1. Introduction
Ischemic cerebral stroke is due to an interruption in cerebral blood flow, which is induced by thrombosis or embolism. There was an increased incidence of ischemic cerebral stroke with increasing age worldwide, and the treatment cost of ischemic cerebral stroke is the largest economic burden in East Asia [
1]. Restoring blood flow is essential for the treatment of ischemic cerebral stroke, but reperfusion itself may lead to additional neurological function damage and the formation of cerebral infarction, called cerebral ischemia/reperfusion (I/R) injury [
2]. Protecting the brain from a cerebral I/R injury is crucial for the treatment of cerebral ischemic stroke. Antithrombotic therapies, including antiplatelet or anticoagulant agents, are recommended for nearly all ischemic stroke patients with no contraindication [
3,
4]. However, pharmacological approaches against ischemic cerebral stroke remain limited, suggesting the need for new treatments.
Hydrogen sulfide (H
2S), a novel gaseous signaling molecule, is known as a gasotransmitter in mammals in addition to nitric oxide and carbon monoxide [
5]. H
2S is endogenously synthesized by enzyme cystathionine-γ-lyase, cystathionine-β-synthase, or 3-mercaptopiruvate sulfurtransferases and is involved in the physiological function and pathological process in the brain [
6,
7]. H
2S exerts an important neuroprotective effect on the cerebral I/R injury via antioxidant, anti-inflammatory, and anti-apoptotic actions [
8], but its precise mechanism is still unclear.
RhoA, a small GTPase of the Rho family, is involved in the regulation of multiple cellular signal transduction pathways [
9]. RhoA and its downstream effector Rho kinase (ROCK) is highly expressed in the nervous system and associated with various neuronal functions and numerous central nervous system diseases [
10]. The RhoA-ROCK pathway participates in astrocyte-mediated angiogenesis and neurogenesis; inhibition of the RhoA-ROCK pathway can alleviate neuroinflammation, apoptosis, and oxidative stress, which are beneficial to neural recovery after an ischemic stroke [
11]. We previously found that exogenous and endogenous H
2S had vascular and neuroprotective effects on cerebral I/R-induced dysfunction in mice and rats via the inhibition of the RhoA/ROCK pathway [
12,
13,
14,
15,
16,
17]. It was reported that exogenous and endothelial H
2S protects rat hippocampal neurons against hypoxia/reoxygenation (H/R) injury by promoting the phosphorylation of RhoA at the Ser188 site [
18]. However, there is little research on the effect of H
2S on ROCK and its underlying mechanism.
ROCK has two types of isoforms, ROCK
1 and ROCK
2, which are identified in cells. ROCK
1 is mainly expressed in non-neuronal tissues, while ROCK
2 is more abundantly distributed in the brain and skeletal muscles [
19]. A previous study demonstrated that the upregulation of ROCK
2 aggravated H/R injury in the nerve cells but the downregulation of ROCK
2 improved the H/R injury [
20]. Our recent studies demonstrated that H
2S promotes the phosphorylation of ROCK
2 at Tyr722 and inhibits ROCK
2 activity to protect rat hippocampal neurons from H/R injury [
21]. However, as a macromolecular protein with more than 1300 amino acid residues, ROCK
2 might have multiple dominant sites that can be phosphorylated and affect its activation. Therefore, the present study was designed to investigate the regulation of H
2S on the phosphorylation of ROCK
2 at other potential sites except Tyr722 and to explore whether this regulation mediates the activation of ROCK
2 and the neuroprotective effect of H
2S against H/R injury in rat hippocampal neurons (RHNs).
3. Discussion
The present study focused on the roles of ROCK2 phosphorylation at related sites in the inhibitory effects of H2S on ROCK2 activation and against H/R injury in RHNs. The main findings include: (1) H2S inhibits the phosphorylation of ROCK2 at the Thr436 and Ser575 sites in vitro; (2) the Thr436 and Ser575 sites of ROCK2 mediate the inhibition of H2S on ROCK2 activation in RHNs; and (3) Thr436 and Ser575 of ROCK2 participate in the protective effect of H2S against an H/R injury via attenuating [Ca2+]i in RHNs.
Phosphorylation refers to the transfer of phosphorylation of adenosine triphosphate (ATP) or GTP at the γ position to amino acid residues of substrate protein catalyzed by protein kinase. Phosphorylation regulation mainly occurs on serine, threonine, and tyrosine [
22]. In the process of phosphorylation modification, the strongly negatively charged phosphate group is transferred to the specific amino acid of the protein, which will cause the overall configuration of the protein to change and the activity of the protein and the interaction between other molecules will also change. Phosphorylation plays different roles in the regulation of cellular physiological processes, such as signal transduction, gene expression, cell division, etc. [
23,
24,
25]. Therefore, the identification and analysis of the phosphorylation of related sites in protein are helpful to find novel therapeutic targets for some diseases. In order to explore the effect of H
2S on the phosphorylation of ROCK
2 at some sites, prokaryotic plasmid ROCK
2wild-pET-32a(+) was constructed and transfected into
E. colis to express His-ROCK
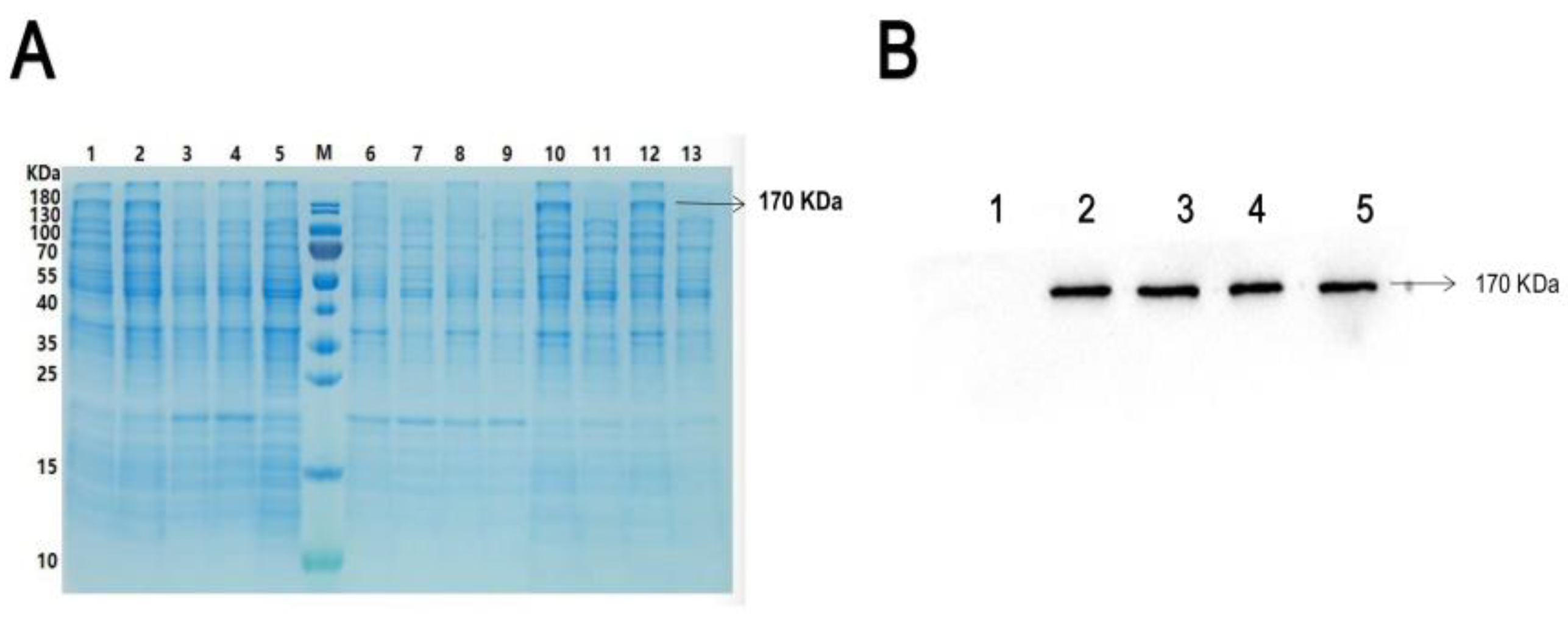
2wild protein in the present study. By using Coomassie brilliant blue staining and Western blot assays, His-ROCK
2wild protein was detected in the lysate of the
E. colis and the lysate precipitation but not in the supernatant, suggesting that His-ROCK
2wild protein exists in an insoluble form, which may be because ROCK
2 is a macromolecular protein composed of more than 1300 amino acid residues.
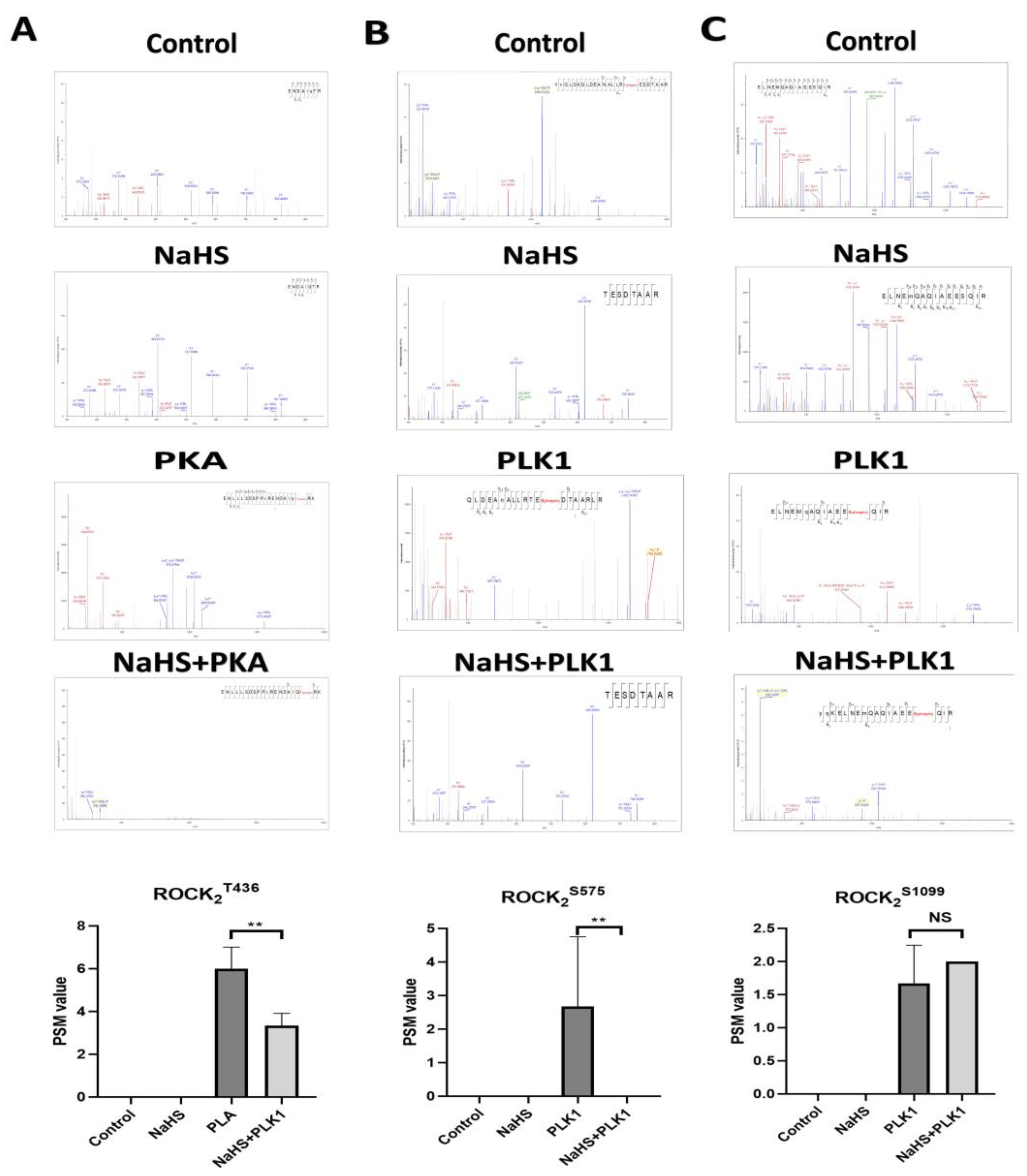
By using an LC–MS/MS assay, it was observed that, in the presence of serine kinase PLK1 or threonine kinase PKA, phosphorylation occurred at three sites, Thr436, Ser575 and Ser1099, in His-ROCK
2wild protein in vitro. However, the phosphorylation of these three sites did not take place in the absence of PLK1 or PKA, indicating that the phosphorylation of His-ROCK
2wild at the three sites cannot happen by autophosphorylation. As far as we know, the phosphorylation of ROCK
2 at Ser1099 was reported previously [
26], but the phosphorylation of ROCK
2 at Thr436 or Ser575 was found for the first time in the present study. Furthermore, our study observed that the phosphorylation of His-ROCK
2wild at Thr436 or Ser575 was significantly inhibited by treatment with H
2S donor NaHS, demonstrating that H
2S could inhibit the phosphorylation of ROCK
2 at both Thr436 and Ser575.
The inhibition of H
2S on the phosphorylation of His-ROCK
2wild at Thr436 or Ser575 seems contrary to our recent report on the promotion of H
2S on the phosphorylation of ROCK
2 at Tyr722 [
27]. It was already known that the phosphorylation of different sites of ROCK
2 had different effects on its activity; the phosphorylation of some sites can enhance the activity of ROCK
2, but the phosphorylation of other sites can inhibit its activity. This study used eukaryotic recombinant ROCK
2 wild-type plasmid (ROCK
2wild-pEGFP-N1) and plasmids mutated at Thr436 and Ser575 (ROCK
2T436A-pEGFP-N1 and ROCK
2S575F-pEGFP-N1) to transfect RHNs for the detection of ROCK
2 expression and activity. In the empty plasmid-transfected RHNs, only ROCK
2 was examined. Nevertheless, in the ROCK
2wild-pEGFP-N1-, the ROCK
2T436A-pEGFP-N1-, or the ROCK
2S575F-pEGFP-N1-transfected RHNs, in addition to ROCK
2, GFP-ROCK
2wild, GFP-ROCK
2T436A, and GFP-ROCK
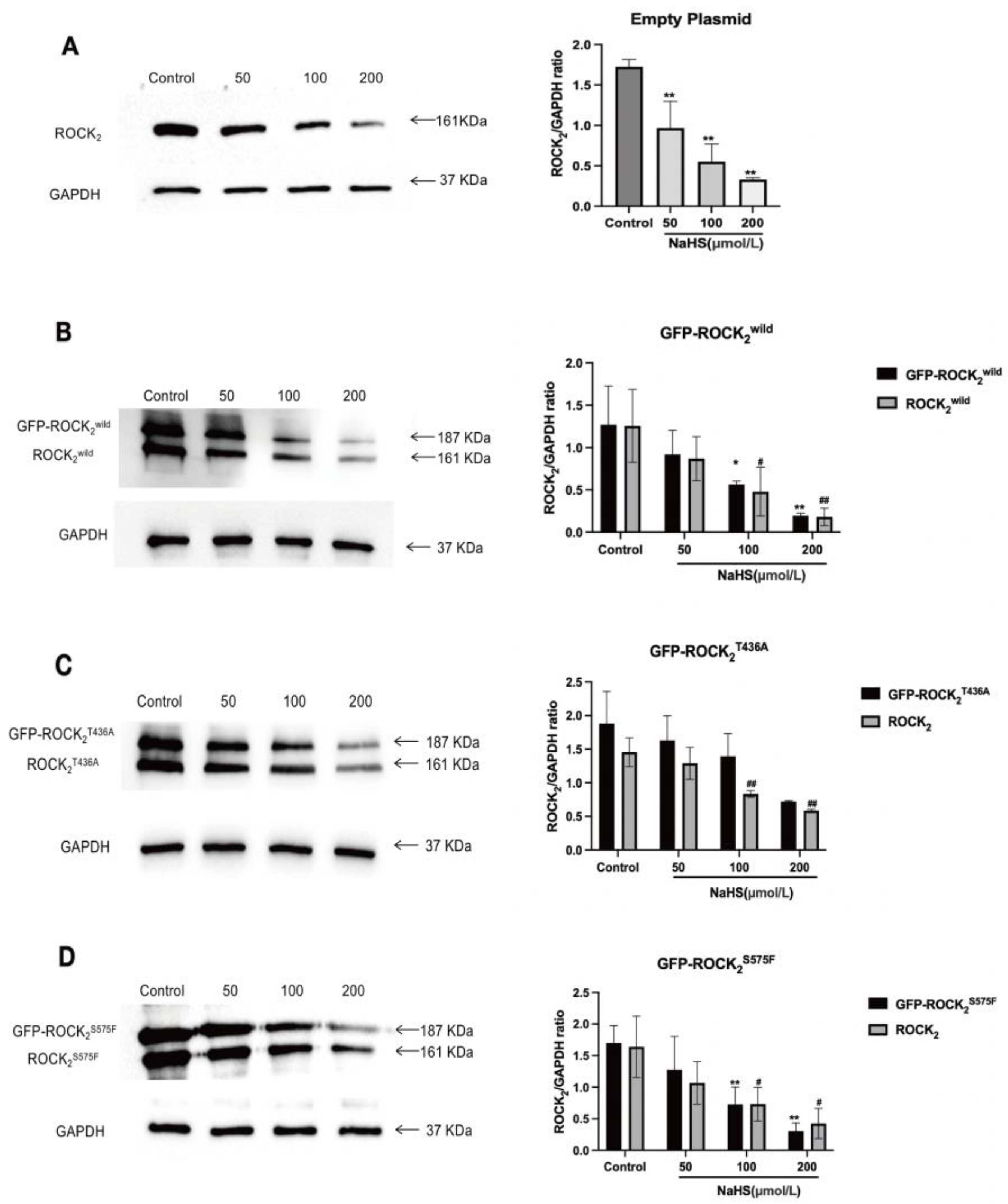
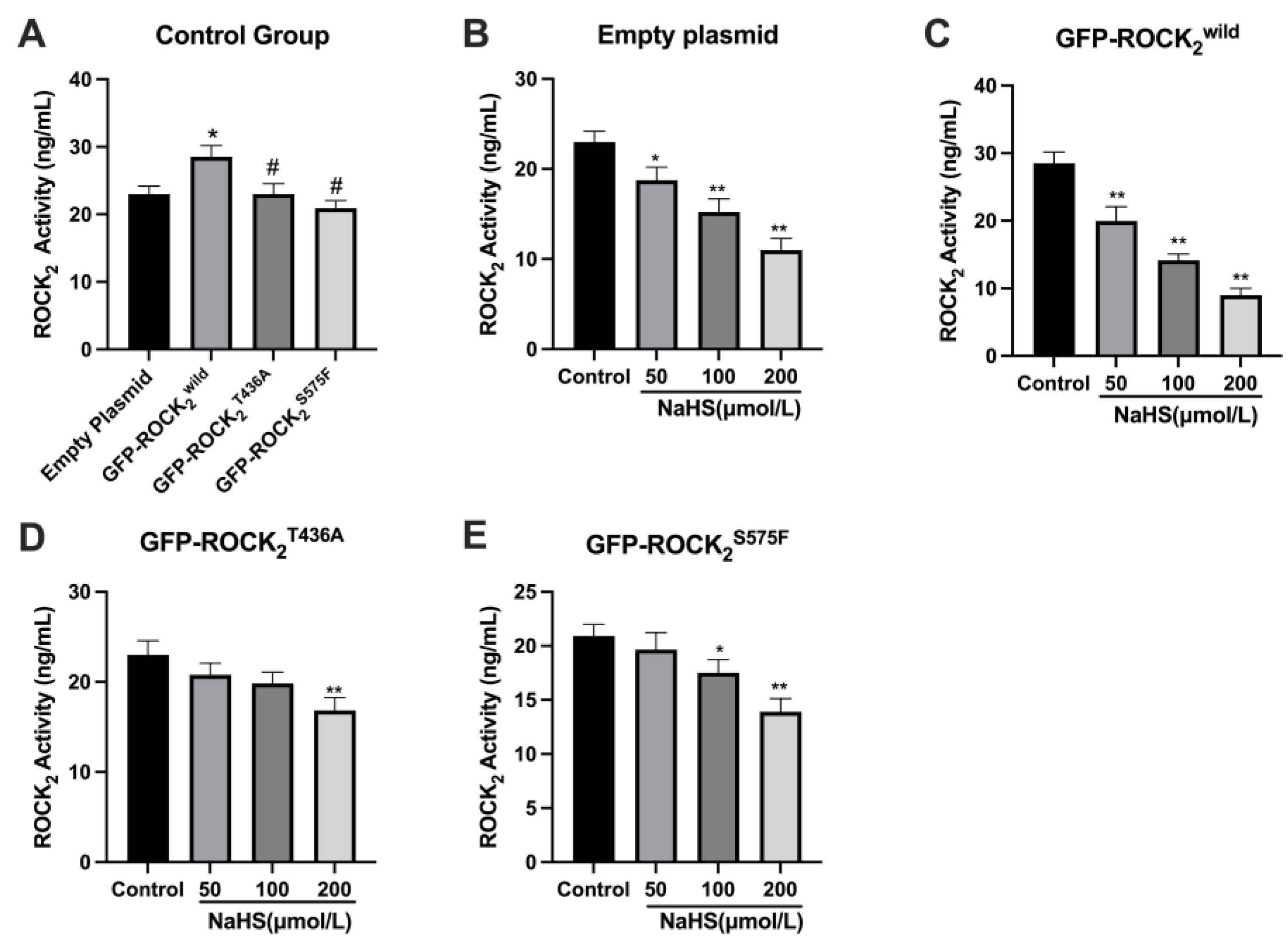
2S575F were detected, respectively, confirming that the transfection was successful. Our result showed that NaHS significantly inhibited ROCK
2 protein expression in each type of plasmid-transfected RHN. This is consistent with a previous study that NaHS inhibits cerebral I/R injury-increased ROCK
2 protein expression in mouse hippocampus [
28]. Our results also indicated that NaHS not only attenuated GFP-ROCK
2wild protein expression but also decreased GFP-ROCK
2T436A and GFP-ROCK
2S575F protein expressions, indicating that both Thr436 and Ser575 sites did not take part in the inhibitory effect of H
2S on ROCK
2 expression in RHNs. However, the present study revealed that both Thr436 and Ser575 participate in the activation of ROCK
2 and mediate the inhibitory effect of H
2S on ROCK
2 activity in RHNs. Combining with the aforementioned result of NaHS inhibiting the phosphorylation of ROCK
2 at Thr436 and Ser575 in vitro, our study demonstrated that H
2S could inhibit the activation of ROCK
2 via attenuating the phosphorylation of ROCK
2 at Thr436 and Ser575. This is not contradictory to its inhibition on ROCK
2 activity by promoting the phosphorylation of ROCK
2 at Tyr722.
The over-activation of ROCK
2 is involved in the pathological process of cerebral stroke. It was reported that KD-025, a ROCK
2 selective inhibitor, has a good efficacy on cerebral ischemic injury in mice [
29]. Therefore, the role of H
2S regulating the phosphorylation of ROCK
2 at Thr436 and Ser575 in its neuroprotection against H/R injury was investigated in this study. LDH is an important intracellular enzyme in glycolysis, with a tetrameric structure, which can catalyze the conversion of pyruvate to lactic acid and oxidize nicotinamide adenine dinucleotide dehydrogenase (NADH) to nicotinamide adenine dinucleotide (NAD+) [
30]. NSE is also an intracellular enzyme, highly specific for nerve cells, and can be used to quantitatively detect brain injury and provide improved diagnosis and outcome evaluation for patients with ischemic stroke, cerebral hemorrhage, seizures, cardiac arrest, coma after cardiopulmonary resuscitation, and traumatic brain injury [
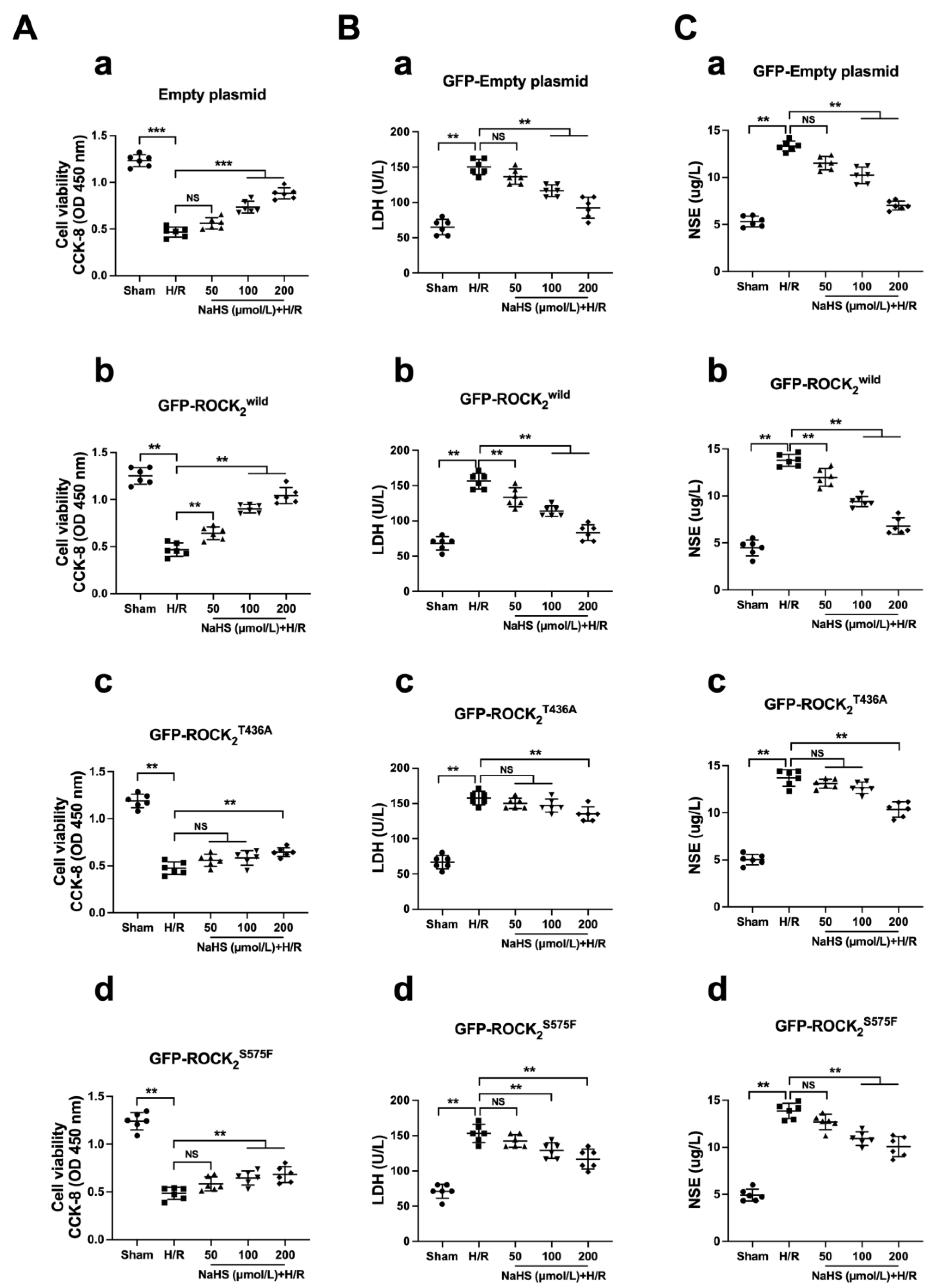
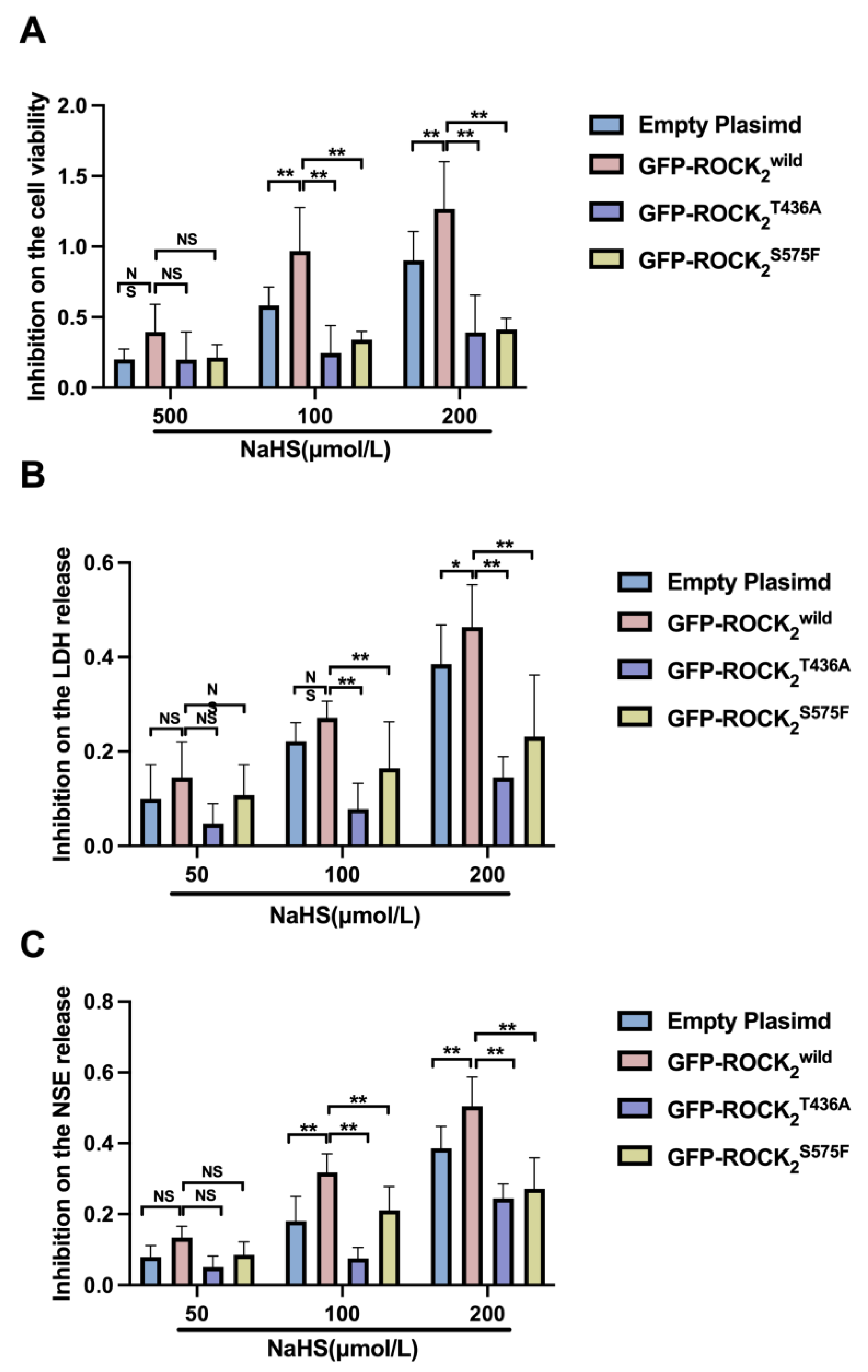
31]. When nerve cells are injured, LDH and NSE are released into the cellular environment. Thus, the release of both LDH and NSE is a reliable index of cerebral damage detection. In the present study, H/R injury in RHNs was indicated by a significant decrease in cellular viability and an obvious release of LDH and NSE (increase in supernatant LDH and NSE). Unlike the Ca
2+ channel blocker Nif, NaHS had an obvious difference in protecting against H/R injury in the empty plasmid-, the ROCK
2wild-pEGFP-N1-, the ROCK
2T436A-pEGFP-N1-, and the ROCK
2S575F-pEGFP-N1-transfected RHNs. The results showed that the protection of NaHS in the ROCK
2wild-pEGFP-N1-transfected RHNs was more patent than that in the empty plasmid-, the ROCK
2T436A-pEGFP-N1-, and the ROCK
2S575F-pEGFP-N1-transfected RHNs, demonstrating that Thr436 and Ser575 of ROCK
2 mediate the neuroprotection of H
2S against an H/R injury in RHNs. Therefore, it was possible to conclude that H
2S attenuates the phosphorylation of ROCK
2 at Thr436 and Ser575 to inhibit ROCK activation so as to protect RHNs from H/R injury.
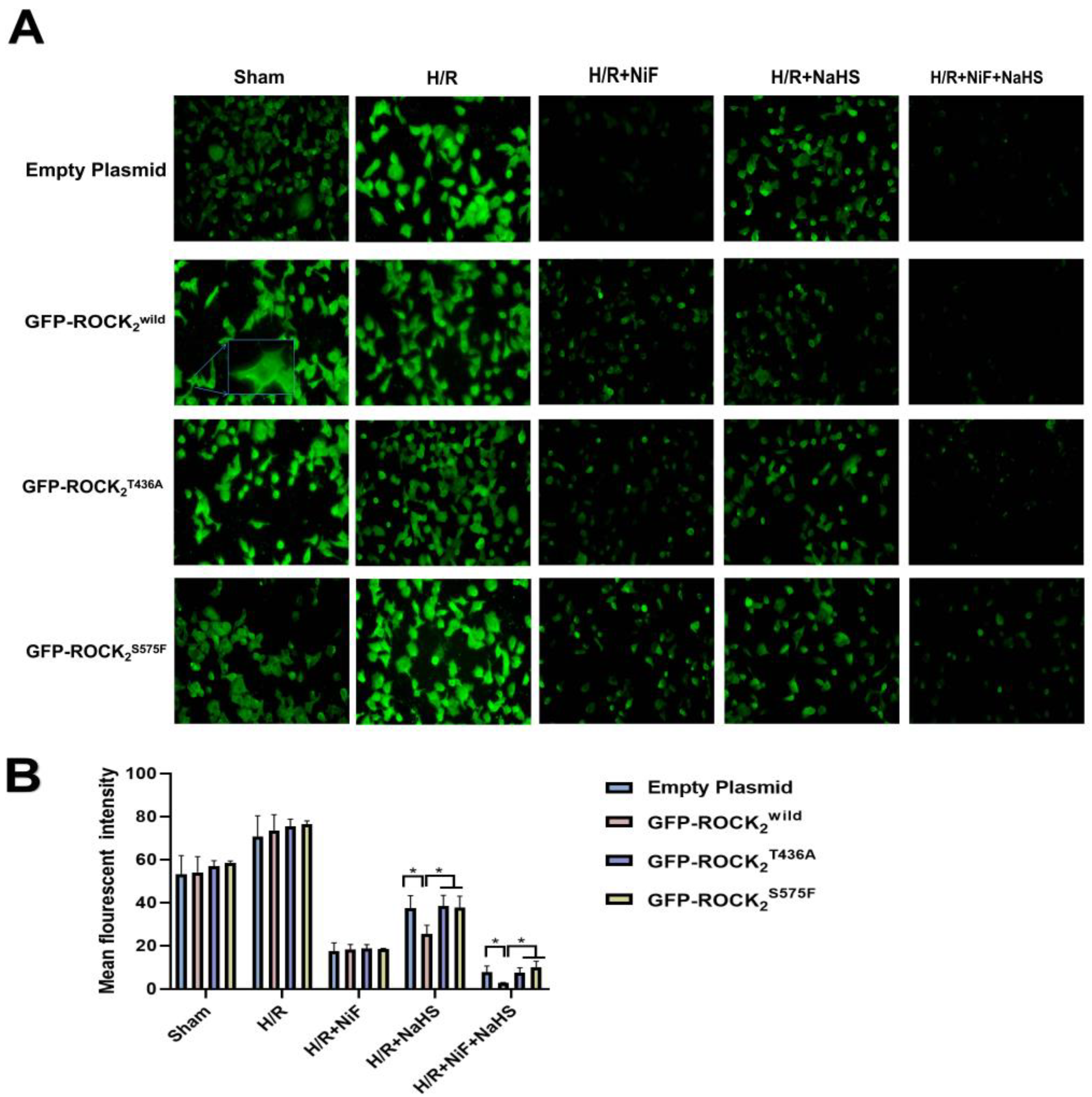
The [Ca
2+]
i is a key factor with a regulatory role in a variety of cell functions. Intracellular [Ca
2+]
i overload plays a major role in the cerebral I/R process [
32]. It was reported that an H/R-induced increase in [Ca
2+]
i was significantly inhibited by treatment with the ROCK inhibitor fasudil or Y-27632 [
33]. In the present study, the change in [Ca
2+]
i in each type of the transfected RHNs in the absence and presence of NaHS was detected. The result indicated that Thr436 and Ser575 of ROCK
2 are engaged with the inhibitory effect of H
2S on H/R-increased [Ca
2+]
i in RHNs. Taken together with the result of the inhibition of H
2S on the activation of ROCK
2, our results suggested that the inhibition of H
2S on the increased [Ca
2+]
i may be owed to its attenuation on the phosphorylation of ROCK
2 at Thr436 and Ser575. This may be related to the protection of H
2S against H/R injury in RHNs.
However, this experiment still had some limitations. For example, we did not obtain phosphorylated antibodies for p-ROCK2(Thr436) and p-ROCK2 (Ser575) so that the phosphorylation of ROCK2 in the cell model could be observed. Meanwhile, after we changed the two labels, the ROCK2 was still insoluble; other expression systems may exist to promote the solubility of the protein. Based on the interesting findings, we intend to further explore whether these sites can mediate the protective effect of H2S against I/R injury in animal models.
4. Materials and Methods
4.1. Reagents
NaHS was obtained from Sigma Chemical (St. Louis, MO, USA). Tris-Base was purchased from Amersco (St. Louis, MO, USA). Ethanol, glacial acetic acid, Na2S2O3, sodium acetate, glutaraldehyde, silver nitrate, formaldehyde, sodium carbonate, and dithiothreitol (DTT) were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Cell counting kit-8, ATP, PLK1, PKA, and IPTG were purchased from Abcam (San Francisco, CA, USA). The LDH assay kit was purchased from Servicebio (Wuhan, China). The NSE assay kit was obtained from Jiangsu Meimian Industrial, Co., Ltd. (Jiangsu, China). Flou-8 AM was purchased from biofount (Beijing, China).
4.2. Plasmids and Bacterium
We commissioned Gene Create Biological Engineering, Co. (Wuhan, China) to construct the prokaryotic plasmids ROCK
2wild-pET-32a(+) and eukaryotic recombinant plasmids ROCK
2wild-pEGFP-N1, ROCK
2T436A-pEGFP-N1, and ROCK
2S575F-pEGFP-N1.
E. coli (BL21) was obtained from Gene Create Biological Engineering, Co. (Wuhan, China). ROCK
2T436A means Thr436 was mutated to an alanine (Ala or A). ROCK
2S575F means Ser575 was mutated to a phenylalanine (Phe or F). All the mutations were at the mRNA level. The sequencing results showed that the plasmids and the mutations were successfully constructed. (See
Supplementary Material.)
4.3. Expression of the Prokaryotic Recombinant Proteins His-ROCK2wild
A total of 10 μL of glycerol bacteria containing the prokaryotic plasmid ROCK
2wild-pGEX-6P-1 was added to the solid Luria-Bertani (LB) medium. These were spread evenly and quickly by using a coating rod. The coated dishes were inverted overnight in an incubator at 37 °C. Single colonies with a good growth status using a pipette gun were inoculated into a centrifuge tube containing 20 mL of liquid LB medium in a shaker (37 °C, 230 rpm). Using medium as a blank control and looking at the OD solution of about 0.7, IPTG-induced expression protein was added at final concentrations of 0.2 mM and 1.0 mM, respectively. The fluids supplemented with different final concentrations of IPTG were induced overnight or for 4 h at 15 °C and 37 °C. The solution was collected at 10,000 rpm for 10 min and concentrated by adding 2 mL of PBS. The concentrated bacterial solution was crushed for 30 min using an ultrasonic crusher (100 W). The crushed bacterial solution was centrifuged at 10,000 rpm for 10 min. An equal volume of PBS solution was added into the precipitate and the supernatant. Some of the bacterial solution after ultrasonic crushing was sent to Shanghai Sangong Biological Co., Ltd. (Shanghai, China) for base sequencing. An upper buffer was added to the supernatant and precipitated samples; we denatured the protein at 100 °C. Electrophoresis was performed using an 8% SDS-PAGE gel for 40 min, with an electrophoresis meter voltage set to 120 V. After the electrophoresis, the gel was removed and the protein expression was visualized by staining using a Coomassie blue rapid staining solution for 10 min in a shaker. Sequencing of the eukaryotic plasmids ROCK2T436A-pEGFP-N1 showed that ACA (Thr) was mutated into GCG (Ala) at 436 and TCT (Ser) was mutated into TTT (Phe) at 575 of ROCK
2S575F-pEGFP-N1 (see
Supplementary Material).
4.4. Culture of Primary RHNS
The hippocampus was carefully removed with eye tweezers. Peripheral blood vessels were removed as far as possible and transferred to a solution containing 0.125% trypsin (0.25% trypsin:PBS = 1:1). The tissue was completely cut up and put into a 37 °C constant temperature cell incubator for digestion. The centrifuge tube containing the tissue was shaken for 20 mins. The preheated DMEM medium was added after digestion and then filtered after blowing evenly. The solution was centrifuged at 1400 rpm for 5 min. The supernatant was poured off, the complete medium was added, and the cells were gently blown and resuspended. After a cell count, the cells were inoculated into the 24-well plate (poly-DL-lysine coated) and cultured in a constant temperature cell incubator. After 24 h, we performed half-dose fluid replacement with Neurobasal medium and performed full fluid replacement every 1 to 2 days for incubation for 5 to 6 days.
4.5. Phosphorylation Test In Vitro
A total of 15 μL of lysate of the ROCK2wild-pGEX-6P-1-transfected E. colis induced by IPTG and 10 μL of 100 μmol/L ATP was added to 30 μL of 10×Kinase buffer (20 mM Tris-Cl, 100 mM KCl, 2 mM EGTA, 5 mM MgCl2, pH 7.4) without or with 3 μL of kinases PLK1 0.52 mg/mL (or PKA 2500 KU/mL) and mixed fully. The mixture was shaken at 180 rpm under 37 °C for 30 min. The phosphorylation reaction was terminated by adding 7 μL of 5× loading buffer (250 mM Tris-HCl, 10% SDS, 0.5% BPB, 50% glycerol, 5% β-mercaptoethanol). The mixture was kept at 100 °C for 10 min to denature protein. A small amount of the mixture was loaded onto an 8% SDS-PAGE gel to perform Western blot assay for detecting His-ROCK2wild protein expression; the rest was also loaded onto the 8% SDS-PAGE gel for electrophoresis and then stained with the Coomassie brilliant blue. Using the phosphorylation reaction mixture of lysates of E. coli induced without IPTG as a control, the differential protein band in the Coomassie bright blue-stained SDS-PAGE gel was determined; this should have had the same position as the His-ROCK band in the Western blot assay. A differential protein band was excised and cut into patches of approximately 1 mm × 1 mm. The patches were cleaned with a glue cleaning solution, dehydrated using acetonitrile, and then sent to Hooper Biotechnology Co., Ltd. (Shanghai, China) for LC–MS/MS assay to detect the phosphorylation of ROCK2 at related sites.
4.6. LC–MS/MS
A mixed patch with 8 M of UA in the 10 K filter unit was centrifuged at 14,000× g for 15 min. We added 200 μL of UA and centrifuged at 14,000 g for 15 min. We discarded the flow-through from the collection tube. We alkylated the proteins with IAA and incubated them for 45 min in the dark. We discarded the flow-through. We added 100 μL of UA and centrifuged at 14,000× g for 15 min; this step was repeated once. We added 200 μL of 50 mM ABC and centrifuged at 14,000× g for 15 min; this step was repeated once. We changed to a new collection tube, added 4 μg of trypsin, and incubated at 37 °C for 16 h. We centrifuged this at 14,000× g for 10 min and collected the flow-through in a new tube. We added 50 mM of ABC, centrifuged at 14,000× g for 10 min, collected the flow-through to the above tube, and dried the sample with SpeedVac. The sample was dissolved in 0.1% TFA, desalted with C18 ZipTips, and dried with SpeedVac. The sample was resuspended with 0.1% formic acid for mass spectrometry analysis. The peptide samples were analyzed on a Thermo Fisher LTQ Obitrap ETD mass spectrometry. Briefly, we loaded the sample onto an HPLC chromatography system called a Thermo Fisher Easy-nLC 1000 equipped with a C18 column (1.8 mm, 0.15 × 100 mm). Solvent A contained 0.1% formic acid and solvent B contained 100% acetonitrile. The elution gradient was from 4% to 18% in 182 min and 18% to 90% in 13 min for solvent B at a flow rate of 300 nL/min. Mass spectrometry analyses were carried out at AIMSMASS Co., Ltd. (Shanghai, China) in the positive-ion mode with an automated data-dependent MS/MS analysis with full scans (350–1600 m/z) acquired using FTMS at a mass resolution of 30,000; the 10 most intense precursor ions were selected for MS/MS. The MS/MS was acquired using higher-energy collision dissociation at 35% collision energy at a mass resolution of 15,000.
4.7. Lentiviral Transfection
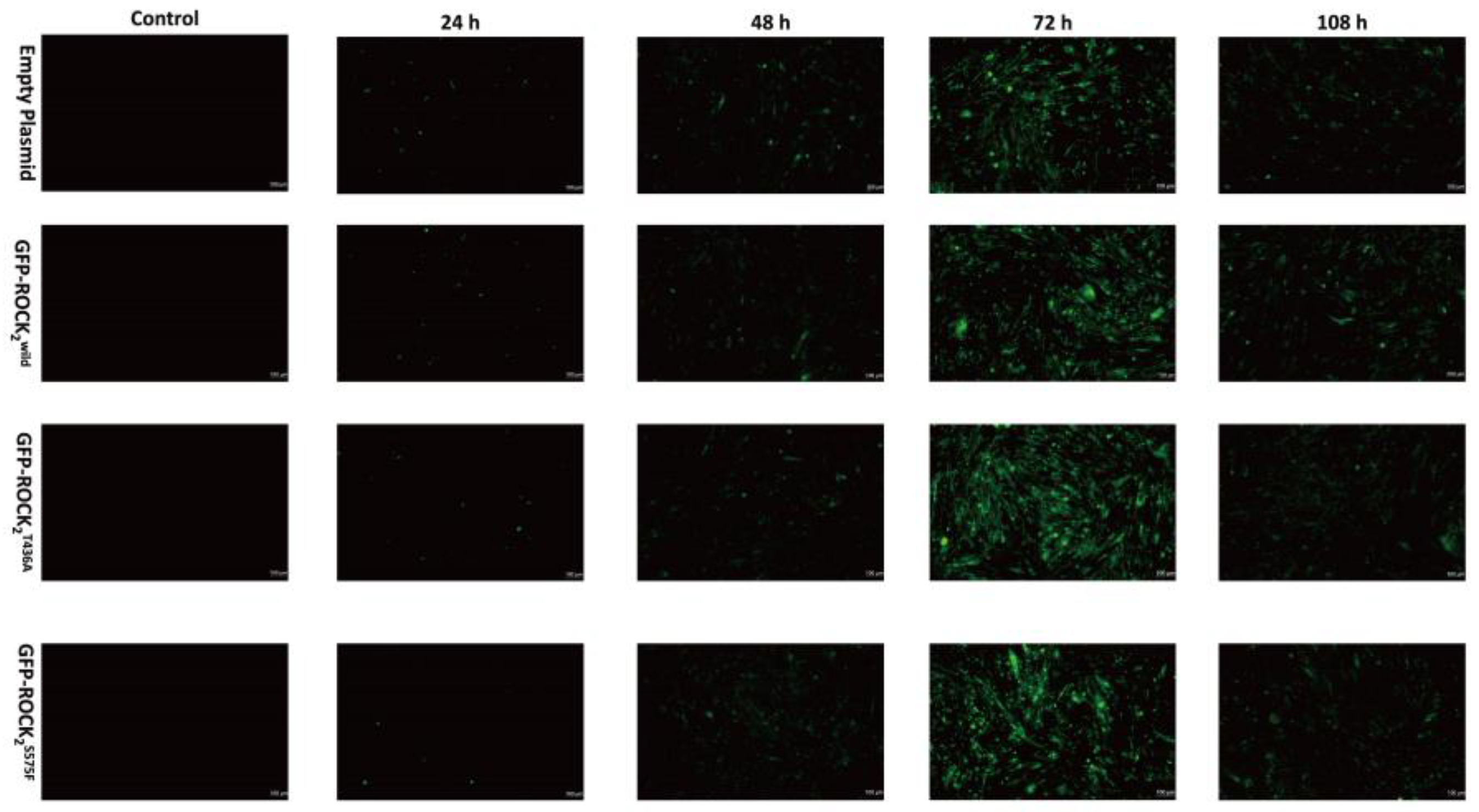
The lentivirus vector was a second-generation vector that was cotransfected into 293T cells by empty, ROCK2wild-pEGFP-N1, ROCK2T436A-pEGFP-N1, and ROCK2S575F-pEGFP-N1 plasmids; lentivirus packaging plasmid pCD/NLBH*DDD; and membrane protein expression plasmid PLTR-G. RHNs in a good growth state were selected and centrifuged at 10,000 rpm for 10 min. Then, the cells were resuspended with a fresh cell culture medium and counted. The suspension of RHNs (5 × 105/mL) was inoculated into 24 wells of a medium; the lentivirus expressing ROCK2wild, ROCK2T436A, and ROCK2S575F (1 × 108 uA/mL) was added to the well plate when the degree of cell fusion reached 70%. After 24 h of culture, the medium was replaced with a fresh medium. The expression of GFP protein at 24 h, 48 h, 72 h, and 108 h was respectively observed under an inverted fluorescence microscope to evaluate the transfection efficiency.
4.8. Western Blot
The 8% SDS-PAGE (7.5 mL top glue premix, 7.5 mL bottom glue premix, 2 μL tetramethylethylene-diamine) was prepared according to the molecular weight of ROCK2. Then, 15 μL of cell lysate was added to each well. After electrophoresis for 180 min at 120 V, the target protein and GAPDH were transferred to a PVDF membrane using wet membrane transfer, blocked at room temperature using 5% skim milk for 1 h, and then transferred to TBST buffer and washed twice for 10 min each. The membranes were individually incubated overnight in the anti-ROCK2 antibody solution for 4 °C and then incubated in the secondary antibody solution for 1 h at room temperature. The ECL-plus development solution was prepared and applied evenly on the PVDF membrane. The chemiluminescence was visualized using a Fluor-S-max imager. Gray values of the different bands were analyzed using Image J software; the ratios of the ROCK2 and GAPDH were calculated and counted using Graphpad prism 8.0 software.
4.9. Determination of ROCK2 Activity in RHNs
According to the manufacturer’s instructions, ROCK2 activity in RHNs was measured. The prepared RHNs were crushed with a 100 W ultrasonic crusher and centrifuged at 10,000 rpm for 10 min. The supernatant was added to the 96-well plate and incubated at 37 °C for 30 min. Then, 50 µL of the PBS was added to each well, followed by 50 µL of chromogenic agent A and chromogenic agent B each. The mixture was gently shaken, mixed well, and incubated at 37 °C for 15 min in the dark. Then, 50 µL of the PBS was added to each well to terminate the reaction. The activity of ROCK2 was measured at a wavelength of 450 nm using a microplate reader.
4.10. Establishment of H/R Injury
RHNs were incubated in a hypoxia incubator (1% O
2, 95% N
2, 4% CO
2) at 37 °C for 4 h and then cultured at 37 °C under normoxic conditions (37 °C, 95% O
2, 5% CO
2) for 12 h. The RHNs in the control group were kept under normoxic conditions without hypoxia [
18].
4.11. Determination of Cell Viability
Cell viability was determined using a cell counting kit (CCK-8). Briefly, the RHNs’ suspension was transferred into a 96-well plate, and a total of 10 µL of the CCK-8 solution was added to each well. Then, the RHNs were incubated in an incubator (37 °C, 5% CO2) for 24 h. The absorbance at 450 nm was measured with a microplate reader.
4.12. Determination of the LDH and NSE Activities
LDH and NSE levels in the RHNs’ culture supernatant were respectively determined using the commercial assay kits. Briefly, the prepared RHNs’ suspension was centrifuged at 10,000 rpm for 10 min using a centrifuge (Eppendorf, Germany); a microplate reader was used to respectively detect LDH and NSE activities in the supernatant at 450 nm, according to the protocol of the LDH and NSE assay kits.
4.13. Determination of [Ca2+]i
RHNs were incubated with a final concentration of 10 μM of Fluo-8 AM for 10 min at room temperature and then washed three times with PBS buffer. The [Ca2+]i fluorescence intensity in the RHNs was measured using a Ca2+ imaging system fluorescence microscope.
Mature RHNs cultured for 6–8 days were selected, incubated with 10 μM of Fluo-8 AM for 10 min at room temperature, washed three times with PBS buffer, and preincubated with a normal physiological saline solution (NPSS) that contained 140 mM of NaCl, 10 mM of glucose, 5 mM of KCl, 5 mM of Hepes, 1 mM of CaCl2, and 1 mM MgCl2; the fluorescence intensity of Ca2+ in the different groups of RHNs was observed using a Ca2+ imaging system fluorescence microscope (excitation wavelength: 488 nm, emission wavelength: 514 nm).
4.14. Statistical Analysis
All data are represented as the mean ± SD. The t-test was used to compare the differences between the two groups. One-way analysis of variance (one-way ANOVA) and two-way ANOVA were used for multi-group comparisons. A p < 0.05 was considered as a statistically significant difference.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







