Abstract
Anti-cancer drug design has been acknowledged as a complicated, expensive, time-consuming, and challenging task. How to reduce the research costs and speed up the development process of anti-cancer drug designs has become a challenging and urgent question for the pharmaceutical industry. Computer-aided drug design methods have played a major role in the development of cancer treatments for over three decades. Recently, artificial intelligence has emerged as a powerful and promising technology for faster, cheaper, and more effective anti-cancer drug designs. This study is a narrative review that reviews a wide range of applications of artificial intelligence-based methods in anti-cancer drug design. We further clarify the fundamental principles of these methods, along with their advantages and disadvantages. Furthermore, we collate a large number of databases, including the omics database, the epigenomics database, the chemical compound database, and drug databases. Other researchers can consider them and adapt them to their own requirements.
1. Introduction
In recent years, many companies have ramped up their R&D (research and development) efforts for anti-cancer drugs [1]. There is a growing number of large and long-term clinical trials providing a possible therapeutic opportunity for more cancer patients [2,3]. Recently, the American Cancer Society announced that the three-year survival rate for lung cancer from 2014 to 2021 was raised from 21% to almost 31% [4]. The efficacy of targeted therapies and immunotherapeutics has been investigated in a variety of solid tumors [5]. Thus, a greater investment in targeted therapies and immunotherapeutics to realize the benefits of precision medicine will benefit the long-term survival of cancer patients [6,7,8].
The anti-cancer drug design and discovery workflow comprises target recognition, hit exploration, hit-to-lead development, lead optimization, preclinical drug candidate identification, and preclinical and clinical research [9,10,11]. Despite the improvements in tumor biotechnology and the advances in cancer mechanism research, the development of novel and effective anti-cancer drugs from scratch remains an arduous, expensive, and time-consuming process [12] that will require close multidisciplinary collaborations, including medicinal chemistry, computational chemistry, biology, pharmacology, and clinical research [13]. Statistically, it can take more than 10–17 years and almost 2.8 billion dollars to bring a new drug into clinical practice [14,15]. Apart from that, only 10% of the tested compounds in clinical trials reach the market [16].
It is especially difficult to design anti-cancer drugs due to challenges such as undruggable targets [17], chemoresistance in oncology [18], tumor heterogeneity [19], and metastasis [20]. The conventional drug design approaches may seem poorly effective. With so many challenges still to be faced, the treatment effects among cancer patients are actually suboptimal. Thus, more effective anti-cancer drug design strategies are urgently needed. They will reduce the cost of drug development and the time required for clinical trials. They can also help increase the global life expectancy and improve human health [4].
Computer-aided drug design (CADD) is a method that began in the early 1980s [21]. The use of computer-aided methods to guide drug screening is emerging as an important component in the practice of drug design [22,23,24,25]. This approach enabled medicinal chemists to calculate the interactions between a ligand and receptors and to design and optimize lead compounds by computer simulation [26]. The typical role of CADD in drug design is to screen out large compound libraries into smaller clusters of predicted active compounds based on computational chemistry. It can greatly speed up the process of anti-drug design and save a huge amount of time and money [27].
In the context of the rapid development of computer hardware and artificial intelligence techniques, researchers in academia and the pharmaceutical industry are turning to artificial intelligence to improve drug design processes [28]. Artificial intelligence (AI) refers to the simulation of human intelligence in machines that are programmed to think and act like humans [15]. A common presumption about artificial intelligence is that its goal is to build machines with a similar capacity for “understanding” [29]. Artificial intelligence is now used in many applications for cancer research, such as image classification of abnormal cancer cells [30], prediction of target protein structures [31], and prediction of drug–protein interactions [32]. These studies demonstrate that artificial intelligence techniques have the power to revolutionize anti-cancer drug design processes. Some applications using artificial intelligence in anti-cancer drug design processes are illustrated in Figure 1. This paper reviewed some of the advances in anti-cancer drug design based on artificial intelligence, presented some of the most classic examples, and clarified the fundamental principles of these methods.
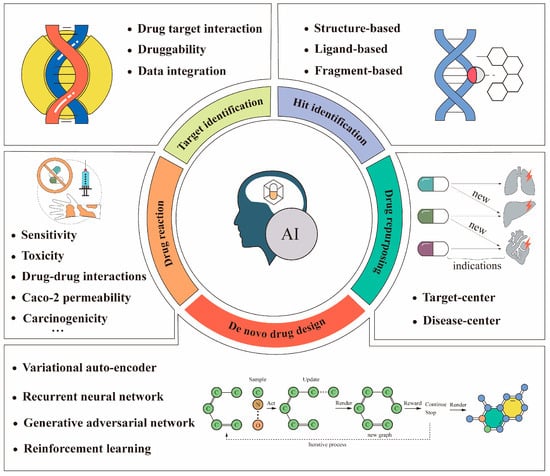
Figure 1.
Some applications of artificial intelligence in anti-cancer drug design. The bottom (de novo drug design) is usually implemented using the deep learning-based models listed above. Recently, reinforcement learning has been used often. The above workflow example of a graphical chemical structure with an O–C–O connection is an iterative chemical graph generation process [33].
2. Method
The present study is a narrative review of the literature. We performed searches in the US National Library of Medicine (PubMed) to find original articles. The search strategy used in PubMed is shown in Table 1. We mainly focused on the articles and reviews published in the past decade. The last search of the present narrative review was performed on 10 December 2022.

Table 1.
Search strategies used in the US National Library of Medicine (PubMed), according to selected descriptors.
3. Artificial Intelligence in Anti-Cancer Drug Target Identification
The identification of drug–target interactions (DTIs) is the initial step in anti-cancer drug design. The strength of drug–target binding is often described by binding affinity constants, including indicators such as a dissociation constant (Kd), an inhibition constant (Ki), and a half-maximal inhibitory concentration (IC50) [34]. Since the experimental determination of DTIs is a time-consuming and expensive process, its computational prediction is of great interest. Accurate and effective DTI predictions can greatly aid drug development and accelerate lead or hit compound discovery.
3.1. Artificial Intelligence Efficiently Elevates the Prediction Accuracy of DTI
Traditionally, the computational methods for DTI predictions have included molecular docking simulation and machine learning-based methods. However, these studies would be expensive, time-consuming, and difficult to conduct without knowing the 3D structures of the drug targets. Peng et al. developed a novel end-to-end learning framework based on heterogeneous graph convolutional networks (EEG)-DTI for DTI predictions. A graph convolutional network-based model was used to learn the low-dimensional feature representations of drugs and targets and predict the DTI based on the learned features. It achieved a promising DTI prediction performance even when the 3D structures of the drug targets were not used [35]. To further improve the prediction performance, Shao et al. considered the DTI prediction as a link prediction problem and proposed an end-to-end model based on the heterogeneous graph with attention mechanism (DTI-HETA), which outperformed the state-of-the-art models [36]. Meanwhile, to address the explanation problem of deep learning, Yang et al. proposed a drug–target interaction prediction method based on mutual learning mechanisms without 3D structural information and with explanation [37].
3.2. Artificial Intelligence Could Integrate Data from Multiple Sources to Help with Anti-Drug Target Identification
Drug target identification is a key step in drug development. However, most previous studies were confined to a single data type and did not integrate multiple data types. Thus, they were vulnerable to data-specific noise and needed to be improved in terms of practicality and accuracy [38]. Recently, there has been a growing number of methods within similarity-based or data-driven frameworks that attempt to use artificial intelligence to improve the predictive power by integrating multiple different data types. Madhukar et al. developed a Bayesian-based machine learning method (BANDIT), which achieved approximately 90% target prediction accuracy on more than 2000 small molecules by integrating six types of data, including growth inhibition data, gene expression data, adverse reaction data, chemical structure data, and drug data [39]. Olayan et al. proposed a method named DDR to investigate how to predict drug–target interactions more efficiently by using data from different sources, which included eight drug similarity networks and eight target similarity networks. The drug similarity networks included the following: gene expression similarity, disease-based similarity, drug side effect-based similarity, chemical structure fingerprint-based similarity, etc. The target similarity networks included the following: gene ontology-based similarity, protein sequence-based similarity, etc. [40]. The above studies illustrated that integrating data from multiple sources through artificial intelligence could increase the biological explanation of drug target prediction and prediction accuracy.
3.3. Artificial Intelligence Could Help Predict the Druggability of Anti-Cancer Drug Targets
The selection of drug targets is also a very critical step in the cancer drug design process, and it has a great impact on the success rate of later clinical trials. Therefore, many related methods were developed. Raies et al. proposed a prediction model called DrugnomeAI to address the problem of targeted drug synthesis. The stochastic semi-supervised machine learning framework was used to develop DrugnomeAI for predicting the druggability of drug targets in the human exome. It also demonstrated how the application of DrugnomeAI can predict the druggability of drug targets in oncology diseases [41]. In recent years, an increasing number of studies have identified synthetic lethality (SL) as a promising approach for the discovery of anticancer drug targets [42]. However, the wet experimental screening for SL has problems, including high costs, batch effects, and off-target results. Wang et al. designed a new model based on a graph neural network (GNN) called KG4SL. It incorporates knowledge graph (KG) messaging into a graph neural network prediction. The experimental results demonstrated a significant beneficial effect of incorporating KG into the GNN for SL predictions [43]. The Table 2 below lists some of the methods for anti-cancer drug target identification based on artificial intelligence that have been developed in recent years.
Table 2.
Methods for anti-cancer drug target identification based on artificial intelligence.
4. Artificial Intelligence in the Screening of Anti-Cancer Drug Hit Compounds
After the identification of therapeutic targets for anti-cancer drugs, we need to screen for anti-cancer drug hit compounds, which are molecules with initial activities against a specific target or linkage of action [49]. The discovery of computer-aided hit compounds is mainly through high-throughput screening. High-throughput screening can be performed in the following two ways: structure-based screening and ligand-based screening [50]. Fragment-based screening methods are also effective for the discovery of hit compounds, as shown in recent studies [51]. High-throughput screening techniques have been highly successful in many R&D projects, but the efficiency of screening compounds by the millions has reached a bottleneck, and the cost is also significant [52]. With the proliferation of GPUs, increased computer power, and the rapid development of artificial intelligence technologies, more virtual hit compound screening tools have been developed to enrich the drug design toolkit.
4.1. Structure-Based Screening of Hit Compounds Using Artificial Intelligence
Structure-based virtual screening uses docking and scoring to select molecules that have good binding affinity with a target protein [53]. This strategy is an important tool for anti-cancer drug design, but many of the current docking procedures are time-consuming and pose challenges for large-scale virtual screening. Lu et al. accelerated the evaluation process through structure screening with the help of deep learning models. They constructed a deep learning model to predict molecular docking scoring [54]. Yasuo et al. used artificial intelligence to propose a new structure-based virtual screening method for hit compounds, called SIEVE-Score, which provided substantial improvements over other state-of-the-art virtual screening methods [55].
4.2. Ligand-Based Screening of Hit Compounds Using Artificial Intelligence
Ligand-based screening is based on taking small molecules with known activities and searching for structures with similar physical or chemical characteristics in a compound library as candidates. Krasoulis et al. proposed an end-to-end method called DENVIS, a scalable and novel algorithm for high-throughput screening using graphical neural networks with atomic and surface protein pocket features. By conducting experiments on two benchmark databases, DENVIS was much faster than other models [56]. This method was not only advantageous in terms of speed and had an impressive success rate, but it was also easy to use. Generally, most of these methods could only receive one representative molecular structure as a search template [57], which may result in data waste. To address this problem, Hutter developed a cumulative molecular fingerprinting algorithm that can take all structure data into account in the calculation, effectively improving the utilization of experimental data and achieving an organic combination of molecular fingerprinting and experimental data. It inherited the speed advantage of the former method with higher information utilization [58].
4.3. Fragment-Based Screening of Hit Compounds Using Artificial Intelligence
In recent years, the rise of emerging technologies such as high-throughput screening (HTS) and combinatorial chemistry (CC) has led to the gradual systematization of drug discovery from the randomized screening of known drugs [59]. These methods can significantly increase the speed of drug discovery and shorten the process of new drug development, but the high cost of screening has also increased the research burden on small drug development companies and research institutions. Therefore, many researchers are focusing on fragment-based drug design (FBDD) [60]. Compared with the traditional screening methods, FBDD starts with small molecular fragments, which greatly reduces the size of the required screening compound library, circumvents the undesirable ADMET properties of molecules, and enhances the diversity of the designed structures [61]. In addition, FBDD has potential advantages for the drug design of difficult targets and has gradually developed into a mainstream drug design method in small drug development companies and research units [62]. To ligate fragments rationally, it is necessary to know where the fragments bind in a pocket. Currently, the main computational prediction methods are molecular docking, functional group mapping, and molecular structure splitting and reconstruction. These methods are more or less limited by computational costs and manual judgement and cannot fully utilize the structural data of protein–ligand complexes. To solve this problem, Didier Rognan’s group proposed the method POEM, which is based on the recognition and matching of the pocket environment in which the fragments are located [63]. Another challenge of FBDD is linking fragments to generate interest libraries of compounds for specific drug targets. To address this issue, Yang et al. proposed a model based on automatic fragment linking with deep conditional transformer neural networks called SyntaLinker [64]. Caburet et al. screened the activity of NDM-1 β-lactamase inhibitors using the FBDD method. They finally found 37 fragments for pharmacophore establishment, which was proven to be accurate and efficient. Table 3 lists all of these methods [65].
Table 3.
Artificial intelligence-based screening methods of anti-cancer drug hit compounds.
5. Artificial Intelligence in De Novo Anti-Cancer Drug Design
The chemical space of drug-like molecules is extremely vast; the number is estimated to be 1023~1060 [66]. Therefore, it is nearly impossible to completely mine the entire chemical space using computational methods. In this context, finding specific lead compounds in the vast chemical space is a major challenge. With the rapid development of computational power and experimental techniques, high-throughput screening (HTS) and virtual screening (VS) methods can effectively evaluate molecules in large compound libraries with a wide variety of filters [67,68].
However, both traditional HTS and vs. methods that are based on molecular docking can only screen the known compound library to find molecules that satisfy specific properties [69]. De novo drug design and virtual screening are very similar in the sense that they both search for molecules that meet specific requirements in the chemical space. However, their processes are very different. Instead, de novo drug design is a molecule generation method that generates and optimizes a molecule by ultimately using artificial intelligence [70]. Molecular generation methods include variational auto-encoders (VAEs), the recurrent neural network (RNN), the generative adversarial network (GAN), and deep reinforcement learning (DRL) [71].
5.1. Application of Variational Auto-Encoder to De Novo Design of Anticancer Drugs
The variational auto-encoder (VAE) is an important type of generative model that was proposed by Diederik P. Kingma and Max Welling in 2013 [72]. Born et al. constructed a hybrid VAE model to generate candidate molecules with anti-cancer drug properties. The model was able to generate molecules with strong inhibitory effects against specific diseases. The generated molecules were similar to existing drugs in terms of structure, synthesizability, and solubility [73]. Hong et al. proposed a molecular structure tree generation model in which the molecules were generated by gradually adding substructures [74]. The proposed model was based on a VAE architecture, which used an encoder to map molecules into the latent vector space and then built an autoregressive generative model as a decoder to generate new molecules from a Gaussian distribution. It showed that the model can generate efficient and new molecules and that the optimized model can effectively improve the properties of the molecules. Samanta et al. proposed the NEVAE method, which solved the problems of current methods. For instance, existing models can only generate molecules with the same number of atoms but fail to utilize a large number of macromolecules in the training process, limiting the diversity of the generated molecules. In addition, they cannot provide the spatial coordinates of the generated atoms [75].
5.2. Application of the Recurrent Neural Network to De Novo Design of Anti-Cancer Drugs
The recurrent neural network (RNN) model uses basic units, such as atoms or fragments of molecules, as the basic vocabulary and generates molecules in a temporal order. The output probability of the next atom character generated by the RNN model depends on the previous generated atom. The RNN-based model has been widely used to process time-series-related data, such as language, text, video, etc. [76]. Grisoni et al. proposed a new bidirectional RNN molecular generation model, or BIMODAL, that can be used for SMILES generation and data enhancement [77]. The model performed bidirectional molecular design by alternate learning, and the model was compared with other bidirectional RNNs. BIMODAL was promising in terms of molecular novelty, backbone diversity, and chemical and biological relevance of the generated molecules and was superior to the state-of-the-art methods [78,79]. To address problems such as the poor performance of DL on small training datasets, Krishnan et al. designed a de novo drug design method based on RNN generative models and migration learning to generate molecules with not only the desired drug-like properties but also target specificity [80]. In addition, Moret et al. combined the RNN generation model with three optimization methods, namely data augmentation, temperature sampling, and transfer learning. This method can generate new molecules with the desired properties with a small amount of data [81].
5.3. Application of Generative Adversarial Network to De Novo Design of Anti-Cancer Drugs
The generative adversarial network (GAN) is an unsupervised learning method proposed by Goodfellow in 2014. It consists of the following two networks: the generative network G, which is used to fit the data distribution, and the discriminative network D, which is used to determine whether the input is “real” or not. In the training process, the generative network G tries to “cheat” D by accepting random noise to imitate the real images in the training set, while D tries to distinguish the real data from the output of the generative network as much as possible, thus forming a game process between the two networks. Ideally, the game results in a generative model that can be “faked” [82]. Maziarka et al. proposed the Mol-Cycle GAN method. Mol-Cycle GAN is a conditional generative adversarial network-based method for de novo drug design and synthesis optimization of molecules through a generative model. It can solve the problem of difficult-to-synthesize compounds given a starting molecule. It can also generate molecules with similar structures and desired properties [83]. ABbbasi et al. proposed a feedback-based GAN framework that implemented an optimization strategy by connecting an encoder–decoder, a GAN, and a predictor depth model with a feedback loop. The results showed that molecules with high binding affinity can be generated by the GAN optimization model [84].
5.4. Application of Deep Reinforcement Learning to De Novo Design of Anti-Cancer Drugs
Even though a variety of drug generation models have been developed, they all focus on the following two points: molecular representation and optimization strategies [71]. Deep reinforcement learning (DRL) is an artificial intelligence technique that combines the perceptual capabilities of deep learning with the decision-making capabilities of reinforcement learning to solve decision-making problems in high dimensional and state spaces [85]. A novel computational strategy, called ReLeaSE, was proposed by Tropsha for designing molecules with desired properties from scratch. ReLeaSE was built on deep learning (DL) and reinforcement learning (RL) methods by integrating two deep neural networks (generative and predictive), which were trained to generate novel libraries of molecules with specified properties [86]. Goel et al. combined RNN and reinforcement learning to propose a molecule generation model named MoleGuLAR that can perform multi-objective optimization of molecules in terms of drug-like properties and binding affinity. In particular, they proposed a new alternating reward strategy where the reward function changes dynamically as different molecules are generated, allowing the model to alternately explore different chemical intervals and sample more reasonable molecules [87]. Table 4 shows some of these methods.
Table 4.
Methods for de novo anti-cancer drug design through artificial intelligence.
6. Artificial Intelligence in Anti-Cancer Drug Repurposing
Effective identification of new indications from approved or established clinical drugs plays a critical role in drug discovery. Such a process is also known as drug repositioning. Despite tremendous efforts in academic and pharmacological research worldwide, current anti-cancer therapies have achieved success in only a few tumor types. The application of drug repositioning in tumor therapies is a hot topic in current research. In theory, repurposing is faster, safer, easier, and less expensive than the known barriers to developing new molecular entities. Opportunities for drug repurposing are often based on incidental observations or time-consuming preclinical drug screens that are not usually hypothesis-driven. Indeed, the widespread use of histology technologies, improved electronic medical record systems, improved data storage, data meaning, machine learning algorithms, and computational modeling have provided unprecedented knowledge of the biological mechanisms of cancer and drug modes of action, providing broad availability of both disease-related and drug-related data. Drug repositioning strategies are often categorized as “target-center” and “disease-center” methods for predicting unknown drug–target and drug–disease interactions.
6.1. Artificial Intelligence in Anti-Drug Repositioning Based on the Interaction between a Drug and a Target
Many artificial intelligence-based methods have been used to predict drug–target relationships, as described above. At present, predicting drug–target relationships is one of the main approaches for drug repurposing. To achieve personalized drug repurposing using genomic information, Cheng et al. developed a genome-wide localization system network algorithm (GPSnet) [88]. This method uses patient-specific DNA and RNA sequencing profiles of specific targets to obtain disease modules for repurposing drugs. They validated that the approved arrhythmia and heart failure drug Ouabain specifically targets the HIF1α/LEO1-mediated cellular metabolic pathways in lung adenocarcinomas, showing potential anti-tumor activities. Wang et al. proposed a deep learning framework through kernel-based data integration, known as DeepDRK [89]. The model was trained on over 20 000 pairs of pan-cancer cell line anti-cancer drug pairs. These pairs were characterized by using kernel-based similarity matrices that integrate multi-source and multi-omics data, including genomics, transcriptomics, epigenomics, chemical properties of compounds, and known drug–target interactions. They provided a computational approach to predict cancer cell responses to drugs by integrating pharmacogenomic data, offering an alternative approach to repurposing drugs in cancer precision therapy.
6.2. Artificial Intelligence in Anti-Drug Repositioning Based on the Interaction between Drugs and Diseases
Predicting drug–disease interactions is essential for disease-centric drug repurposing. The current identification of drug–disease interactions is mainly based on similarity and network, respectively. For the similarity-based methods, Zhang et al. proposed a multiscale drug–disease topology learning framework (MTRD). By learning the representative properties of drug–disease, this method explored a new therapeutic effect of existing drugs based on the relevant similarity and association information of drug–disease node pairs. [90]. Jarada et al. proposed a novel framework based on deep learning, known as SNF-NN, to predict new drug–disease interactions using drug-related similarity information, disease-related similarity information, and known drug–disease interactions [91]. Luo et al. proposed a new computational method named MBiRW [92], which uses combined similarity measurements and a birandom walk (BiRW) algorithm to identify potential new indications for known drugs. This method was based on the assumption that similar drugs are usually associated with similar diseases. Moreover, Sadeghi et al. proposed a new model named DR-HGNN for drug repositioning using multiple labeling of heterogeneous graph neural networks [93]. Doshi et al. proposed a graph neural network-based drug repositioning model called GDRnet [94], which was able to efficiently screen the database for existing drugs and predict their unknown therapeutic effects. Table 5 shows some of the methods mentioned above.
Table 5.
Methods for anti-cancer drug repurposing based on artificial intelligence.
7. Artificial Intelligence-Assisted Accurate Prediction of Anti-Cancer Drug Reactions
Drug reactions are related to their ADMET properties, which may influence drug sensitivity, drug toxicity, and drug–drug interactions [95,96]. The accurate prediction of drug reactions can effectively increase the success rate of clinical trials and improve patient outcomes. With the rapid development of artificial intelligence technologies, more and more related studies are being proposed at the drug design stage using artificial intelligence techniques.
7.1. Artificial Intelligence Aids in Predicting the ADMET Properties of Anti-Cancer Drugs
To explore drug reactions, the ADMET properties should be accurately predicted first. Several ADMET properties, including Caco-2 permeability, carcinogenicity, blood–brain barrier permeability, and plasma protein binding, are included in previous studies. For instance, Selvaraj et al. reviewed the applications of various machine learning models, such as SVM regression and partial least squares (PLSs), for the prediction of the Caco-2 permeability coefficient [97]. Li et al. proposed a DeepCarc model to predict the carcinogenicity of small molecules using deep learning-based model-level representations [98]. Vatansever et al. reviewed the current state-of-the-art methods in AI-guided central nervous system (CNS) drug discovery, focusing on the blood–brain barrier permeability prediction [99]. To predict the plasma protein binding of a drug, Mulpuru et al. built a prediction model of a fraction of unbound drug in human plasma using a chemical fingerprint and a freely available AutoML framework [100].
7.2. Artificial Intelligence Aids in Predicting Anti-Cancer Drug Sensitivity
Anti-cancer drug sensitivity predictions are important in guiding the enrollment of those patients who may benefit from specific treatments. Chawla et al. developed a deep neural network named Precily, which uses gene expression data to predict drug sensitivity for cancer therapy. The model combines the structural properties of drugs with the pathway specificity of gene expression as features to train the model [101]. Eliseo Papa et al. built a recommendation system based on the BIKG knowledge graph to predict drug sensitivity and identified effective patient subgroups early in clinical trials [102]. Gerdes et al. proposed a model called DRUML, which uses omics data to rank over 400 drugs based on their anti-tumor cell proliferation efficacy. The results showed that DRUML can accurately rank anti-cancer drugs based on their efficacy [103].
7.3. Artificial Intelligence Aids in Predicting Toxicity of Anti-Cancer Drugs
Drug toxicity is a central issue to be considered in the drug development process. Recently, Wang et al. proposed a machine learning classifier that combines chemical structure (CS) and gene expression (GE) features. In addition, they prioritized the adverse effects of approved drugs and preclinical small-molecule compounds. The results showed that integrating GE data with drug CSs can significantly improve the predictability of adverse effects [104]. However, most of the current studies only predict the occurrence of adverse drug reactions, not their intensity or frequency. To address this issue, Zhao et al. designed a novel graphical attention model for predicting drug side effect frequency from multi-view data. The computational results showed the best performance on the benchmark dataset, illustrating effectiveness in predicting the frequency of drug side effects [105].
7.4. Artificial Intelligence Can Predict Drug–Drug Interactions
Zhu et al. proposed a unified multi-attribute discriminative representation learning (MADRL) model for DDI predictions. MADRL uses a generative adversarial network (GAN) to capture intra-attribute specificity information of DDI attributes and uses them for DDI predictions. The effectiveness of the MADRL algorithm was validated on a publicly available dataset [106]. Most methods for predicting drug–drug interactions only predict whether there is an interaction between two drugs, but it is more relevant to investigate the hidden mechanisms behind DDIs. Therefore, Zhang et al. proposed a deep learning method (DDIMDL) that used multiple drug features to predict the types of drug–drug interaction events and explored their hidden mechanisms [107]. To further increase the model’s accuracy and biological explanation, Chen et al. developed 3DGT-DDI, which consists of a 3D graph neural network and a pre-trained textual attention module. The innovation of the method is that it utilizes a 3D molecular graph structure and location information to enhance the prediction ability of DDIs. The experiments showed that the prediction performance of 3DGT-DDI outperformed other baseline models [108]. Table 6 table shows some of the methods mentioned above.
Table 6.
Methods for prediction of cancer drug reactions based on artificial intelligence.
8. Data Sources of Artificial Intelligence to Anti-Cancer Drug Designs
A large number of artificial intelligence-based algorithms, including deep learning, have become powerful tools in AI-assisted anti-cancer drug design [110,111]. Scientists are developing algorithms that can learn and analyze large amounts of data with superhuman efficiency to speed up the anti-cancer drug design process [112]. However, artificial intelligence is not universal and requires large amounts of reliable data or training experiences [113]. Nowadays, there are some specific databases for artificial intelligence-based anti-cancer drug design. They are listed in Table 7.
Table 7.
Different data sources for anti-cancer drug design.
9. Successful Cases Applying AI in Anti-Cancer Drug Design
To depict how AI facilitates the development of anticancer drugs, we list some of the anticancer drugs that have successfully entered human phase 2/3 clinical trials in the last 5 years in Table 8. For instance, Recursion identified REC-2282 as a potential candidate for the treatment of diseases caused by mutations in the NF2 gene through its proprietary AI-driven drug discovery platform, Recursion OS. REC-2282 is a permeable, orally bioavailable, small-molecule HDAC inhibitor that is being developed for the treatment of meningiomas with mutations in the NF2 gene. This molecule appears to be well tolerated, including in patients that have been administering it over several years, and different from other HDAC inhibitors in that it may reduce cardiotoxicity. It was granted both orphan drug status and fast-track status by the U.S. FDA [114]. Relay Therapeutics developed the FGFR2-specific inhibitor RLY-4008 by analyzing the dynamic balance of protein conformations through an artificial intelligence platform. Preclinical studies have shown that RLY-4008 exhibits high selectivity for FGFR2 targets in cancer cell lines, shrinking tumors with minimal impact on other targets [115]. Breg developed a new drug, BPM 31510, through an artificial intelligence platform that is currently in clinical testing. The drug restructures the metabolism of cancer cells so that patients do not have to undergo chemotherapy, allowing cancer cells to die naturally [116]. EXS-21546 is an AI-designed A2A receptor antagonist. Some tumors produce high levels of adenosine, which binds to and activates the A2A receptors on immune cells, thereby inhibiting the anti-tumor activity of the immune system [117]. PHI-101 is an orally available, selective checkpoint kinase 2 (Chk2) inhibitor designed by an AI-driven drug discovery platform [118].
Table 8.
Some of the AI-designed anti-cancer drugs that have successfully entered human phase 2/3 clinical trials in the last 5 years.
10. Conclusions and Prospects of Future Challenges
This review focuses on work that has been performed in the past decade on anti-cancer drug design based on artificial intelligence. Compared to other reviews, our study collated a large number of databases and source codes. It will offer some guidelines for other researchers to apply to their own research. This means our review has great practicality.
Artificial intelligence (AI) has strong logical reasoning and independent learning abilities that can simulate the thinking process of a human brain. AI technologies, such as machine learning, can profoundly optimize the existing anti-cancer drug research paradigm. In recent years, AI has already made unique contributions to the development and treatment of anti-cancer drugs. Artificial intelligence can accelerate the discovery of new drug molecules and the synthesis of more desirable drug molecules. This process may greatly accelerate the development of anti-cancer drugs. It is believed that artificial intelligence will be a powerful driving force for human cancer research and treatment in the future. However, AI also has several limitations, including a high dependence on data and a limited explanation. The “black box” behind traditional AI models prevents scientists from using algorithms for hypothesis validation and mining the logic behind the data. Moreover, in the drug development process, predicting the underlying logic behind a model is critical to designing the right drug molecules. In the future, interpretable AI models will be the new development direction, and the close combination of data and computation will be a feature of AI-assisted cancer drug development. We believe that AI will bring profound changes to anti-cancer drug designs.
Our study is also subject to certain limitations. For instance, we only focused on articles published in the last ten years. In addition, the search was limited to the database of PubMed. We will address these limitations in future studies.
Author Contributions
Conceptualization, L.W., K.L. and L.C.; methodology, L.W. and Y.S.; investigation, L.W., H.W. and X.Z.; resources, M.W. and J.H.; writing—original draft preparation, L.W. and L.C.; writing—review and editing, L.W., S.L. and L.Z.; supervision, K.L. and L.C.; project administration, L.C. All authors contributed to conceptualization, writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the National Natural Science Foundation of China (81973149).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Moreau, P.; Garfall, A.L.; van de Donk, N.W.C.J.; Nahi, H.; San-Miguel, J.F.; Oriol, A.; Nooka, A.K.; Martin, T.; Rosinol, L.; Chari, A.; et al. Teclistamab in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2022, 387, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Avery, R.K.; Alain, S.; Alexander, B.D.; Blumberg, E.A.; Chemaly, R.F.; Cordonnier, C.; Duarte, R.F.; Florescu, D.F.; Kamar, N.; Kumar, D.; et al. Maribavir for Refractory Cytomegalovirus Infections With or Without Resistance Post-Transplant: Results From a Phase 3 Randomized Clinical Trial. Clin. Infect. Dis. 2022, 75, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Cercek, A.; Lumish, M.; Sinopoli, J.; Weiss, J.; Shia, J.; Lamendola-Essel, M.; El Dika, I.H.; Segal, N.; Shcherba, M.; Sugarman, R.; et al. PD-1 Blockade in Mismatch Repair-Deficient, Locally Advanced Rectal Cancer. N. Engl. J. Med. 2022, 386, 2363–2376. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Spetz, M.R.; Li, D.; Ho, M. Advances in immunotherapeutic targets for childhood cancers: A focus on glypican-2 and B7-H3. Pharmacol. Ther. 2021, 223, 107892. [Google Scholar] [CrossRef]
- Corti, C.; Cobanaj, M.; Marian, F.; Dee, E.C.; Lloyd, M.R.; Marcu, S.; Dombrovschi, A.; Biondetti, G.P.; Batalini, F.; Celi, L.A.; et al. Artificial intelligence for prediction of treatment outcomes in breast cancer: Systematic review of design, reporting standards, and bias. Cancer Treat. Rev. 2022, 108, 102410. [Google Scholar] [CrossRef]
- Chiu, H.-Y.; Chao, H.-S.; Chen, Y.-M. Application of Artificial Intelligence in Lung Cancer. Cancers 2022, 14, 1370. [Google Scholar] [CrossRef]
- Hamilton, W.; Walter, F.M.; Rubin, G.; Neal, R.D. Improving early diagnosis of symptomatic cancer. Nat. Rev. Clin. Oncol. 2016, 13, 740–749. [Google Scholar] [CrossRef]
- Fountzilas, E.; Tsimberidou, A.M.; Vo, H.H.; Kurzrock, R. Clinical trial design in the era of precision medicine. Genome. Med. 2022, 14, 101. [Google Scholar] [CrossRef]
- Bannigan, P.; Aldeghi, M.; Bao, Z.; Häse, F.; Aspuru-Guzik, A.; Allen, C. Machine learning directed drug formulation development. Adv. Drug Deliv. Rev. 2021, 175, 113806. [Google Scholar] [CrossRef]
- Olgen, S. Overview on Anticancer Drug Design and Development. Curr. Med. Chem. 2018, 25, 1704–1719. [Google Scholar] [CrossRef]
- Grandori, C.; Kemp, C.J. Personalized Cancer Models for Target Discovery and Precision Medicine. Trends Cancer 2018, 4, 634–642. [Google Scholar] [CrossRef]
- Hoelder, S.; Clarke, P.A.; Workman, P. Discovery of small molecule cancer drugs: Successes, challenges and opportunities. Mol. Oncol. 2012, 6, 155–176. [Google Scholar] [CrossRef]
- Schneider, P.; Walters, W.P.; Plowright, A.T.; Sieroka, N.; Listgarten, J.; Goodnow, R.A.; Fisher, J.; Jansen, J.M.; Duca, J.S.; Rush, T.S.; et al. Rethinking drug design in the artificial intelligence era. Nat. Rev. Drug Discov. 2020, 19, 353–364. [Google Scholar] [CrossRef]
- Pandiyan, S.; Wang, L. A comprehensive review on recent approaches for cancer drug discovery associated with artificial intelligence. Comput. Biol. Med. 2022, 150, 106140. [Google Scholar] [CrossRef]
- Wouters, O.J.; McKee, M.; Luyten, J. Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009–2018. JAMA 2020, 323, 844–853. [Google Scholar] [CrossRef]
- Dang, C.V.; Reddy, E.P.; Shokat, K.M.; Soucek, L. Drugging the ‘undruggable’ cancer targets. Nat. Rev. Cancer 2017, 17, 502–508. [Google Scholar] [CrossRef]
- Bleker de Oliveira, M.; Koshkin, V.; Liu, G.; Krylov, S.N. Analytical Challenges in Development of Chemoresistance Predictors for Precision Oncology. Anal. Chem. 2020, 92, 12101–12110. [Google Scholar] [CrossRef]
- Dentro, S.C.; Leshchiner, I.; Haase, K.; Tarabichi, M.; Wintersinger, J.; Deshwar, A.G.; Yu, K.; Rubanova, Y.; Macintyre, G.; Demeulemeester, J.; et al. Characterizing genetic intra-tumor heterogeneity across 2658 human cancer genomes. Cell 2021, 184. [Google Scholar] [CrossRef]
- Suhail, Y.; Cain, M.P.; Vanaja, K.; Kurywchak, P.A.; Levchenko, A.; Kalluri, R.; Kshitiz. Systems Biology of Cancer Metastasis. Cell Syst. 2019, 9, 109–127. [Google Scholar] [CrossRef]
- Fleming, N. How artificial intelligence is changing drug discovery. Nature 2018, 557, S55–S57. [Google Scholar] [CrossRef] [PubMed]
- Boniolo, F.; Dorigatti, E.; Ohnmacht, A.J.; Saur, D.; Schubert, B.; Menden, M.P. Artificial intelligence in early drug discovery enabling precision medicine. Expert. Opin. Drug Discov. 2021, 16. [Google Scholar] [CrossRef] [PubMed]
- Huwaimel, B.; Alobaida, A. Anti-Cancer Drug Solubility Development within a Green Solvent: Design of Novel and Robust Mathematical Models Based on Artificial Intelligence. Molecules 2022, 27, 5140. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, Y.; Byrne, R.; Schneider, G.; Yang, S. Concepts of Artificial Intelligence for Computer-Assisted Drug Discovery. Chem. Rev. 2019, 119, 10520–10594. [Google Scholar] [CrossRef] [PubMed]
- Kapetanovic, I.M. Computer-aided drug discovery and development (CADDD): In silico-chemico-biological approach. Chem. Biol. Interact. 2008, 171, 165–176. [Google Scholar] [CrossRef]
- Hansen, S.G.; Wu, H.L.; Burwitz, B.J.; Hughes, C.M.; Hammond, K.B.; Ventura, A.B.; Reed, J.S.; Gilbride, R.M.; Ainslie, E.; Morrow, D.W.; et al. Broadly targeted CD8⁺ T cell responses restricted by major histocompatibility complex E. Science 2016, 351, 714–720. [Google Scholar] [CrossRef]
- Campos, K.R.; Coleman, P.J.; Alvarez, J.C.; Dreher, S.D.; Garbaccio, R.M.; Terrett, N.K.; Tillyer, R.D.; Truppo, M.D.; Parmee, E.R. The importance of synthetic chemistry in the pharmaceutical industry. Science 2019, 363. [Google Scholar] [CrossRef]
- Woo, M. An AI boost for clinical trials. Nature 2019, 573, S100–S102. [Google Scholar] [CrossRef]
- Zwicker, M. Understanding spatial environments from images. Science 2018, 360, 1188. [Google Scholar] [CrossRef]
- Cao, L.; Yang, J.; Rong, Z.; Li, L.; Xia, B.; You, C.; Lou, G.; Jiang, L.; Du, C.; Meng, H.; et al. A novel attention-guided convolutional network for the detection of abnormal cervical cells in cervical cancer screening. Med. Image Anal. 2021, 73, 102197. [Google Scholar] [CrossRef]
- Abriata, L.A.; Dal Peraro, M. State-of-the-art web services for de novo protein structure prediction. Brief. Bioinform. 2021, 22. [Google Scholar] [CrossRef]
- Xuan, P.; Zhang, Y.; Cui, H.; Zhang, T.; Guo, M.; Nakaguchi, T. Integrating multi-scale neighbouring topologies and cross-modal similarities for drug-protein interaction prediction. Brief. Bioinform. 2021, 22. [Google Scholar] [CrossRef]
- You, J.; Liu, B.; Ying, Z.; Pande, V.; Leskovec, J. Graph convolutional policy network for goal-directed molecular graph generation. Adv. Neural Inform. Process. Syst. 2018, 31. [Google Scholar] [CrossRef]
- Ghimire, A.; Tayara, H.; Xuan, Z.; Chong, K.T. CSatDTA: Prediction of Drug-Target Binding Affinity Using Convolution Model with Self-Attention. Int. J. Mol. Sci. 2022, 23, 8453. [Google Scholar] [CrossRef]
- Peng, J.; Wang, Y.; Guan, J.; Li, J.; Han, R.; Hao, J.; Wei, Z.; Shang, X. An end-to-end heterogeneous graph representation learning-based framework for drug-target interaction prediction. Brief. Bioinform. 2021, 22, bbaa430. [Google Scholar] [CrossRef]
- Shao, K.; Zhang, Y.; Wen, Y.; Zhang, Z.; He, S.; Bo, X. DTI-HETA: Prediction of drug-target interactions based on GCN and GAT on heterogeneous graph. Brief. Bioinform. 2022, 23. [Google Scholar] [CrossRef]
- Yang, Z.; Zhong, W.; Zhao, L.; Chen, C.Y.-C. ML-DTI: Mutual Learning Mechanism for Interpretable Drug-Target Interaction Prediction. J. Phys. Chem. Lett. 2021, 12, 4247–4261. [Google Scholar] [CrossRef]
- Dara, S.; Dhamercherla, S.; Jadav, S.S.; Babu, C.M.; Ahsan, M.J. Machine Learning in Drug Discovery: A Review. Artif. Intell. Rev. 2022, 55, 1947–1999. [Google Scholar] [CrossRef]
- Madhukar, N.S.; Khade, P.K.; Huang, L.; Gayvert, K.; Galletti, G.; Stogniew, M.; Allen, J.E.; Giannakakou, P.; Elemento, O. A Bayesian machine learning approach for drug target identification using diverse data types. Nat. Commun. 2019, 10, 5221. [Google Scholar] [CrossRef]
- Olayan, R.S.; Ashoor, H.; Bajic, V.B. DDR: Efficient computational method to predict drug-target interactions using graph mining and machine learning approaches. Bioinformatics 2018, 34, 1164–1173. [Google Scholar] [CrossRef]
- Raies, A.; Tulodziecka, E.; Stainer, J.; Middleton, L.; Dhindsa, R.S.; Hill, P.; Engkvist, O.; Harper, A.R.; Petrovski, S.; Vitsios, D. DrugnomeAI is an ensemble machine-learning framework for predicting druggability of candidate drug targets. Commun. Biol. 2022, 5, 1291. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Garraway, L.A.; Ashworth, A.; Weber, B. Synthetic lethality as an engine for cancer drug target discovery. Nat. Rev. Drug Discov. 2020, 19, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xu, F.; Li, Y.; Wang, J.; Zhang, K.; Liu, Y.; Wu, M.; Zheng, J. KG4SL: Knowledge graph neural network for synthetic lethality prediction in human cancers. Bioinformatics 2021, 37, i418–i425. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zhao, X.; Zhou, J.; Yang, J.; Zhang, Y.; Kuang, W.; Peng, J.; Chen, L.; Zeng, J. A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information. Nat. Commun. 2017, 8, 573. [Google Scholar] [CrossRef]
- Yamanishi, Y.; Araki, M.; Gutteridge, A.; Honda, W.; Kanehisa, M. Prediction of drug-target interaction networks from the integration of chemical and genomic spaces. Bioinformatics 2008, 24, i232–i240. [Google Scholar] [CrossRef]
- Tang, J.; Szwajda, A.; Shakyawar, S.; Xu, T.; Hintsanen, P.; Wennerberg, K.; Aittokallio, T. Making sense of large-scale kinase inhibitor bioactivity data sets: A comparative and integrative analysis. J. Chem. Inf. Model 2014, 54, 735–743. [Google Scholar] [CrossRef]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef]
- Metz, J.T.; Johnson, E.F.; Soni, N.B.; Merta, P.J.; Kifle, L.; Hajduk, P.J. Navigating the kinome. Nat. Chem. Biol. 2011, 7, 200–202. [Google Scholar] [CrossRef]
- Chen, H.; Engkvist, O. Has Drug Design Augmented by Artificial Intelligence Become a Reality? Trends Pharmacol. Sci. 2019, 40, 806–809. [Google Scholar] [CrossRef]
- Lu, S.-H.; Wu, J.W.; Liu, H.-L.; Zhao, J.-H.; Liu, K.-T.; Chuang, C.-K.; Lin, H.-Y.; Tsai, W.-B.; Ho, Y. The discovery of potential acetylcholinesterase inhibitors: A combination of pharmacophore modeling, virtual screening, and molecular docking studies. J. Biomed. Sci. 2011, 18, 8. [Google Scholar] [CrossRef]
- Paricharak, S.; Méndez-Lucio, O.; Chavan Ravindranath, A.; Bender, A.; Ijzerman, A.P.; van Westen, G.J.P. Data-driven approaches used for compound library design, hit triage and bioactivity modeling in high-throughput screening. Brief. Bioinform. 2018, 19, 277–285. [Google Scholar] [CrossRef]
- Blay, V.; Tolani, B.; Ho, S.P.; Arkin, M.R. High-Throughput Screening: Today’s biochemical and cell-based approaches. Drug Discov. Today 2020, 25, 1807–1821. [Google Scholar] [CrossRef]
- Sheng, C.; Dong, G.; Miao, Z.; Zhang, W.; Wang, W. State-of-the-art strategies for targeting protein-protein interactions by small-molecule inhibitors. Chem. Soc. Rev. 2015, 44, 8238–8259. [Google Scholar] [CrossRef]
- Lu, C.; Liu, S.; Shi, W.; Yu, J.; Zhou, Z.; Zhang, X.; Lu, X.; Cai, F.; Xia, N.; Wang, Y. Systemic evolutionary chemical space exploration for drug discovery. J. Cheminform. 2022, 14, 19. [Google Scholar] [CrossRef]
- Yasuo, N.; Sekijima, M. Improved Method of Structure-Based Virtual Screening via Interaction-Energy-Based Learning. J. Chem. Inf. Model. 2019, 59, 1050–1061. [Google Scholar] [CrossRef]
- Krasoulis, A.; Antonopoulos, N.; Pitsikalis, V.; Theodorakis, S. DENVIS: Scalable and High-Throughput Virtual Screening Using Graph Neural Networks with Atomic and Surface Protein Pocket Features. J. Chem. Inf. Model. 2022, 62, 4642–4659. [Google Scholar] [CrossRef]
- Li, B.-H.; Ge, J.-Q.; Wang, Y.-L.; Wang, L.-J.; Zhang, Q.; Bian, C. Ligand-Based and Docking-Based Virtual Screening of MDM2 Inhibitors as Potent Anticancer Agents. Comput. Math. Methods Med. 2021, 2021, 3195957. [Google Scholar] [CrossRef]
- Hutter, M.C. Differential Multimolecule Fingerprint for Similarity Search─Making Use of Active and Inactive Compound Sets in Virtual Screening. J. Chem. Inf. Model. 2022, 62, 2726–2736. [Google Scholar] [CrossRef]
- Liu, R.; Li, X.; Lam, K.S. Combinatorial chemistry in drug discovery. Curr. Opin. Chem. Biol. 2017, 38, 117–126. [Google Scholar] [CrossRef]
- Wang, Z.-Z.; Shi, X.-X.; Huang, G.-Y.; Hao, G.-F.; Yang, G.-F. Fragment-based drug design facilitates selective kinase inhibitor discovery. Trends Pharmacol. Sci. 2021, 42, 551–565. [Google Scholar] [CrossRef]
- Erlanson, D.A. Introduction to fragment-based drug discovery. Top. Curr. Chem. 2012, 317. [Google Scholar] [CrossRef]
- Bon, M.; Bilsland, A.; Bower, J.; McAulay, K. Fragment-based drug discovery-the importance of high-quality molecule libraries. Mol. Oncol. 2022, 16, 3761–3777. [Google Scholar] [CrossRef] [PubMed]
- Eguida, M.; Schmitt-Valencia, C.; Hibert, M.; Villa, P.; Rognan, D. Target-Focused Library Design by Pocket-Applied Computer Vision and Fragment Deep Generative Linking. J. Med. Chem. 2022, 65, 13771–13783. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zheng, S.; Su, S.; Zhao, C.; Xu, J.; Chen, H. SyntaLinker: Automatic fragment linking with deep conditional transformer neural networks. Chem. Sci. 2020, 11, 8312–8322. [Google Scholar] [CrossRef]
- Caburet, J.; Boucherle, B.; Bourdillon, S.; Simoncelli, G.; Verdirosa, F.; Docquier, J.-D.; Moreau, Y.; Krimm, I.; Crouzy, S.; Peuchmaur, M. A fragment-based drug discovery strategy applied to the identification of NDM-1 β-lactamase inhibitors. Eur. J. Med. Chem. 2022, 240, 114599. [Google Scholar] [CrossRef]
- Yang, T.; Li, Z.; Chen, Y.; Feng, D.; Wang, G.; Fu, Z.; Ding, X.; Tan, X.; Zhao, J.; Luo, X.; et al. DrugSpaceX: A large screenable and synthetically tractable database extending drug space. Nucleic. Acids Res. 2021, 49, D1170–D1178. [Google Scholar] [CrossRef]
- Vamathevan, J.; Clark, D.; Czodrowski, P.; Dunham, I.; Ferran, E.; Lee, G.; Li, B.; Madabhushi, A.; Shah, P.; Spitzer, M.; et al. Applications of machine learning in drug discovery and development. Nat. Rev. Drug. Discov. 2019, 18, 463–477. [Google Scholar] [CrossRef]
- Paul, D.; Sanap, G.; Shenoy, S.; Kalyane, D.; Kalia, K.; Tekade, R.K. Artificial intelligence in drug discovery and development. Drug. Discov. Today 2021, 26, 80–93. [Google Scholar] [CrossRef]
- Henson, A.B.; Gromski, P.S.; Cronin, L. Designing Algorithms To Aid Discovery by Chemical Robots. ACS Cent. Sci. 2018, 4, 793–804. [Google Scholar] [CrossRef]
- Mouchlis, V.D.; Afantitis, A.; Serra, A.; Fratello, M.; Papadiamantis, A.G.; Aidinis, V.; Lynch, I.; Greco, D.; Melagraki, G. Advances in de Novo Drug Design: From Conventional to Machine Learning Methods. Int. J. Mol. Sci. 2021, 22, 1676. [Google Scholar] [CrossRef]
- Tong, X.; Liu, X.; Tan, X.; Li, X.; Jiang, J.; Xiong, Z.; Xu, T.; Jiang, H.; Qiao, N.; Zheng, M. Generative Models for De Novo Drug Design. J. Med. Chem. 2021, 64, 14011–14027. [Google Scholar] [CrossRef]
- Kingma, D.P.; Welling, M. Auto-encoding variational bayes. arXiv 2013. [Google Scholar] [CrossRef]
- Born, J.; Manica, M.; Oskooei, A.; Cadow, J.; Markert, G.; Rodríguez Martínez, M. PaccMann: De novo generation of hit-like anticancer molecules from transcriptomic data via reinforcement learning. iScience 2021, 24, 102269. [Google Scholar] [CrossRef]
- Hong, S.H.; Ryu, S.; Lim, J.; Kim, W.Y. Molecular Generative Model Based on an Adversarially Regularized Autoencoder. J. Chem. Inf. Model. 2020, 60, 29–36. [Google Scholar] [CrossRef]
- Samanta, B.; De, A.; Jana, G.; Gómez, V.; Chattaraj, P.K.; Ganguly, N.; Gomez-Rodriguez, M. Nevae: A deep generative model for molecular graphs. J. Mach. Learn. Res. 2020, 21, 1–33. [Google Scholar] [CrossRef]
- Kriegeskorte, N.; Golan, T. Neural network models and deep learning. Curr. Biol. 2019, 29, R231–R236. [Google Scholar] [CrossRef]
- Grisoni, F.; Moret, M.; Lingwood, R.; Schneider, G. Bidirectional Molecule Generation with Recurrent Neural Networks. J. Chem. Inf. Model. 2020, 60, 1175–1183. [Google Scholar] [CrossRef]
- Mou, L.; Yan, R.; Li, G.; Zhang, L.; Jin, Z. Backward and forward language modeling for constrained sentence generation. arXiv 2015. [Google Scholar] [CrossRef]
- Berglund, M.; Raiko, T.; Honkala, M.; Kärkkäinen, L.; Vetek, A.; Karhunen, J.T. Bidirectional recurrent neural networks as generative models. Adv. Neural Inform. Process. Syst. 2015, 28. [Google Scholar] [CrossRef]
- Krishnan, S.R.; Bung, N.; Bulusu, G.; Roy, A. Accelerating Drug Design against Novel Proteins Using Deep Learning. J. Chem. Inf. Model. 2021, 61, 621–630. [Google Scholar] [CrossRef]
- Moret, M.; Friedrich, L.; Grisoni, F.; Merk, D.; Schneider, G. Generative molecular design in low data regimes. Nature Mach. Intell. 2020, 2, 171–180. [Google Scholar] [CrossRef]
- Goodfellow, I.; Pouget-Abadie, J.; Mirza, M.; Xu, B.; Warde-Farley, D.; Ozair, S.; Courville, A.; Bengio, Y. Generative adversarial networks. Commun. ACM 2020, 63, 139–144. [Google Scholar] [CrossRef]
- Maziarka, Ł.; Pocha, A.; Kaczmarczyk, J.; Rataj, K.; Danel, T.; Warchoł, M. Mol-CycleGAN: A generative model for molecular optimization. J. Cheminform. 2020, 12, 2. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, M.; Santos, B.P.; Pereira, T.C.; Sofia, R.; Monteiro, N.R.C.; Simões, C.J.V.; Brito, R.M.M.; Ribeiro, B.; Oliveira, J.L.; Arrais, J.P. Designing optimized drug candidates with Generative Adversarial Network. J. Cheminform. 2022, 14, 40. [Google Scholar] [CrossRef] [PubMed]
- Arulkumaran, K.; Deisenroth, M.P.; Brundage, M.; Bharath, A.A. Deep reinforcement learning: A brief survey. IEEE Signal Process. Magazine 2017, 34, 26–38. [Google Scholar] [CrossRef]
- Popova, M.; Isayev, O.; Tropsha, A. Deep reinforcement learning for de novo drug design. Sci. Adv. 2018, 4, eaap7885. [Google Scholar] [CrossRef]
- Goel, M.; Raghunathan, S.; Laghuvarapu, S.; Priyakumar, U.D. MoleGuLAR: Molecule Generation Using Reinforcement Learning with Alternating Rewards. J. Chem. Inf. Model. 2021, 61, 5815–5826. [Google Scholar] [CrossRef]
- Cheng, F.; Lu, W.; Liu, C.; Fang, J.; Hou, Y.; Handy, D.E.; Wang, R.; Zhao, Y.; Yang, Y.; Huang, J.; et al. A genome-wide positioning systems network algorithm for in silico drug repurposing. Nat. Commun. 2019, 10, 3476. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Y.; Chen, S.; Wang, J. DeepDRK: A deep learning framework for drug repurposing through kernel-based multi-omics integration. Brief. Bioinform. 2021, 22, bbab048. [Google Scholar] [CrossRef]
- Zhang, H.; Cui, H.; Zhang, T.; Cao, Y.; Xuan, P. Learning multi-scale heterogenous network topologies and various pairwise attributes for drug-disease association prediction. Brief. Bioinform. 2022, 23, bbac009. [Google Scholar] [CrossRef]
- Jarada, T.N.; Rokne, J.G.; Alhajj, R. SNF-NN: Computational method to predict drug-disease interactions using similarity network fusion and neural networks. BMC Bioinform. 2021, 22, 28. [Google Scholar] [CrossRef]
- Luo, H.; Wang, J.; Li, M.; Luo, J.; Peng, X.; Wu, F.-X.; Pan, Y. Drug repositioning based on comprehensive similarity measures and Bi-Random walk algorithm. Bioinformatics 2016, 32, 2664–2671. [Google Scholar] [CrossRef]
- Sadeghi, S.; Lu, J.; Ngom, A. An Integrative Heterogeneous Graph Neural Network-Based Method for Multi-Labeled Drug Repurposing. Front. Pharmacol. 2022, 13, 908549. [Google Scholar] [CrossRef]
- Doshi, S.; Chepuri, S.P. A computational approach to drug repurposing using graph neural networks. Comput. Biol. Med. 2022, 150, 105992. [Google Scholar] [CrossRef]
- Hodgson, J. ADMET--turning chemicals into drugs. Nat. Biotechnol. 2001, 19, 722–726. [Google Scholar] [CrossRef]
- Niu, J.; Straubinger, R.M.; Mager, D.E. Pharmacodynamic Drug-Drug Interactions. Clin. Pharmacol. Ther. 2019, 105, 1395–1406. [Google Scholar] [CrossRef]
- Selvaraj, C.; Chandra, I.; Singh, S.K. Artificial intelligence and machine learning approaches for drug design: Challenges and opportunities for the pharmaceutical industries. Mol. Divers 2022, 26, 1893–1913. [Google Scholar] [CrossRef]
- Li, T.; Tong, W.; Roberts, R.; Liu, Z.; Thakkar, S. DeepCarc: Deep Learning-Powered Carcinogenicity Prediction Using Model-Level Representation. Front. Artif. Intell. 2021, 4, 757780. [Google Scholar] [CrossRef]
- Vatansever, S.; Schlessinger, A.; Wacker, D.; Kaniskan, H.Ü.; Jin, J.; Zhou, M.-M.; Zhang, B. Artificial intelligence and machine learning-aided drug discovery in central nervous system diseases: State-of-the-arts and future directions. Med. Res. Rev. 2021, 41, 1427–1473. [Google Scholar] [CrossRef]
- Mulpuru, V.; Mishra, N. In Silico Prediction of Fraction Unbound in Human Plasma from Chemical Fingerprint Using Automated Machine Learning. ACS Omega 2021, 6, 6791–6797. [Google Scholar] [CrossRef]
- Chawla, S.; Rockstroh, A.; Lehman, M.; Ratther, E.; Jain, A.; Anand, A.; Gupta, A.; Bhattacharya, N.; Poonia, S.; Rai, P.; et al. Gene expression based inference of cancer drug sensitivity. Nat. Commun. 2022, 13, 5680. [Google Scholar] [CrossRef] [PubMed]
- Gogleva, A.; Polychronopoulos, D.; Pfeifer, M.; Poroshin, V.; Ughetto, M.; Martin, M.J.; Thorpe, H.; Bornot, A.; Smith, P.D.; Sidders, B.; et al. Knowledge graph-based recommendation framework identifies drivers of resistance in EGFR mutant non-small cell lung cancer. Nat. Commun. 2022, 13, 1667. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, H.; Casado, P.; Dokal, A.; Hijazi, M.; Akhtar, N.; Osuntola, R.; Rajeeve, V.; Fitzgibbon, J.; Travers, J.; Britton, D.; et al. Drug ranking using machine learning systematically predicts the efficacy of anti-cancer drugs. Nat. Commun. 2021, 12, 1850. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Clark, N.R.; Ma’ayan, A. Drug-induced adverse events prediction with the LINCS L1000 data. Bioinformatics 2016, 32, 2338–2345. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zheng, K.; Li, Y.; Wang, J. A novel graph attention model for predicting frequencies of drug-side effects from multi-view data. Brief. Bioinform. 2021, 22, bbab239. [Google Scholar] [CrossRef]
- Zhu, J.; Liu, Y.; Zhang, Y.; Chen, Z.; Wu, X. Multi-Attribute Discriminative Representation Learning for Prediction of Adverse Drug-Drug Interaction. IEEE Trans. Pattern Anal. Mach. Intell. 2022, 44, 10129–10144. [Google Scholar] [CrossRef]
- Deng, Y.; Xu, X.; Qiu, Y.; Xia, J.; Zhang, W.; Liu, S. A multimodal deep learning framework for predicting drug-drug interaction events. Bioinformatics 2020, 36, 4316–4322. [Google Scholar] [CrossRef]
- He, H.; Chen, G.; Yu-Chian Chen, C. 3DGT-DDI: 3D graph and text based neural network for drug-drug interaction prediction. Brief Bioinform. 2022, 23, bbac134. [Google Scholar] [CrossRef]
- Wunnava, S.; Qin, X.; Kakar, T.; Sen, C.; Rundensteiner, E.A.; Kong, X. Adverse Drug Event Detection from Electronic Health Records Using Hierarchical Recurrent Neural Networks with Dual-Level Embedding. Drug Saf. 2019, 42, 113–122. [Google Scholar] [CrossRef]
- Zhang, T.; Leng, J.; Liu, Y. Deep learning for drug-drug interaction extraction from the literature: A review. Brief. Bioinform. 2020, 21, 1609–1627. [Google Scholar] [CrossRef]
- Basile, A.O.; Yahi, A.; Tatonetti, N.P. Artificial Intelligence for Drug Toxicity and Safety. Trends Pharmacol. Sci. 2019, 40, 624–635. [Google Scholar] [CrossRef]
- Jing, Y.; Bian, Y.; Hu, Z.; Wang, L.; Xie, X.-Q. Deep Learning for Drug Design: An Artificial Intelligence Paradigm for Drug Discovery in the Big Data Era. AAPS J. 2018, 20, 58. [Google Scholar] [CrossRef]
- Harari, Y.N. Reboot for the AI revolution. Nature 2017, 550, 324–327. [Google Scholar] [CrossRef]
- Welling, D.B.; Collier, K.A.; Burns, S.S.; Oblinger, J.L.; Shu, E.; Miles-Markley, B.A.; Hofmeister, C.C.; Makary, M.S.; Slone, H.W.; Blakeley, J.O.; et al. Early phase clinical studies of AR-42, a histone deacetylase inhibitor, for neurofibromatosis type 2-associated vestibular schwannomas and meningiomas. Laryngoscope Investig. Otolaryngol. 2021, 6, 1008–1019. [Google Scholar] [CrossRef]
- Casaletto, J.; Maglic, D.; Toure, B.B.; Taylor, A.; Schoenherr, H.; Hudson, B.; Bruderek, K.; Zhao, S.; O’Hearn, P.; Gerami-Moayed, N. RLY-4008, a novel precision therapy for FGFR2-driven cancers designed to potently and selectively inhibit FGFR2 and FGFR2 resistance mutations. Cancer Res. 2021, 81, 1455. [Google Scholar] [CrossRef]
- Sun, J.; Patel, C.B.; Jang, T.; Merchant, M.; Chen, C.; Kazerounian, S.; Diers, A.R.; Kiebish, M.A.; Vishnudas, V.K.; Gesta, S.; et al. High levels of ubidecarenone (oxidized CoQ10) delivered using a drug-lipid conjugate nanodispersion (BPM31510) differentially affect redox status and growth in malignant glioma versus non-tumor cells. Sci. Rep. 2020, 10, 13899. [Google Scholar] [CrossRef]
- Vladimer, G.; Alt, I.; Sehlke, R.; Lobley, A.; Baumgärtler, C.; Stulic, M.; Hackner, K.; Dzurillova, L.; Petru, E.; Hadjari, L. 23P Enriching for response: Patient selection criteria for A2AR inhibition by EXS-21546 through ex vivo modelling in primary patient material. Immuno-Oncol. Technol. 2022, 16, 100128. [Google Scholar] [CrossRef]
- Park, S.J.; Chang, S.-J.; Suh, D.H.; Kong, T.W.; Song, H.; Kim, T.H.; Kim, J.-W.; Kim, H.S.; Lee, S.-J. A phase IA dose-escalation study of PHI-101, a new checkpoint kinase 2 inhibitor, for platinum-resistant recurrent ovarian cancer. BMC Cancer 2022, 22, 28. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).