2002, 66, 181–190. [Google Scholar] [PubMed]
- Mittal, L.; Srivastava, M.; Asthana, S. Conformational Characterization of Linker Revealed the Mechanism of Cavity Formation by 227G in BVDV RDRP. J. Phys. Chem. B 2019, 123, 6150–6160. [Google Scholar] [CrossRef] [PubMed]
- Asthana, S.; Shukla, S.; Ruggerone, P.; Ceccarelli, M.; Giliberti, G.; La Colla, P.; Vargiu, A.V. Point Mutation I261M Affects the Dynamics of BVDV and its Interaction with Benzimidazole Antiviral 227G. Biophys. J. 2011, 100, 395a–396a. [Google Scholar] [CrossRef]
- Scharnböck, B.; Roch, F.-F.; Richter, V.; Funke, C.; Firth, C.L.; Obritzhauser, W.; Baumgartner, W.; Käsbohrer, A.; Pinior, B. A meta-analysis of bovine viral diarrhoea virus (BVDV) prevalences in the global cattle population. Sci. Rep. 2018, 8, 14420. [Google Scholar] [CrossRef]
- Al-Kubati, A.A.G.; Hussen, J.; Kandeel, M.; Al-Mubarak, A.I.A.; Hemida, M.G. Recent Advances on the Bovine Viral Diarrhea Virus Molecular Pathogenesis, Immune Response, and Vaccines Development. Front. Vet. Sci. 2021, 8, 665128. [Google Scholar] [CrossRef]
- Ibba, R.; Riu, F.; Delogu, I.; Lupinu, I.; Carboni, G.; Loddo, R.; Piras, S.; Carta, A. Benzimidazole-2-Phenyl-Carboxamides as Dual-Target Inhibitors of BVDV Entry and Replication. Viruses 2022, 14, 1300. [Google Scholar] [CrossRef]
- Pathania, S.; Rawal, R.K.; Singh, P.K. RdRp (RNA-dependent RNA polymerase): A key target providing anti-virals for the management of various viral diseases. J. Mol. Struct. 2022, 1250, 131756. [Google Scholar] [CrossRef]
- Carta, A.; Briguglio, I.; Piras, S.; Corona, P.; Ibba, R.; Laurini, E.; Fermeglia, M.; Pricl, S.; Desideri, N.; Atzori, E.M.; et al. A combined in silico/in vitro approach unveils common molecular requirements for efficient BVDV RdRp binding of linear aromatic N-polycyclic systems. Eur. J. Med. Chem. 2016, 117, 321–334. [Google Scholar] [CrossRef]
- Fernández, G.A.; Castro, E.F.; Rosas, R.A.; Fidalgo, D.M.; Adler, N.S.; Battini, L.; España de Marco, M.J.; Fabiani, M.; Bruno, A.M.; Bollini, M.; et al. Design and Optimization of Quinazoline Derivatives: New Non-nucleoside Inhibitors of Bovine Viral Diarrhea Virus. Front. Chem. 2020, 8, 590235. [Google Scholar] [CrossRef] [PubMed]
- Musiu, S.; Pürstinger, G.; Stallinger, S.; Vrancken, R.; Haegeman, A.; Koenen, F.; Leyssen, P.; Froeyen, M.; Neyts, J.; Paeshuyse, J. Substituted 2,6-bis(benzimidazol-2-yl)pyridines: A novel chemical class of pestivirus inhibitors that targets a hot spot for inhibition of pestivirus replication in the RNA-dependent RNA polymerase. Antivir. Res. 2014, 106, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Carta, A.; Briguglio, I.; Piras, S.; Corona, P.; Boatto, G.; Nieddu, M.; Giunchedi, P.; Marongiu, M.E.; Giliberti, G.; Iuliano, F.; et al. Quinoline tricyclic derivatives. Design, synthesis and evaluation of the antiviral activity of three new classes of RNA-dependent RNA polymerase inhibitors. Biorg. Med. Chem. 2011, 19, 7070–7084. [Google Scholar] [CrossRef] [PubMed]
- Musiu, S.; Castillo, Y.P.; Muigg, A.; Pürstinger, G.; Leyssen, P.; Froeyen, M.; Neyts, J.; Paeshuyse, J. Quinolinecarboxamides Inhibit the Replication of the Bovine Viral Diarrhea Virus by Targeting a Hot Spot for the Inhibition of Pestivirus Replication in the RNA-Dependent RNA Polymerase. Molecules 2020, 25, 1283. [Google Scholar] [CrossRef] [PubMed]
- Loddo, R.; Francesconi, V.; Laurini, E.; Boccardo, S.; Aulic, S.; Fermeglia, M.; Pricl, S.; Tonelli, M. 9-Aminoacridine-based agents impair the bovine viral diarrhea virus (BVDV) replication targeting the RNA-dependent RNA polymerase (RdRp). Biorg. Med. Chem. 2018, 26, 855–868. [Google Scholar] [CrossRef]
- Castro, E.F.; Fabian, L.E.; Caputto, M.E.; Gagey, D.; Finkielsztein, L.M.; Moltrasio, G.Y.; Moglioni, A.G.; Campos, R.H.; Cavallaro, L.V. Inhibition of Bovine Viral Diarrhea Virus RNA Synthesis by Thiosemicarbazone Derived from 5,6-Dimethoxy-1-Indanone. J. Virol. 2011, 85, 5436–5445. [Google Scholar] [CrossRef]
- He, D.; Li, X.; Wang, S.; Wang, C.; Liu, X.; Zhang, Y.; Cui, Y.; Yu, S. Mechanism of drug resistance of BVDV induced by F224S mutation in RdRp: A case study of VP32947. Comput. Biol. Chem. 2022, 99, 107715. [Google Scholar] [CrossRef]
- Asthana, S.; Shukla, S.; Vargiu, A.V.; Ceccarelli, M.; Ruggerone, P.; Paglietti, G.; Marongiu, M.E.; Blois, S.; Giliberti, G.; La Colla, P. Different Molecular Mechanisms of Inhibition of Bovine Viral Diarrhea Virus and Hepatitis C Virus RNA-Dependent RNA Polymerases by a Novel Benzimidazole. Biochemistry 2013, 52, 3752–3764. [Google Scholar] [CrossRef]
- Asthana, S.; Shukla, S.; Ruggerone, P.; Vargiu, A.V. Molecular Mechanism of Viral Resistance to a Potent Non-nucleoside Inhibitor Unveiled by Molecular Simulations. Biochemistry 2014, 53, 6941–6953. [Google Scholar] [CrossRef]
- Harris, R.; Olson, A.J.; Goodsell, D.S. Automated prediction of ligand-binding sites in proteins. Proteins Struct. Funct. Bioinform. 2007, 70, 1506–1517. [Google Scholar] [CrossRef]
- Srivastava, M.; Suri, C.; Singh, M.; Mathur, R.; Asthana, S. Molecular dynamics simulation reveals the possible druggablehot-spotsof USP7. Oncotarget 2018, 9, 34289–34305. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.H.; Groarke, J.M.; Young, D.C.; Kuhn, R.J.; Smith, J.L.; Pevear, D.C.; Rossmann, M.G. The structure of the RNA-dependent RNA polymerase from bovine viral diarrhea virus establishes the role of GTP in de novo initiation. Proc. Natl. Acad. Sci. USA 2004, 101, 4425–4430. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.; Mittal, L.; Kumari, A.; Agrahari, A.K.; Singh, M.; Mathur, R.; Asthana, S. Characterizing (un)binding mechanism of USP7 inhibitors to unravel the cause of enhanced binding potencies at allosteric checkpoint. Protein Sci. 2022, 31, e4398. [Google Scholar] [CrossRef] [PubMed]
- Tummino, P.J.; Copeland, R.A. Residence Time of Receptor−Ligand Complexes and Its Effect on Biological Function. Biochemistry 2008, 47, 5481–5492. [Google Scholar] [CrossRef] [PubMed]
- Rinken, A.; Veiksina, S.; Kopanchuk, S. Dynamics of ligand binding to GPCR: Residence time of melanocortins and its modulation. Pharmacol. Res. 2016, 113, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Li, Y.; Shen, M.; Li, D.; Kang, Y.; Hou, T. Characterizing Drug–Target Residence Time with Metadynamics: How to Achieve Dissociation Rate Efficiently without Losing Accuracy against Time-Consuming Approaches. J. Chem. Inf. Model. 2017, 57, 1895–1906. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Hillger, J.M.; Ijzerman, A.P.; Heitman, L.H. Drug-Target Residence Time-A Case for G Protein-Coupled Receptors. Med. Res. Rev. 2014, 34, 856–892. [Google Scholar] [CrossRef]
- Dror, R.O.; Pan, A.C.; Arlow, D.H.; Borhani, D.W.; Maragakis, P.; Shan, Y.; Xu, H.; Shaw, D.E. Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc. Natl. Acad. Sci. USA 2011, 108, 13118–13123. [Google Scholar] [CrossRef]
- Keserü, G.; Swinney, D.C.; Mannhold, R.; Kubinyi, H.; Folkers, G. Thermodynamics and Kinetics of Drug Binding: Keserü/Thermodynamics and Kinetics of Drug Binding; Keseru, G.S., David, C., Eds.; Wiley-VCH Verlag: Weinheim, Germany, 2015. [Google Scholar] [CrossRef]
- Choi, K.H.; Gallei, A.; Becher, P.; Rossmann, M.G. The Structure of Bovine Viral Diarrhea Virus RNA-Dependent RNA Polymerase and Its Amino-Terminal Domain. Structure 2006, 14, 1107–1113. [Google Scholar] [CrossRef]
- Venkataraman, S.; Prasad, B.; Selvarajan, R. RNA Dependent RNA Polymerases: Insights from Structure, Function and Evolution. Viruses 2018, 10, 76. [Google Scholar] [CrossRef]
- Smertina, E.; Urakova, N.; Strive, T.; Frese, M. Calicivirus RNA-Dependent RNA Polymerases: Evolution, Structure, Protein Dynamics, and Function. Front. Microbiol. 2019, 10, 1280. [Google Scholar] [CrossRef]
- Mittal, L.; Kumari, A.; Suri, C.; Bhattacharya, S.; Asthana, S. Insights into structural dynamics of allosteric binding sites in HCV RNA-dependent RNA polymerase. J. Biomol. Struct. Dyn. 2020, 38, 1612–1625. [Google Scholar] [CrossRef] [PubMed]
- Paeshuyse, J.; Letellier, C.; Froeyen, M.; Dutartre, H.; Vrancken, R.; Canard, B.; De Clercq, E.; Gueiffier, A.; Teulade, J.-C.; Herdewijn, P.; et al. A pyrazolotriazolopyrimidinamine inhibitor of bovine viral diarrhea virus replication that targets the viral RNA-dependent RNA polymerase. Antivir. Res. 2009, 82, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Niyomrattanakit, P.; Chen, Y.-L.; Dong, H.; Yin, Z.; Qing, M.; Glickman, J.F.; Lin, K.; Mueller, D.; Voshol, H.; Lim, J.Y.H.; et al. Inhibition of Dengue Virus Polymerase by Blocking of the RNA Tunnel. J. Virol. 2010, 84, 5678–5686. [Google Scholar] [CrossRef]
- Lang, D.M.; Zemla, A.T.; Zhou, C.L.E. Highly similar structural frames link the template tunnel and NTP entry tunnel to the exterior surface in RNA-dependent RNA polymerases. Nucleic Acids Res. 2013, 41, 1464–1482. [Google Scholar] [CrossRef]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2015, 12, 281–296. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Tonelli, M.; Boido, V.; Colla, P.L.; Loddo, R.; Posocco, P.; Paneni, M.S.; Fermeglia, M.; Pricl, S. Pharmacophore modeling, resistant mutant isolation, docking, and MM-PBSA analysis: Combined experimental/computer-assisted approaches to identify new inhibitors of the bovine viral diarrhea virus (BVDV). Biorg. Med. Chem. 2010, 18, 2304–2316. [Google Scholar] [CrossRef] [PubMed]
- Paeshuyse, J.; Leyssen, P.; Mabery, E.; Boddeker, N.; Vrancken, R.; Froeyen, M.; Ansari, I.H.; Dutartre, H.; Rozenski, J.; Gil, L.H.V.G.; et al. A Novel, Highly Selective Inhibitor of Pestivirus Replication That Targets the Viral RNA-Dependent RNA Polymerase. J. Virol. 2006, 80, 149–160. [Google Scholar] [CrossRef]
- Giliberti, G.; Ibba, C.; Marongiu, E.; Loddo, R.; Tonelli, M.; Boido, V.; Laurini, E.; Posocco, P.; Fermeglia, M.; Pricl, S. Synergistic experimental/computational studies on arylazoenamine derivatives that target the bovine viral diarrhea virus RNA-dependent RNA polymerase. Biorg. Med. Chem. 2010, 18, 6055–6068. [Google Scholar] [CrossRef] [PubMed]
- Ördög, R.; Grolmusz, V. Evaluating Genetic Algorithms in Protein-Ligand Docking. In Bioinformatics Research and Applications; Springer: Berlin/Heidelberg, Germany, 2008; pp. 402–413. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Melis, P.; Genna, V.; Cocco, E.; Marrosu, M.G.; Pieroni, E. Antigenic peptide molecular recognition by the DRB1–DQB1 haplotype modulates multiple sclerosis susceptibility. Mol. Biosyst. 2014, 10, 2043–2054. [Google Scholar] [CrossRef]
- Kumar, A.; Sechi, L.A.; Caboni, P.; Marrosu, M.G.; Atzori, L.; Pieroni, E. Dynamical insights into the differential characteristics of Mycobacterium avium subsp. paratuberculosis peptide binding to HLA-DRB1 proteins associated with multiple sclerosis. New J. Chem. 2015, 39, 1355–1366. [Google Scholar] [CrossRef]
- Kumar, A.; Delogu, F. Dynamical footprint of cross-reactivity in a human autoimmune T-cell receptor. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Davidchack, R.L.; Handel, R.; Tretyakov, M.V. Langevin thermostat for rigid body dynamics. J. Chem. Phys. 2009, 130, 234101. [Google Scholar] [CrossRef]
- Davidchack, R.L.; Ouldridge, T.E.; Tretyakov, M.V. New Langevin and gradient thermostats for rigid body dynamics. J. Chem. Phys. 2015, 142, 144114. [Google Scholar] [CrossRef]
- Evans, D.J.; Holian, B.L. The Nose—Hoover thermostat. J. Chem. Phys. 1985, 83, 4069–4074. [Google Scholar] [CrossRef]
- Pyrkov, T.V.; Chugunov, A.O.; Krylov, N.A.; Nolde, D.E.; Efremov, R.G. PLATINUM: A web tool for analysis of hydrophobic/hydrophilic organization of biomolecular complexes. Bioinformatics 2009, 25, 1201–1202. [Google Scholar] [CrossRef] [PubMed]
- Tiwary, P.; Limongelli, V.; Salvalaglio, M.; Parrinello, M. Kinetics of protein–ligand unbinding: Predicting pathways, rates, and rate-limiting steps. Proc. Natl. Acad. Sci. USA 2015, 112, E386–E391. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, A.; Spitaleri, A.; Saladino, G.; Gervasio, F.L. Investigating Drug–Target Association and Dissociation Mechanisms Using Metadynamics-Based Algorithms. Acc. Chem. Res. 2014, 48, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Leone, V.; Marinelli, F.; Carloni, P.; Parrinello, M. Targeting biomolecular flexibility with metadynamics. Curr. Opin. Struct. Biol. 2010, 20, 148–154. [Google Scholar] [CrossRef]
- Pfaendtner, J. Metadynamics to Enhance Sampling in Biomolecular Simulations. In Biomolecular Simulations; Springer: Berlin/Heidelberg, Germany, 2019; pp. 179–200. [Google Scholar] [CrossRef]
- Provasi, D. Ligand-Binding Calculations with Metadynamics. In Biomolecular Simulations; Springer: Berlin/Heidelberg, Germany, 2019; pp. 233–253. [Google Scholar] [CrossRef]
- Jin, L.; Ye, F.; Zhao, D.; Chen, S.; Zhu, K.; Zheng, M.; Jiang, R.-W.; Jiang, H.; Luo, C. Metadynamics Simulation Study on the Conformational Transformation of HhaI Methyltransferase: An Induced-Fit Base-Flipping Hypothesis. BioMed Res. Int. 2014, 2014, 304563. [Google Scholar] [CrossRef] [PubMed]
- Saladino, G.; Gauthier, L.; Bianciotto, M.; Gervasio, F.L. Assessing the Performance of Metadynamics and Path Variables in Predicting the Binding Free Energies of p38 Inhibitors. J. Chem. Theory Comput. 2012, 8, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Nicholls, A.; Honig, B. A rapid finite difference algorithm, utilizing successive over-relaxation to solve the Poisson-Boltzmann equation. J. Comput. Chem. 1991, 12, 435–445. [Google Scholar] [CrossRef]
- Zhu, K.; Day, T.; Warshaviak, D.; Murrett, C.; Friesner, R.; Pearlman, D. Antibody structure determination using a combination of homology modeling, energy-based refinement, and loop prediction. Proteins Struct. Funct. Bioinform. 2014, 82, 1646–1655. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Loop Pairs | Ligand 2h | Ligand 5m | ||
|---|---|---|---|---|
| Mini-1 | Mini-2 | Mini-1 | Mini-2 | |
| L1–L2 | 11.5(1.6) | 13.3(1.7) | 9.2(1.6) | 11.6(1.1) |
| L1–L3 | 19.0(1.7) | 21.0(1.9) | 16.0(1.1) | 23.1(1.8) |
| L1–L4 | 14.3(1.3) | 22.1(1.4) | 17.8(1.3) | 29.8(1.6) |
| L2–L3 | 13.4(1.5) | 17.5(1.6) | 13.7(1.9) | 16.2(2.4) |
| L2–L4 | 19.6(1.5) | 26.3(1.7) | 14.5(2.2) | 25.7(1.4) |
| L3–L4 | 8.5(1.5) | 11.3(1.9) | 10.8(1.2) | 15.9(1.7) |
| First shell water | ||||
| 5.0(0.9) | 8.0(2.1) | 9.0(1.4) | 13.0(1.7) | |
| System | With Ligand 2h | With Ligand 5m |
|---|---|---|
| ΔEVDW | −30.0(2.7) | −28.3(3.2) |
| ΔEELE | −18.6(5.2) | −20.2(2.3) |
| ΔGPB | 24.3(5.5) | 22.9(5.1) |
| ΔGNP | −4.6(0.7) | −4.5(0.19) |
| PBtot | −28.8(3.1) | −26.6(2.8) |
| ΔGELE+PB | 5.7(5.2) | 2.9(3.2) |
| ΔGvdw+NP | −34.6(2.8) | −32.8(1.2) |
| TΔSsolute | −18.4(2.5) | −18.3(2.8) |
| ΔG a/ΔG b | −10.6/−9.8 | −8.9/−8.1 |
| IC50 a/IC50 b | 0.02/0.06 | 0.2/1.0 |
| Energy | Ligand 2h | Ligand 5m |
|---|---|---|
| Docking energy | −11.8 | −10.5 |
| Interaction energy | −34.8 | −29.3 |
| Association energy (MM/PBSA) | −10.6 | −8.9 |
| Disassociation energy (metadynamics) | −11 | −9.0 |
| Calculated binding free energy | −9.8 | −8.1 |
| Experimental IC50 (μM) | 0.06 | 1.0 |
| Calculated IC50 (μM) | 0.02 | 0.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srivastava, M.; Mittal, L.; Sarmadhikari, D.; Singh, V.K.; Fais, A.; Kumar, A.; Asthana, S. Template Entrance Channel as Possible Allosteric Inhibition and Resistance Site for Quinolines Tricyclic Derivatives in RNA Dependent RNA Polymerase of Bovine Viral Diarrhea Virus. Pharmaceuticals 2023, 16, 376. https://doi.org/10.3390/ph16030376
Srivastava M, Mittal L, Sarmadhikari D, Singh VK, Fais A, Kumar A, Asthana S. Template Entrance Channel as Possible Allosteric Inhibition and Resistance Site for Quinolines Tricyclic Derivatives in RNA Dependent RNA Polymerase of Bovine Viral Diarrhea Virus. Pharmaceuticals. 2023; 16(3):376. https://doi.org/10.3390/ph16030376
Chicago/Turabian StyleSrivastava, Mitul, Lovika Mittal, Debapriyo Sarmadhikari, Vijay Kumar Singh, Antonella Fais, Amit Kumar, and Shailendra Asthana. 2023. "Template Entrance Channel as Possible Allosteric Inhibition and Resistance Site for Quinolines Tricyclic Derivatives in RNA Dependent RNA Polymerase of Bovine Viral Diarrhea Virus" Pharmaceuticals 16, no. 3: 376. https://doi.org/10.3390/ph16030376
APA StyleSrivastava, M., Mittal, L., Sarmadhikari, D., Singh, V. K., Fais, A., Kumar, A., & Asthana, S. (2023). Template Entrance Channel as Possible Allosteric Inhibition and Resistance Site for Quinolines Tricyclic Derivatives in RNA Dependent RNA Polymerase of Bovine Viral Diarrhea Virus. Pharmaceuticals, 16(3), 376. https://doi.org/10.3390/ph16030376