

Application of Design of Experiments in the Development of Self-Microemulsifying Drug Delivery Systems


Abstract
1. Introduction
2. Lipid-Based Formulation for Oral Administration
2.1. Lipid Formulation Classification System
2.2. The Compositions of Lipid-Based Formulations and Their Role in Enhancement of Bioavailability
2.2.1. Triglycerides and Mixed Glycerides Used as Lipid Phase in Lipid-Based Formulations
2.2.2. Surfactants
2.2.3. Co-Surfactants/Co-Solvents
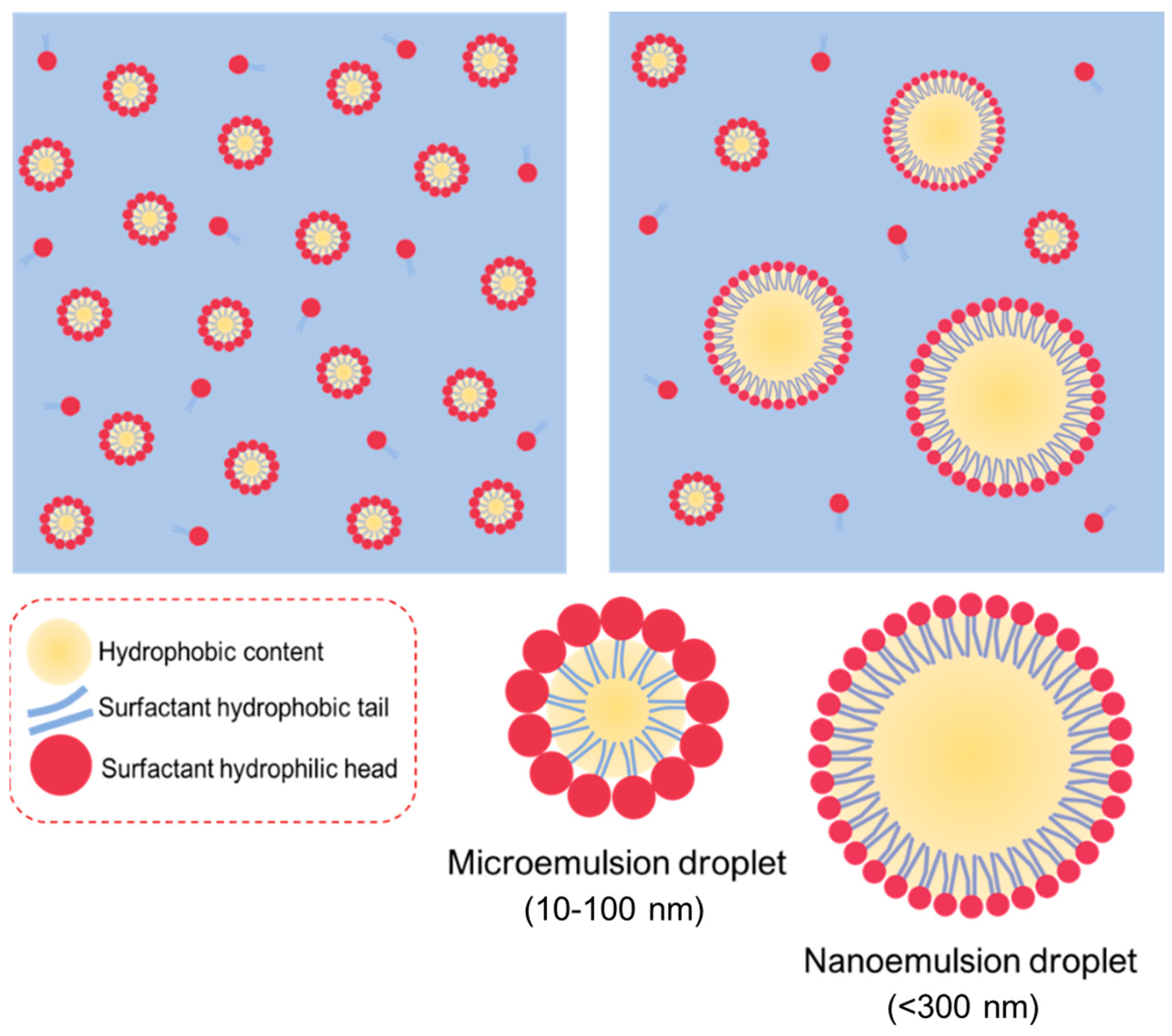
2.3. Macroemulsions, Microemulsions and Nanoemulsions
2.4. Self-Microemulsifying Drug Delivery System (SMEDDS)
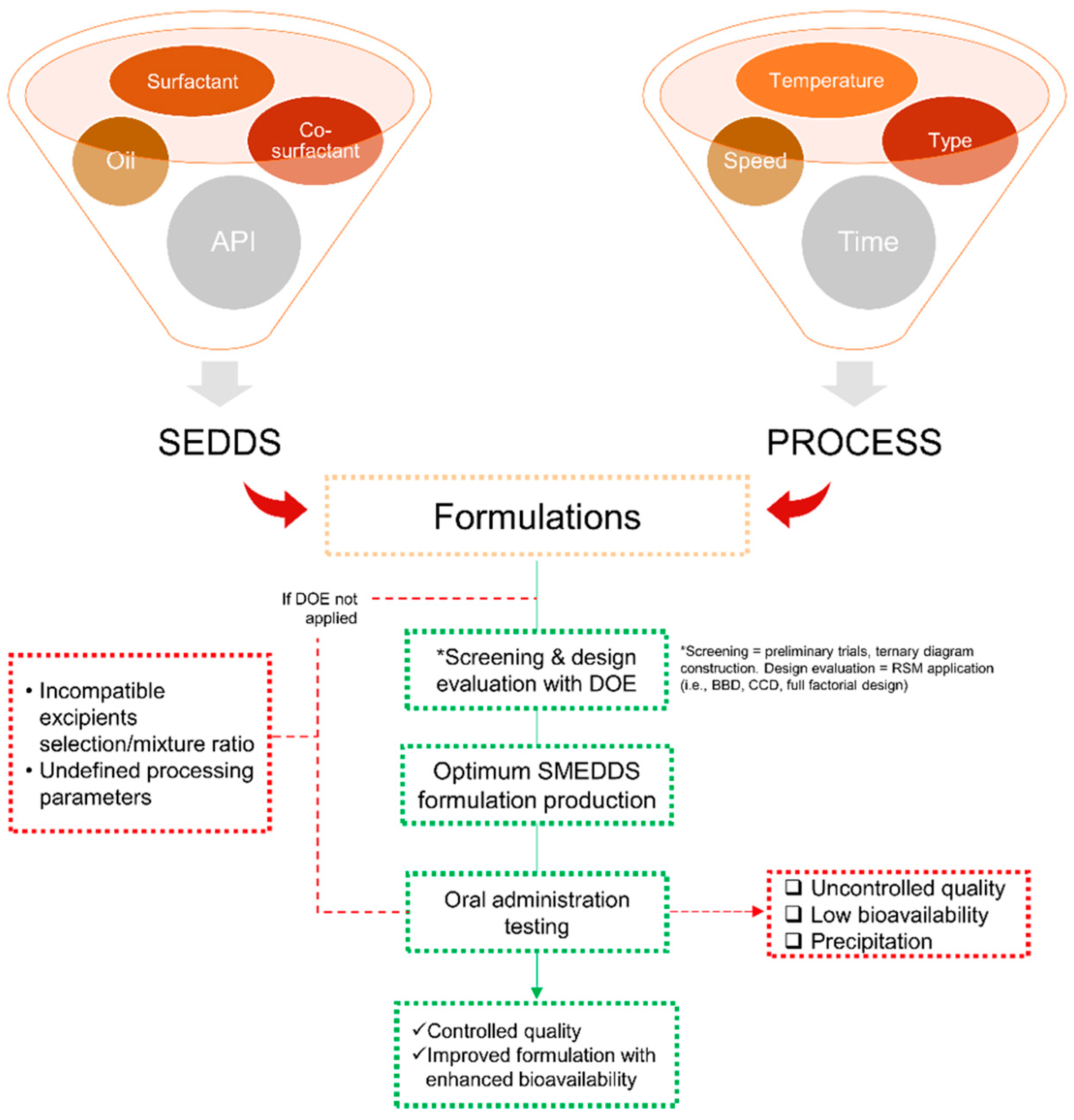
2.4.1. Formulation Design and Factors Affecting SMEDDS Formulations
Screening of Excipients
Active Pharmaceutical Ingredient (API) Dose
Polarity of the Lipid Phase
2.4.2. Characterization and Evaluation Methods for SMEDDS Formulations
2.4.3. New Strategy for SMEDDS Development
3. Overview of the Quality by Design (QbD) and Design of Experiment (DoE) for Pharmaceutical Development
3.1. Quality by Design (QbD)
3.2. Design of Experiment (DoE)
3.3. Screening Experiment and Factorial Design
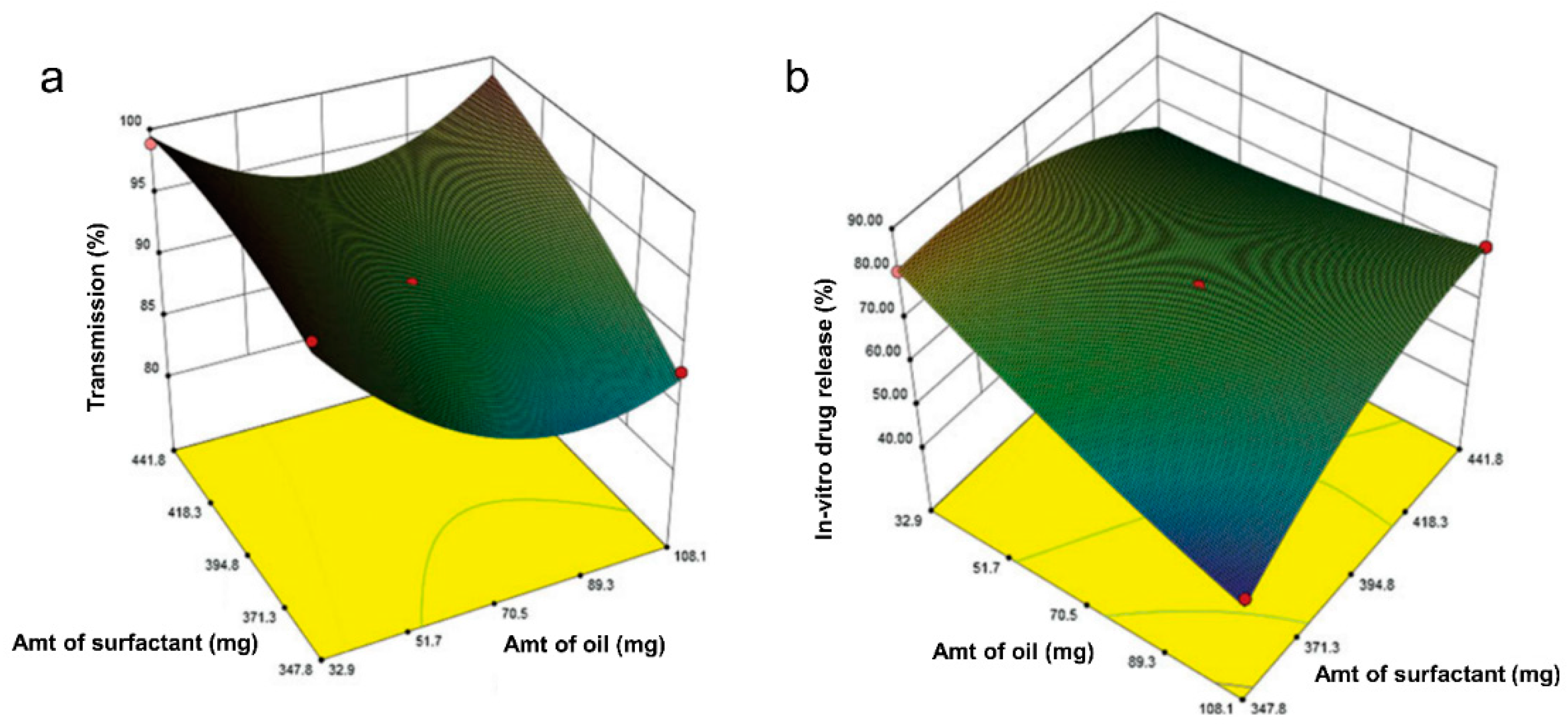
3.4. Response Surface Methodology
3.5. Optimization Methodology
4. Advantages of Applying DoE Techniques for the Development of SMEDDS Formulations
4.1. Box-Behnken Design (BBD)
4.2. Central Composite Design (CCD)
4.3. The Mixture Design
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ghadi, R.; Dand, N. BCS class IV drugs: Highly notorious candidates for formulation development. J. Control. Release 2017, 248, 71–95. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, Y.; Wada, K.; Nakatani, M.; Yamada, S.; Onoue, S. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: Basic approaches and practical applications. Int. J. Pharm. 2011, 420, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: Importance and enhancement techniques. ISRN Pharm 2012, 2012, 195727. [Google Scholar] [CrossRef] [PubMed]
- Bhalani, D.V.; Nutan, B.; Kumar, A.; Singh Chandel, A.K. Bioavailability Enhancement Techniques for Poorly Aqueous Soluble Drugs and Therapeutics. Biomedicines 2022, 10, 2055. [Google Scholar] [CrossRef]
- Sharma, V.K.; Koka, A.; Yadav, J.; Sharma, A.K.; Keservani, R.K. Self-micro emulsifying drug delivery systems: A strategy to improve oral bioavailability. Ars Pharm. 2016, 57, 97–109. [Google Scholar] [CrossRef]
- Desai, P.M.; Tan, B.M.; Liew, C.V.; Chan, L.W.; Heng, P.W. Impact of Electrostatics on Processing and Product Performance of Pharmaceutical Solids. Curr. Pharm. Des. 2015, 21, 5923–5929. [Google Scholar] [CrossRef]
- Pedrosa, V.M.; Sanches, A.G.; da Silva, M.B.; Gratao, P.L.; Isaac, V.L.; Gindri, M.; Teixeira, G.H. Production of mycosporine-like amino acid (MAA)-loaded emulsions as chemical barriers to control sunscald in fruits and vegetables. J. Sci. Food Agric. 2022, 102, 801–812. [Google Scholar] [CrossRef]
- Koli, A.R.; Ranch, K.M.; Patel, H.P.; Parikh, R.K.; Shah, D.O.; Maulvi, F.A. Oral bioavailability improvement of felodipine using tailored microemulsion: Surface science, ex vivo and in vivo studies. Int. J. Pharm. 2021, 596, 120202. [Google Scholar] [CrossRef]
- Baek, J.S.; Cho, C.W. Surface modification of solid lipid nanoparticles for oral delivery of curcumin: Improvement of bioavailability through enhanced cellular uptake, and lymphatic uptake. Eur. J. Pharm. Biopharm. 2017, 117, 132–140. [Google Scholar] [CrossRef]
- Murthy, A.; Ravi, P.R.; Kathuria, H.; Malekar, S. Oral Bioavailability Enhancement of Raloxifene with Nanostructured Lipid Carriers. Nanomaterials 2020, 10, 1085. [Google Scholar] [CrossRef]
- Sharma, A.; Streets, J.; Bhatt, P.; Patel, P.; Sutariya, V.; Varghese Gupta, S. Formulation and Characterization of Raloxifene Nanostructured Lipid Carriers for Permeability and Uptake Enhancement Applications. Assay Drug. Dev. Technol. 2022, 20, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Yamazoe, E.; Fang, J.Y.; Tahara, K. Oral mucus-penetrating PEGylated liposomes to improve drug absorption: Differences in the interaction mechanisms of a mucoadhesive liposome. Int. J. Pharm. 2021, 593, 120148. [Google Scholar] [CrossRef] [PubMed]
- Pouton, C.W. Lipid formulations for oral administration of drugs: Non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. Eur. J. Pharm. Sci. 2000, 11 (Suppl. 2), S93–S98. [Google Scholar] [CrossRef]
- Pouton, C.W. Formulation of poorly water-soluble drugs for oral administration: Physicochemical and physiological issues and the lipid formulation classification system. Eur. J. Pharm. Sci. 2006, 29, 278–287. [Google Scholar] [CrossRef]
- Huang, Y.; Yu, Q.; Chen, Z.; Wu, W.; Zhu, Q.; Lu, Y. In vitro and in vivo correlation for lipid-based formulations: Current status and future perspectives. Acta Pharm. Sin. B 2021, 11, 2469–2487. [Google Scholar] [CrossRef] [PubMed]
- Tay, E.; Nguyen, T.H.; Ford, L.; Williams, H.D.; Benameur, H.; Scammells, P.J.; Porter, C.J.H. Ionic Liquid Forms of the Antimalarial Lumefantrine in Combination with LFCS Type IIIB Lipid-Based Formulations Preferentially Increase Lipid Solubility, In Vitro Solubilization Behavior and In Vivo Exposure. Pharmaceutics 2019, 12, 17. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Harwansh, R.; Mirza, M.A.; Hussain, S.; Hussain, A. Oral lipid based drug delivery system (LBDDS): Formulation, characterization and application: A review. Curr. Drug Deliv. 2011, 8, 330–345. [Google Scholar] [CrossRef]
- Feng, W.; Qin, C.; Cipolla, E.; Lee, J.B.; Zgair, A.; Chu, Y.; Ortori, C.A.; Stocks, M.J.; Constantinescu, C.S.; Barrett, D.A.; et al. Inclusion of Medium-Chain Triglyceride in Lipid-Based Formulation of Cannabidiol Facilitates Micellar Solubilization In Vitro, but In Vivo Performance Remains Superior with Pure Sesame Oil Vehicle. Pharmaceutics 2021, 13, 1349. [Google Scholar] [CrossRef]
- Gao, H.; Jia, H.; Dong, J.; Yang, X.; Li, H.; Ouyang, D. Integrated in silico formulation design of self-emulsifying drug delivery systems. Acta Pharm. Sin. B 2021, 11, 3585–3594. [Google Scholar] [CrossRef]
- Jadhav, H.; Waghmare, J.; Annapure, U. Study on oxidative stability of deep fat fried food in Canola oil blended with medium chain triglyceride. Indian J. Chem. Technol. 2022, 29, 95–98. [Google Scholar]
- Kaukonen, A.M.; Boyd, B.J.; Porter, C.J.; Charman, W.N. Drug solubilization behavior during in vitro digestion of simple triglyceride lipid solution formulations. Pharm. Res. 2004, 21, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Cerpnjak, K.; Zvonar, A.; Gasperlin, M.; Vrecer, F. Lipid-based systems as a promising approach for enhancing the bioavailability of poorly water-soluble drugs. Acta Pharm. 2013, 63, 427–445. [Google Scholar] [CrossRef]
- Rosen, M.J.; Kunjappu, J.T. Surfactants and Interfacial Phenomena; John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar]
- Wilson, C.G.; Halbert, G.W.; Mains, J. The gut in the beaker: Missing the surfactants? Int. J. Pharm. 2016, 514, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Gelardi, G.; Mantellato, S.; Marchon, D.; Palacios, M.; Eberhardt, A.B.; Flatt, R.J. admixtures. In Science and Technology of Concrete Admixtures; Aïtcin, P.-C., Flatt, R.J., Eds.; Woodhead Publishing: Cambridge, UK, 2016; pp. 149–218. [Google Scholar] [CrossRef]
- Zhu, Y.; Ye, J.; Zhang, Q. Self-emulsifying Drug Delivery System Improve Oral Bioavailability: Role of Excipients and Physico-chemical Characterization. Pharm. Nanotechnol. 2020, 8, 290–301. [Google Scholar] [CrossRef]
- Rowe, R.C.; Sheskey, P.J.; Cook, W.G.; Fenton, M.E. Handbook of Pharmaceutical Excipients, 6th ed.; APhA/Pharmaceutical Press: London, UK, 2012; p. 1033. [Google Scholar]
- Lalanne-Cassou, C.; Carmona, I.; Fortney, L.; Samii, A.; Schechter, R.; Wade, W.; Weerasooriya, U.; Weerasooriya, V.; Yiv, S. Minimizing cosolvent requirements for microemulsion formed with binary surfactant mixtures. J. Dispers. Sci. Technol. 1987, 8, 137–156. [Google Scholar] [CrossRef]
- Dokania, S.; Joshi, A.K. Self-microemulsifying drug delivery system (SMEDDS)—Challenges and road ahead. Drug Deliv. 2015, 22, 675–690. [Google Scholar] [CrossRef]
- Jorgensen, A.M.; Friedl, J.D.; Wibel, R.; Chamieh, J.; Cottet, H.; Bernkop-Schnurch, A. Cosolvents in Self-Emulsifying Drug Delivery Systems (SEDDS): Do They Really Solve Our Solubility Problems? Mol. Pharm. 2020, 17, 3236–3245. [Google Scholar] [CrossRef]
- Prince, L.M. Microemulsions: Theory and Practice; Academic Press: New York, NY, USA, 1977; p. 179. [Google Scholar]
- Anton, N.; Vandamme, T.F. Nano-emulsions and micro-emulsions: Clarifications of the critical differences. Pharm Res. 2011, 28, 978–985. [Google Scholar] [CrossRef] [PubMed]
- McClements, D.J. Nanoemulsions versus microemulsions: Terminology, differences, and similarities. Soft Matter. 2012, 8, 1719–1729. [Google Scholar] [CrossRef]
- Tiwari, P.; Ranjan Sinha, V.; Kaur, R. Chapter 4—Clinical considerations on micro- and nanodrug delivery systems. In Drug Delivery Trends; Shegokar, R., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 77–101. [Google Scholar] [CrossRef]
- Callender, S.P.; Mathews, J.A.; Kobernyk, K.; Wettig, S.D. Microemulsion utility in pharmaceuticals: Implications for multi-drug delivery. Int. J. Pharm. 2017, 526, 425–442. [Google Scholar] [CrossRef]
- Yang, T.L.; Hsieh, C.M.; Meng, L.J.; Tsai, T.; Chen, C.T. Oleic Acid-Based Self Micro-Emulsifying Delivery System for Enhancing Antifungal Activities of Clotrimazole. Pharmaceutics 2022, 14, 478. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Cho, J.H.; Park, J.H.; Kim, J.S.; Song, E.S.; Kwon, J.; Giri, B.R.; Jin, S.G.; Kim, K.S.; Choi, H.G.; et al. Self-microemulsifying drug delivery system (SMEDDS) for improved oral delivery and photostability of methotrexate. Int. J. Nanomed. 2019, 14, 4949–4960. [Google Scholar] [CrossRef]
- Kamboj, S.; Rana, V. Quality-by-design based development of a self-microemulsifying drug delivery system to reduce the effect of food on Nelfinavir mesylate. Int. J. Pharm. 2016, 501, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Han, S.D.; Jung, S.W.; Jang, S.W.; Son, M.; Kim, B.M.; Kang, M.J. Reduced Food-Effect on Intestinal Absorption of Dronedarone by Self-microemulsifying Drug Delivery System (SMEDDS). Biol. Pharm. Bull. 2015, 38, 1026–1032. [Google Scholar] [CrossRef] [PubMed]
- Naccarelli, G.V.; Wolbrette, D.L.; Levin, V.; Samii, S.; Banchs, J.E.; Penny-Peterson, E.; Gonzalez, M.D. Safety and efficacy of dronedarone in the treatment of atrial fibrillation/flutter. Clin. Med. Insights Cardiol. 2011, 5, 103–119. [Google Scholar] [CrossRef]
- Renugopal, P.; Sangeetha, S.; Damodharan, N. An Emerging Trend in Solid Self Micro Emulsifying Drug Delivery System. Res. J. Pharm. Technol. 2020, 13, 3028–3034. [Google Scholar] [CrossRef]
- Salawi, A. Self-emulsifying drug delivery systems: A novel approach to deliver drugs. Drug Deliv. 2022, 29, 1811–1823. [Google Scholar] [CrossRef]
- Caliph, S.M.; Charman, W.N.; Porter, C.J.H. Effect of Short-, Medium-, and Long-Chain Fatty Acid-Based Vehicles on the Absolute Oral Bioavailability and Intestinal Lymphatic Transport of Halofantrine and Assessment of Mass Balance in Lymph-Cannulated and Non-cannulated Rats. J. Pharm. Sci. 2000, 89, 1073–1084. [Google Scholar] [CrossRef]
- Rahman, M.A.; Hussain, A.; Hussain, M.S.; Mirza, M.A.; Iqbal, Z. Role of excipients in successful development of self-emulsifying/microemulsifying drug delivery system (SEDDS/SMEDDS). Drug Dev. Ind. Pharm. 2013, 39, 1–19. [Google Scholar] [CrossRef]
- Kadu, P.J.; Kushare, S.S.; Thacker, D.D.; Gattani, S.G. Enhancement of oral bioavailability of atorvastatin calcium by self-emulsifying drug delivery systems (SEDDS). Pharm. Dev. Technol. 2011, 16, 65–74. [Google Scholar] [CrossRef]
- Heshmati, N.; Cheng, X.; Eisenbrand, G.; Fricker, G. Enhancement of oral bioavailability of E804 by self-nanoemulsifying drug delivery system (SNEDDS) in rats. J. Pharm. Sci. 2013, 102, 3792–3799. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Hui Zhou, C.; Ping Xu, Z. Chapter 14—Self-Nanoemulsifying Drug-Delivery System. In Nanocarriers for Drug Delivery; Mohapatra, S.S., Ranjan, S., Dasgupta, N., Mishra, R.K., Thomas, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 421–449. [Google Scholar] [CrossRef]
- Hamed, R.; Awadallah, A.; Sunoqrot, S.; Tarawneh, O.; Nazzal, S.; AlBaraghthi, T.; Al Sayyad, J.; Abbas, A. pH-Dependent Solubility and Dissolution Behavior of Carvedilol--Case Example of a Weakly Basic BCS Class II Drug. AAPS PharmSciTech 2016, 17, 418–426. [Google Scholar] [CrossRef]
- Abuhelwa, A.Y.; Williams, D.B.; Upton, R.N.; Foster, D.J. Food, gastrointestinal pH, and models of oral drug absorption. Eur. J. Pharm. Biopharm 2017, 112, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Akula, S.; Gurram, A.K.; Devireddy, S.R. Self-Microemulsifying Drug Delivery Systems: An Attractive Strategy for Enhanced Therapeutic Profile. Int. Sch. Res. Not. 2014, 2014, 964051. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, Y.; Cheng, J.; Shan, B.; Wang, Y.; Wang, R.; Hou, L. Solid self-microemulsifying drug delivery system of Sophoraflavanone G: Prescription optimization and pharmacokinetic evaluation. Eur. J. Pharm. Sci 2019, 136, 104953. [Google Scholar] [CrossRef]
- Musakhanian, J.; Rodier, J.D.; Dave, M. Oxidative Stability in Lipid Formulations: A Review of the Mechanisms, Drivers, and Inhibitors of Oxidation. AAPS PharmSciTech 2022, 23, 151. [Google Scholar] [CrossRef]
- Gabric, A.; Hodnik, Z.; Pajk, S. Oxidation of Drugs during Drug Product Development: Problems and Solutions. Pharmaceutics 2022, 14, 325. [Google Scholar] [CrossRef]
- Yeom, D.W.; Chae, B.R.; Son, H.Y.; Kim, J.H.; Chae, J.S.; Song, S.H.; Oh, D.; Choi, Y.W. Enhanced oral bioavailability of valsartan using a polymer-based supersaturable self-microemulsifying drug delivery system. Int. J. Nanomed. 2017, 12, 3533–3545. [Google Scholar] [CrossRef] [PubMed]
- ICH. ICH Harmonised Tripartite Guideline; Q8 (R2) Pharmaceutical Development; ICH: London, UK, 2009. [Google Scholar]
- Yu, L.X.; Amidon, G.; Khan, M.A.; Hoag, S.W.; Polli, J.; Raju, G.; Woodcock, J.J.T.A.j. Understanding pharmaceutical quality by design. AAPS J. 2014, 16, 771–783. [Google Scholar] [CrossRef]
- Astakhov, V.P. Design of experiment methods in manufacturing: Basics and practical applications. In Statistical and Computational Techniques in Manufacturing; Springer: Berlin/Heidelberg, Germany, 2012; pp. 1–54. [Google Scholar]
- Fisher, R.A. Design of experiments. Br. Med. J. 1936, 1, 554. [Google Scholar] [CrossRef]
- Montgomery, D.C. Design and Analysis of Experiments; John Wiley & Sons: Hoboken, NJ, USA, 2017. [Google Scholar]
- Beg, S.; Raza, K. Full Factorial and Fractional Factorial Design Applications in Pharmaceutical Product Development. In Design of Experiments for Pharmaceutical Product Development: Volume I: Basics and Fundamental Principles; Beg, S., Ed.; Springer: Singapore, 2021; pp. 43–53. [Google Scholar] [CrossRef]
- Box, G.E.; Hunter, W.H.; Hunter, S. Statistics for Experimenters; John Wiley and Sons: New York, NY, USA, 1978; Volume 664. [Google Scholar]
- Plackett, R.L.; Burman, J.P. The design of optimum multifactorial experiments. Biometrika 1946, 33, 305–325. [Google Scholar] [CrossRef]
- Mousavi, L.; Tamiji, Z.; Khoshayand, M.R. Applications and opportunities of experimental design for the dispersive liquid–liquid microextraction method—A review. Talanta 2018, 190, 335–356. [Google Scholar] [CrossRef]
- Luiz, M.T.; Viegas, J.S.R.; Abriata, J.P.; Viegas, F.; de Carvalho Vicentini, F.T.M.; Bentley, M.V.L.B.; Chorilli, M.; Marchetti, J.M.; Tapia-Blacido, D.R. Design of experiments (DoE) to develop and to optimize nanoparticles as drug delivery systems. Eur. J. Pharm. Biopharm. 2021, 165, 127–148. [Google Scholar] [CrossRef] [PubMed]
- Sy Mohamad, S.F.; Mohd Said, F.; Abdul Munaim, M.S.; Mohamad, S.; Azizi Wan Sulaiman, W.M.J.C.r.i.b. Application of experimental designs and response surface methods in screening and optimization of reverse micellar extraction. Crit. Rev. Biotechnol. 2020, 40, 341–356. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, G.W. Formulation Approaches for Improving the Dissolution Behavior and Bioavailability of Tolvaptan Using SMEDDS. Pharmaceutics 2022, 14, 415. [Google Scholar] [CrossRef] [PubMed]
- Na, Y.G.; Byeon, J.J.; Wang, M.; Huh, H.W.; Kim, M.K.; Bang, K.H.; Han, M.G.; Lee, H.K.; Cho, C.W. Statistical approach for solidifying ticagrelor loaded self-microemulsifying drug delivery system with enhanced dissolution and oral bioavailability. Mater. Sci. Eng. C Mater. Biol. Appl. 2019, 104, 109980. [Google Scholar] [CrossRef]
- Dalvadi, H.; Patel, N.; Parmar, K. Systematic development of design of experiments (DoE) optimised self-microemulsifying drug delivery system of Zotepine. J. Microencapsul. 2017, 34, 308–318. [Google Scholar] [CrossRef]
- Marasini, N.; Tran, T.H.; Poudel, B.K.; Choi, H.-G.; Yong, C.S.; Kim, J.O.J.C.; Bulletin, P. Statistical modeling, optimization and characterization of spray-dried solid self-microemulsifying drug delivery system using design of experiments. Chem. Pharm. Bull. 2013, 61, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.A.D.; Almeida, S.L.; Alonso, E.C.P.; Rocha, P.B.R.; Martins, F.T.; Freitas, L.A.P.; Taveira, S.F.; Cunha-Filho, M.S.S.; Marreto, R.N. Preparation of a solid self-microemulsifying drug delivery system by hot-melt extrusion. Int. J. Pharm. 2018, 541, 1–10. [Google Scholar] [CrossRef]
- Čerpnjak, K.; Pobirk, A.Z.; Vrečer, F.; Gašperlin, M. Tablets and minitablets prepared from spray-dried SMEDDS containing naproxen. Int. J. Pharm. 2015, 495, 336–346. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, S.; Shen, H.; Li, J.; Gao, C.J.A.P. Controlled release of the Nimodipine-loaded self-microemulsion osmotic pump capsules: Development and characterization. AAPS PharmSciTech 2018, 19, 1308–1319. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Lv, C.; Sun, X.; Wang, J.; Zhao, Z. Preparation of a supersaturatable self-microemulsion as drug delivery system for ellagic acid and evaluation of its antioxidant activities. J. Drug Deliv. Sci. Technol. 2019, 53, 101209. [Google Scholar] [CrossRef]
- Tung, N.-T.; Tran, C.-S.; Nguyen, H.-A.; Nguyen, T.-L.; Chi, S.-C.; Nguyen, D.-D.J.I.j.o.p. Development of solidified self-microemulsifying drug delivery systems containing l-tetrahydropalmatine: Design of experiment approach and bioavailability comparison. Int. J. Pharm. 2018, 537, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Wang, Y.; Ma, Y.; Zhao, J.; Liu, Y.; Wang, L. In vitro and in vivo evaluation of poly (acrylic acid) modified mesoporous silica nanoparticles as pH response carrier for β-elemene self-micro emulsifying. Int. J. Pharm. 2019, 572, 118768. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Dudhat, K.; Soniwala, M.; Kotadiya, N.; Mori, D.J.J.o.P.I. DoE-Based Solid Self-microemulsifying Drug Delivery System (S-SMEDDS) Approach for Improving the Dissolution Properties of Raltegravir Potassium. J. Pharm. Innov. 2022. [Google Scholar] [CrossRef]
- Dhaval, M.; Panjwani, M.; Parmar, R.; Soniwala, M.M.; Dudhat, K.; Chavda, J. Application of Simple Lattice Design and Desirability Function for Formulating and Optimizing SMEDDS of Clofazimine. J. Pharm. Innov. 2021, 16, 504–515. [Google Scholar] [CrossRef]
- Na, Y.G.; Byeon, J.J.; Wang, M.; Huh, H.W.; Son, G.H.; Jeon, S.H.; Bang, K.H.; Kim, S.J.; Lee, H.J.; Lee, H.K.; et al. Strategic approach to developing a self-microemulsifying drug delivery system to enhance antiplatelet activity and bioavailability of ticagrelor. Int. J. Nanomed. 2019, 14, 1193–1212. [Google Scholar] [CrossRef]
- Yang, Q.; Wang, Q.; Feng, Y.; Wei, Q.; Sun, C.; Firempong, C.K.; Adu-Frimpong, M.; Li, R.; Bao, R.; Toreniyazov, E.; et al. Anti-hyperuricemic property of 6-shogaol via self-micro emulsifying drug delivery system in model rats: Formulation design, in vitro and in vivo evaluation. Drug Dev. Ind. Pharm. 2019, 45, 1265–1276. [Google Scholar] [CrossRef]
- Li, F.; Song, S.; Guo, Y.; Zhao, Q.; Zhang, X.; Pan, W.; Yang, X. Preparation and pharmacokinetics evaluation of oral self-emulsifying system for poorly water-soluble drug Lornoxicam. Drug Deliv. 2015, 22, 487–498. [Google Scholar] [CrossRef]
- Qu, Y.; Mu, S.; Song, C.; Zheng, G. Preparation and in vitro/in vivo evaluation of a self-microemulsifying drug delivery system containing chrysin. Drug Dev. Ind. Pharm. 2021, 47, 1127–1139. [Google Scholar] [CrossRef]
- Wang, L.; Yan, W.; Tian, Y.; Xue, H.; Tang, J.; Zhang, L. Self-microemulsifying drug delivery system of phillygenin: Formulation development, characterization and pharmacokinetic evaluation. Pharmaceutics 2020, 12, 130. [Google Scholar] [CrossRef]
- Zhang, N.; Zhang, F.; Xu, S.; Yun, K.; Wu, W.; Pan, W. Formulation and evaluation of luteolin supersaturatable self-nanoemulsifying drug delivery system (S-SNEDDS) for enhanced oral bioavailability. J. Drug Deliv. Sci. Technol. 2020, 58, 101783. [Google Scholar] [CrossRef]
- Xie, M.; Wu, J.; Ji, L.; Jiang, X.; Zhang, J.; Ge, M.; Cai, X. Development of triptolide self-microemulsifying drug delivery system and its anti-tumor effect on gastric cancer xenografts. Front. Oncol. 2019, 9, 978. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.e.; Xu, Z.; Li, X.; Wang, Z.; Li, J.; Yang, Z.; Khattak, S.U.; Liu, Y.; Shi, Y. Development and evaluation of rhubarb free anthraquinones loaded self-nanoemulsifying tablets. J. Drug Deliv. Sci. Technol. 2020, 57, 101737. [Google Scholar] [CrossRef]
- Mahore, J.; Shelar, A.; Deshkar, S.; More, G.J.J.R.P. Conceptual design and optimization of self microemulsifying drug delivery systems for dapsone by using Box-Behnken design. J. Res. Pharm. 2021, 25, 179–195. [Google Scholar]
- Lee, D.W.; Marasini, N.; Poudel, B.K.; Kim, J.H.; Cho, H.J.; Moon, B.K.; Choi, H.-G.; Yong, C.S.; Kim, J.O.J.J.o.m. Application of Box–Behnken design in the preparation and optimization of fenofibrate-loaded self-microemulsifying drug delivery system (SMEDDS). J. Microencapsul. 2014, 31, 31–40. [Google Scholar] [CrossRef]
- Yadav, P.; Rastogi, V.; Verma, A. Application of Box–Behnken design and desirability function in the development and optimization of self-nanoemulsifying drug delivery system for enhanced dissolution of ezetimibe. Future J. Pharm. Sci. 2020, 6, 7. [Google Scholar] [CrossRef]
- Visetvichaporn, V.; Kim, K.-H.; Jung, K.; Cho, Y.-S.; Kim, D.-D. Formulation of self-microemulsifying drug delivery system (SMEDDS) by D-optimal mixture design to enhance the oral bioavailability of a new cathepsin K inhibitor (HL235). Int. J. Pharm. 2020, 573, 118772. [Google Scholar] [CrossRef] [PubMed]
- Vaghela, S.; Chaudhary, S.; Chaudhary, A. Formulation Development and Optimization of Blonanserin Liquid SMEDDS using D-Optimal Mixture Design. Curr. Drug Ther. 2022, 17, 266–280. [Google Scholar] [CrossRef]
- Gahlawat, N.; Verma, R.; Kaushik, D. Application of D-optimal Mixture Design for Development and Optimization of Olmesartan Medoxomil Loaded SMEDDS. Curr. Drug Ther. 2020, 15, 548–560. [Google Scholar] [CrossRef]
- Son, H.Y.; Chae, B.R.; Choi, J.Y.; Shin, D.J.; Goo, Y.T.; Lee, E.S.; Kang, T.H.; Kim, C.H.; Yoon, H.Y.; Choi, Y.W. Optimization of self-microemulsifying drug delivery system for phospholipid complex of telmisartan using D-optimal mixture design. PLoS ONE 2018, 13, e0208339. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Thakkar, V.; Gohel, M.; Baldaniya, L.; Gandhi, T. Optimization of Self Micro Emulsifying Drug Delivery System Containing Curcumin and Artemisinin Using D-Optimal Mixture Design. J Saudi J. Med. Pharm. Sci 2017, 3, 388–398. [Google Scholar]
- Xiong, Y.; Zou, Y.; Chen, L.; Xu, Y.; Wang, S.J.A.P. Development and in vivo evaluation of ziyuglycoside i–loaded self-microemulsifying formulation for activity of increasing leukocyte. AAPS PharmSciTech 2019, 20, 101. [Google Scholar] [CrossRef] [PubMed]
- Goo, Y.T.; Lee, S.; Choi, J.Y.; Kim, M.S.; Sin, G.H.; Hong, S.H.; Kim, C.H.; Song, S.H.; Choi, Y.W. Enhanced oral absorption of insulin: Hydrophobic ion pairing and a self-microemulsifying drug delivery system using a D-optimal mixture design. Drug Deliv. 2022, 29, 2831–2845. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type I | Type II | Type IIIa | Type IIIb | Type IV | ||
|---|---|---|---|---|---|---|
| Composition (w/w %) | Glycerides (mono-, di-, tri-glycerides) | 100 | 40–80 | 40–80 | <20 | 0 |
| Lipophilic surfactants (HLB < 12) | -- | 20–60 | 20–40 | 0 | 0–20 | |
| Hydrophilic surfactants (HLB > 12) | -- | -- | 0 | 20–50 | 20–80 | |
| Co-solvents | -- | -- | 0–40 | 20–50 | 0–80 | |
| Characteristic features | Oil solution | Self-emulsification | Self-emulsification | Self-micro- emulsification | Spontaneous micelle dispersion | |
| Droplet size | Coarse | 0.25–2 µm | 100–250 nm | 50–100 nm | <50 nm | |
| Lipase digestion | Crucial | Not crucial, but likely | Not crucial, but may occur | Not important | Not important | |
| Disadvantages | Poor solvent capacity for the drugs with log p < 2 | Coarser emulsion | Possible loss of solvent capacity on dispersion | May cause partial drug precipitation | Risk of drug precipitation upon dispersion | |
| Class | Example | Characteristics |
|---|---|---|
| Medium chain triglycerides (MCT) | Coconut oil Palm seed oil, Miglyol® 812 Captex® 355 | Good solubilizing capacity for less lipophilic drugs Higher self-dispersing ability |
| Long chain triglycerides (LCT) | Corn oil Soybean oil Olive oil Peanut oil Sesame oil Sunflower oil Castor oil | GRAS status Easily ingested, digested, and absorbed Poor self-dispersing properties Lower loading capacity for drugs with intermediate log p values Higher solubilizing capacity after dispersion and digestion of the formulation |
| Mixed mono-, di- and triglycerides | Imwitor® 988 Imwitor® 308 Maisine® 35-1 Peceol® Plurol Oleique® CC49 Capryol® Myrj® | Higher self-dispersing ability Higher solubilizing capacity for poorly water-soluble drugs |
| Surfactants | HLB | Description | Oral | Topical | Injection | Maximum Potency per Dosage Unit | |
|---|---|---|---|---|---|---|---|
| Polyoxylglycerides | Caprylocaproyl polyoxylglycerides (Labrasol®) | 12 | Pale-yellow oily liquids | √ | √ | -- | Oral = 61.2 mg/mL |
| Lauroyl polyoxylglyceride (Gelucire 44/14®) | 11 | Pale-yellow waxy solids | √ | -- | -- | Oral = 0.15–218 mg | |
| Stearoyl polyoxylglycerides (Gelucire 50/13®) | 11 | Pale-yellow waxy solids | √ | -- | -- | Oral = 23.34 mg | |
| Polyoxyethylene Stearates | Polyoxyl 8 stearate | 11.1 | Waxy cream | √ | √ | √ | Oral = 25 mg/5 mL |
| Polyoxyl 12 stearate | 13.6 | Pasty solid | √ | √ | √ | n/a | |
| Polyoxyl 20 stearate | 14 | Waxy solid | √ | √ | √ | n/a | |
| Polyoxyl 40 stearate | 16.9 | Waxy solid | √ | √ | √ | Oral = 2–8.48 mg; Topical = 3–8.8% w/w | |
| Polyoxyl 50 stearate | 17.9 | Solid | √ | √ | √ | n/a | |
| Polyoxyl 100 stearate | 18.8 | Solid | √ | √ | √ | Topical = 0.5–2.1% w/w | |
| Polyoxyl 12 distearate | 10.6 | Paste | √ | √ | √ | n/a | |
| Polyoxyethylene Sorbitan Fatty Acid Esters | Polyoxyethylene 20 sorbitan monolaurate (Tween 20) | 16.7 | Yellow oily liquid | √ | √ | √ | Oral = 0.35–4.2 mg; Topical= 0.02–8% w/w |
| Polyoxyethylene 20 sorbitan monopalmitate (Tween 40) | 15.6 | Yellow oily liquid | √ | √ | √ | Oral = 0.05 mg/5 mL; Topical = 2–3% w/w | |
| Polyoxyethylene 20 sorbitan monostearate (Tween 60) | 14.9 | Yellow oily liquid | √ | √ | √ | Oral = 5–20 mg/mL; Topical = 0.42–14.55% w/w | |
| Polyoxyethylene 20 sorbitan tristearate (Tween 65) | 10.5 | Tan solid | √ | √ | √ | Topical = 0.5% w/w | |
| Polyoxyethylene 20 sorbitan monooleate (Tween 80) | 15 | Yellow oily liquid | √ | √ | √ | Oral = 0.04–418.37 mg; Topical = 0.1–15% w/w | |
| Polyoxyethylene 20 sorbitan trioleate (Tween 85) | 11 | Amber liquid | √ | √ | √ | Oral = 1.5 mg/5 mL | |
| Polyoxyethylene 20 sorbitan monoisostearate | 14.9 | Yellow oily liquid | √ | √ | √ | n/a | |
| Polyoxyethylene Alkyl Ethers | Polyoxyl 23 lauryl ether (Brij 35®) | 16.9 | White waxy solid | √ | √ | -- | Topical = 0.45–1.08% w/w |
| Polyoxyl 10 cetyl ether (Brij 56®) | 12.9 | White waxy solid | √ | √ | -- | Topical = 2.5% w/w | |
| Polyoxyl 20 cetyl ether (Brij 58®) | 15.7 | Waxy solid | √ | √ | -- | Topical = 2–6% w/w | |
| Polyoxyl 10 stearyl ether (Brij 76®) | 12.4 | White waxy solid | √ | √ | -- | n/a | |
| Polyoxyethylene Castor Oil Derivatives | Polyoxyl 35 castoroil (Cremophor EL®) | 12–14 | Pale yellow oily liquid Clear above 26 °C with faint characteristic odor | √ | √ | √ | Oral = 0.4–515 mg/mL Topical = 4% w/w |
| Poloxyl 35 castoroil, purified (Cremophor ELP®) | 12–14 | White to slightly yellowish paste or cloudy liquid with weak characteristic odor | √ | √ | √ | n/a | |
| Polyoxyl 40 hydrogenated castoroil (Cremophor RH40®) | 14–16 | Viscous liquid or soft paste with very little odor in aqueous solutions, almost tasteless | √ | √ | √ | Oral coated capsule = 101.25 mg Oral solution = 450 mg/mL Topical = 1% w/w | |
| Polyoxyl 60 hydrogenated castor oil | 15–17 | White to yellowish soft or flowing paste with faint odor or taste in aqueous solutions | √ | √ | √ | Topical = 1.9% w/w | |
| D-α-Tocopherol polyethylene glycol 1000 succinate (TPGS) | 13.2 | White to light-brown, waxy solid | √ | √ | -- | n/a | |
| Nanoemulsions | Microemulsions | |
|---|---|---|
| Stability | Kinetic stable system | Thermodynamic stable system |
| Compositions | Oil, Surfactants, Water | Oil, Surfactants, Water |
| Order of mixing | The surfactant should first be mixed with the oil phase, and then titrated with the aqueous | The order of mixing does not affect the size of particle |
| Particle size | 50–300 nm | 10–100 nm |
| Manufacturing process | Specific equipment is required to provide sufficient energy to increase the interfacial area | Spontaneous formation |
| Compound | Screening | RSM | Experiments | Independent Variables | Responses | Program | Optimized Conditions | Reference |
|---|---|---|---|---|---|---|---|---|
| 6-Shogaol (purified alkylphenol from ginger root) | n/a | CCD | p < 0.05 | Ethyl oleate (18.62% w/w), tween 80:PEG 400 (1.73:1 w/w) | Particle size, PDI, cumulative drug release | Design-Expert®, version 8.0.6 | Particle size (20.00 ± 0.26 nm), PDI (0.18 ± 0.02), increased cumulative release compared to free 6-shogaol, oral bioavailability | [72] |
| Lornoxicam | Regular experiment | CCD | p < 0.05 | Labrafil M 1944 CS (25%), Kolliphor HS 15 (56.25%), Transcutol HP (18.75%) | Particle size, PDI, self-emulsifying time | Design-Expert® n/a version | Particle size (70.14 ± 1.06 nm), PDI (0.193 ± 0.010), self-emulsifying time (68 ± 2 s) | [73] |
| Chrysin | Compatibility tests and pseudo-ternary phase diagram studies | CCD | p < 0.05 | Surface morphology, pH, diameter, PDI, zeta potential, and phase type | Maximum drug loading and optimize SMEDDS formation | Design-Expert® n/a version | Medium chain triglyceride:oleic acid:Cremophor RH40: Transcutol HP w/w) (12%:12%:32%:44%), with a drug loading capacity of 5 mg/g | [74] |
| Phillygenin | Compatibility tests and pseudo-ternary phase diagram studies | CCD | p < 0.05 | Oil phase mass% and surfactant/co-surfactant mixture weight ratio | Equilibrium solubility, particle size, PDI | Design-Expert® version 8.0.6 | Optimized Labrafil M1944CS:Cremophor EL:PEG400 = 27.8:33.6:38.6% wt produced 10.2 mg/g equilibrium solubility, 40.11 ± 0.74 nm particle size, and 0.243 ± 0.01 PDI | [75] |
| Luteolin | Compatibility tests and pseudo-ternary phase diagram studies | CCD | p < 0.05 | Weight percent of oil and the mass ratio | Particle size, PDI, self-emulsifying time | Design-Expert® version 8.0 | Optimized Crodamol GTCC:Kolliphor EL:PEG400 = 20.1:48.2:31.7% wt produced LUT loading capacity = 24.66 mg/g; S-SNEDDS showed 2.2-fold increase of bioavailability compared to conventional SNEDDS. | [76] |
| Triptolide | n/a | CCD | n/a | Oil phase mass% and surfactant/co-surfactant mixture weight ratio | Particle size and drug content | Design-Expert® version 8.0.6 | Optimized MCT:EL:PEG400 = 25.3:49.6:25.1 with particle size of 30.46 nm and drug content of 2.91 mg/g. These optimized parameters produced SMEDDS with complete release in 6 h, increased oral bioavailability, and enhanced the tumor inhibitory effect. | [77] |
| Ellagic acid | Ternary phase diagram studies | CCD | p < 0.01 | Oil phase mass% and surfactant/co-surfactant mixture weight ratio | Particle size and solubility | Design-Expert® version 8.0.5 | 10% ethyl oleate, 67.5% Tween 80, 22.5% PEG 400, 0.5% PVP K30 and 4 mg/g ellagic acid. The presence of PVP K30 in the optimized excipients inhibited the precipitation. The in vitro and in vivo showed an improved antioxidant ability of eligilic acid. | [78] |
| Rhubarb free-anthraquinone | n/a | CCD | p < 0.05 | Mass ratio of Neusilin US2/preconcentrated RhA nanoemulsions and contents of PVPP % w/w | Friability, disintegration time, and 4 h cumulative dissolution rate of RhA in SNEDDS tablets | Design-Expert® version 8.0.6 | Optimized 1:1(w/w) Neusilin US2/pre-concentrated RhA nanoemulsions, 5.0% w/w PVPP, 1% w/w Mg stearate produced friability of 0.389 ± 0.007%, disintegration time of 5.13 ± 0.14 min, and 4 h-dissolution rate of 87.91 ± 1.89%. | [79] |
| Compound | Screening | RSM | Experiments | Independent Variables | Responses | Program | Optimized Conditions | Reference |
|---|---|---|---|---|---|---|---|---|
| Furbiprofen | Regular experiment | BBD (33) | p < 0.05 | Inlet temperature, feed rate, and carrier concentration | %moisture, %yield, drug content, and particle size | Design-Expert® version 8.0.5 | %yield (58.5%) and drug content (70.1 mg/g) with minimum moisture content (0.72%) and particle size (166.8 nm). | [69] |
| Zotepine | Pseudo-ternary diagrams studies | BBD (33) | p < 0.05 | Oleic acid (oil), Tween 80 (surfactant), and PEG400 (co-surfactant) | %microemulsions transparency and %cumulative drug release | Design-Expert® version 8.0.5 | %transmittance of 98.75% and an improved 30 min-in vitro drug release of 86.57%. | [68] |
| Dapsone | Pseudo-ternary diagrams studies | BBD (33) | p < 0.05 | Inlet temperature, feed flow rate, carrier concentration | Particle size and %yield | Design-Expert® version 11.0 | The optimized solid SMEDDS with inlet temperature of 130 °C, flow rate of 6 mL/min, and carrier conc. (i.e., neusilin US2) of 0.25% resulted in 87.5 ± 4.95 nm of particle size and yielded 34.06 ± 1.70%. | [83] |
| Carvedilol | Regular experiments on formulation compositions and hot melt extruder conditions | BBD (33) | p < 0.05 | Recirculation time, first heating zone temperature, API concentration | %drug releases (in 0.1 M HCl and 0.4 M phosphate buffer), %efficiency, and particle size | Statistica ® version 7.0 | The optimized formulation of carvedilol in solid SMEDDS using hot-melt extrusion resulted in max. 25.54 ± 0.77% release in HCl followed by max. 85.54 ± 1.79% release in phosphate buffer. | [70] |
| Fenofibrate | Pseudo-ternary diagrams studies | BBD (33) | p < 0.05 | Amount of Labrafil M 1944 (oil), Labrasol (surfactant), and Capryol (co-surfactant) | Particle size, %cumulative release in 30 min, and equilibrium solubility | Design-Expert® version 8.0.4 | The optimized formulation of fenofibrate in solid SMEDDS resulted in 113.13 ± 1.63 mg/g solubility with particle size of 171.4 ± 2.5 nm, %cumulative release of 87.7 ± 1.6%, and 3.6-fold higher bioavailability than its free-form suspension. | [84] |
| Ezetimibe | Pseudo-ternary diagrams studies | BBD (33) | p < 0.05 | Amount of Peceol (oil), Tween 80 (surfactant), Transcutol P (co-surfactant) | Particle size, %transmittance, self-emulsification time, %cumulative releases in 5 and 40 min | Design-Expert® version 11.0 | The optimized ezetimibe in solid SMEDDS resulted in 26.31 ± 2.64 nm particle size, 69.26 ± 2.56 self-emulsification time, and 95.38 ± 3.67% cumulative release in 40 min. | [85] |
| Naproxen | Regular experiment | FFD | p < 0.05, except %yield | Inlet temperature, pressure, and pump speed | Droplet size, PDI, and %yield | Unscrambler1 software(version 10.1, CAMO software) | The inlet temperature of 120 °C, pressure of 50 mmHg, and pump speed of 15 mL/min resulted the optimized solid SMEDDS. | [71] |
| Compound | Screening | RSM | Experiments | Independent Variables | Responses | Program | Optimized Conditions | Reference |
|---|---|---|---|---|---|---|---|---|
| HL235 (i.e., Cathepsin K inhibitor) | Pseudo-ternary diagrams studies | D-optimal mixture | p < 0.05 | Capmul MCM (oil), Tween-20 (surfactant), Carbitol (co-surfactant) | Cumulative drug release in 15 min and solubilization capacity | Design-Expert® version 7.0 | The optimized SMEDDs formulation resulting in 2.34 ± 0.21 µg/mL and solubilization capacity of 6.164 ± 0.06 mg/mL. | [86] |
| Blonanserin | Pseudo-ternary diagrams studies | D-optimal mixture | n/a | Captex 200P: Capmul MCM (1:1) (oil), Tween-20 (surfactant), and ethanol (co-surfactant) | Drug loading, percentage cumulative drug release, particle size | n/a | The optimized Blonanserin in SMEDDS with 1:1 (23% v/v) Captex 200P:Capmul MCM mixture, Tween-80 (57% v/v), and ethanol (20% v/v) produced cumulative drug release of 94.72% in 30 min and particle size of 21 nm | [87] |
| Olmesartan medoxomil | Pseudo-ternary diagrams studies | D-optimal mixture | p < 0.05 | Capmul MCM EP (oil), Kolliphore EL (surfactant), Transcutol P (co-surfactant) | Cumulative drug release and particle size | JMP ver.9.0.0 software | The optimized formulation with Capmul MCM EP (23% v/v), Kolliphore EL (49% v/v) and Transcutol P (28% v/v) resulted in 94.7% of drug release and 105 nm of particle size. | [88] |
| Telmisartan (loaded with phospholipid complex) | Pseudo-ternary diagrams studies | D-optimal mixture | p < 0.05 | Capryol 90 (oil), Tween 80 (surfactant), and tetraglycol (co-surfactant) | Drug loading, drug release, and particle size | Minitab ver.17.0 software | The optimized SMEDDS formulation of telmisartan loaded phospholipid complex resulted in 22.17 nm of globular size, 4.06 mg/mL of solubilization, and 99.4% of drug release in 15 min. | [89] |
| Curcumin and artemisin | Pseudo-ternary diagrams studies | D-optimal mixture | p < 0.05 | Oleic acid (oil), Tween-80 (surfactant), and PEG400 (co-surfactant) | %transmittance, particle size, and polydispersity index | Design-Expert® version 10.0 | The optimized SMEDDS containing curcumin and artemisin produced 98.27% of transmittance, 150.7 nm of particle size, and 0.118 of polydispersity index. | [90] |
| Ziyuglycoside I | Solubility and pseudo-ternary diagrams studies | D-optimal mixture | p < 0.05 | Obleique CC497 (oil), Tween-20 (surfactant), and Transcutol HP (co-surfactant) | Drug loading and particle size | Design Expert version 8.0.4.1 | An enhanced solubility up to 23.93 mg/g and particle size of 207.92 ± 2.13 nm, along with an improved bioavailability (21.94%) as compared to the free drug (3.16%) | [91] |
| Insulin | Solubility and pseudo-ternary diagrams studies | D-optimal mixture | p < 0.05 | Capmul MCM (oil), Labrasol (surfactant), Tetraglycol (co-surfactant) | Particle size, stability, and leakage | Design-Expert version 11.0 | The optimized insulin in SMEDDS formulation resulted in particle size of 115.2 nm, enhanced stability up to 46.75%, and lessened leakage down to 17.67% | [92] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsieh, C.-M.; Yang, T.-L.; Putri, A.D.; Chen, C.-T. Application of Design of Experiments in the Development of Self-Microemulsifying Drug Delivery Systems. Pharmaceuticals 2023, 16, 283. https://doi.org/10.3390/ph16020283
Hsieh C-M, Yang T-L, Putri AD, Chen C-T. Application of Design of Experiments in the Development of Self-Microemulsifying Drug Delivery Systems. Pharmaceuticals. 2023; 16(2):283. https://doi.org/10.3390/ph16020283
Chicago/Turabian StyleHsieh, Chien-Ming, Ting-Lun Yang, Athika Darumas Putri, and Chin-Tin Chen. 2023. "Application of Design of Experiments in the Development of Self-Microemulsifying Drug Delivery Systems" Pharmaceuticals 16, no. 2: 283. https://doi.org/10.3390/ph16020283
APA StyleHsieh, C.-M., Yang, T.-L., Putri, A. D., & Chen, C.-T. (2023). Application of Design of Experiments in the Development of Self-Microemulsifying Drug Delivery Systems. Pharmaceuticals, 16(2), 283. https://doi.org/10.3390/ph16020283