
Natural Bioactive Products as Epigenetic Modulators for Treating Neurodegenerative Disorders




Abstract
1. Introduction
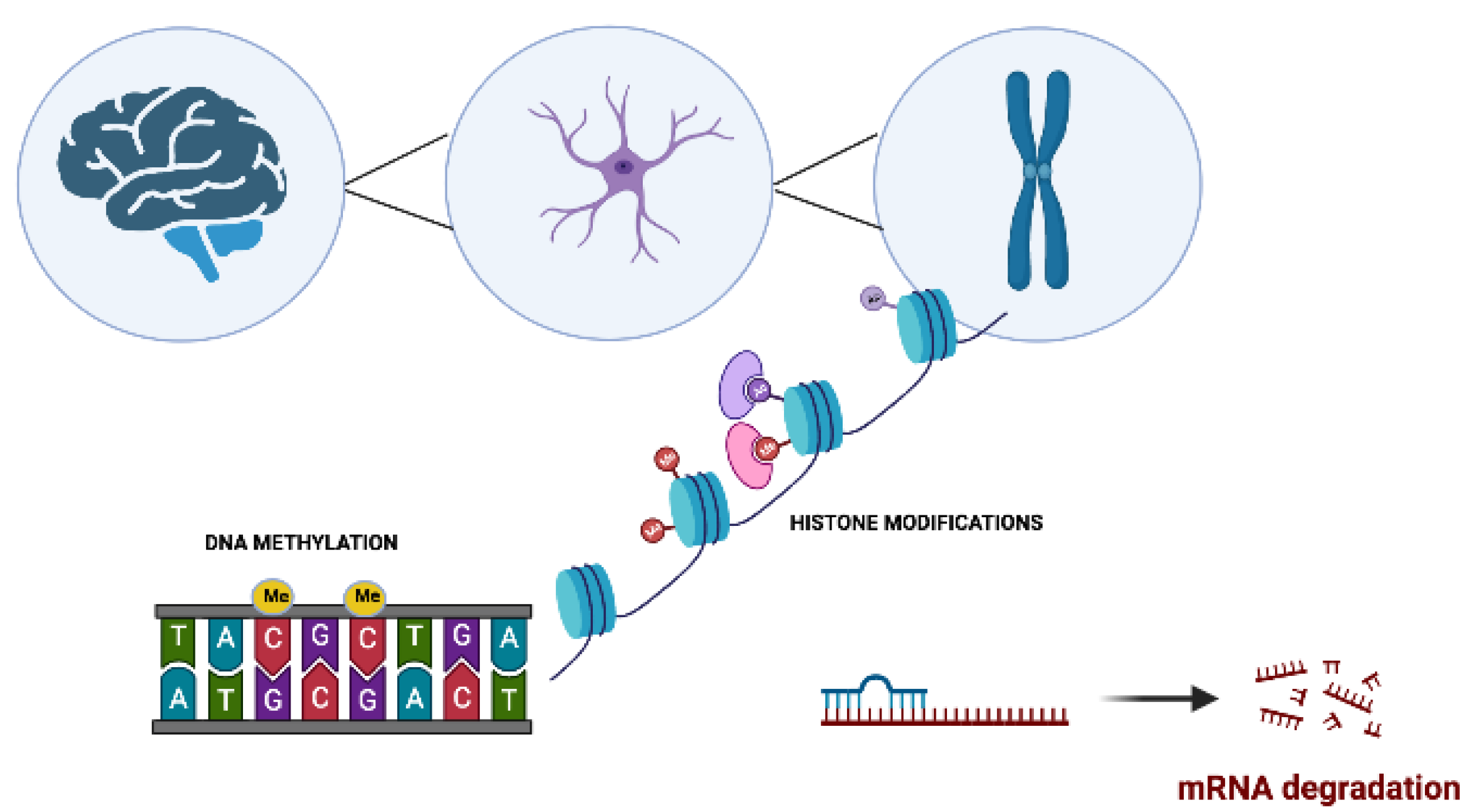
2. Epigenetics in Neurodegenerative Disorders
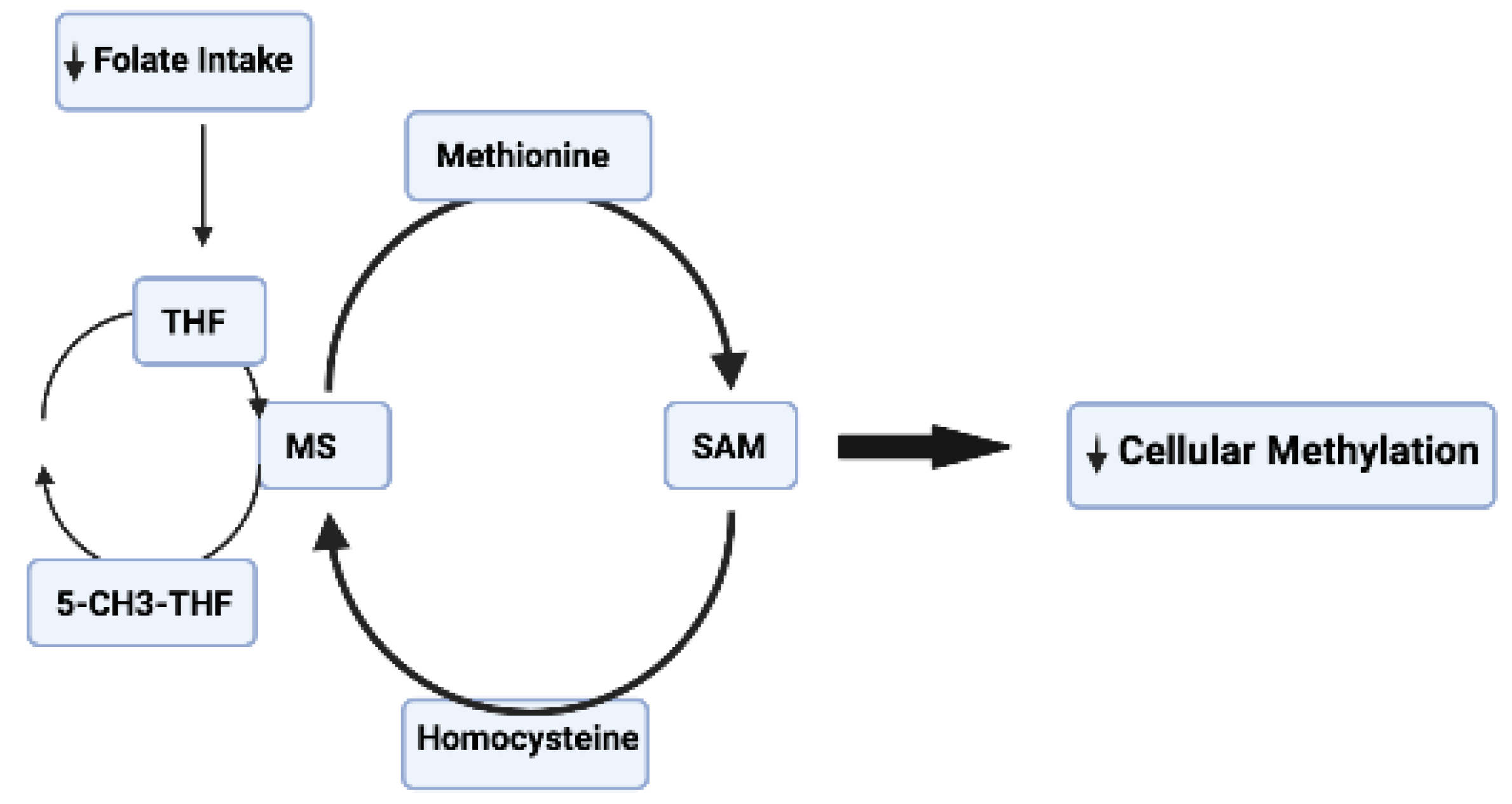
2.1. Role of DNA Methylation in Neurodegenerative Disorders
2.2. Post-Translational Histone Modifications in Neurodegenerative Disorders
2.3. Regulation of micro-RNAs in Neurodegenerative Disorders
3. Treatment of Neurodegenerative Disorders with Epinutraceutical Bioproducts
3.1. Vitamins
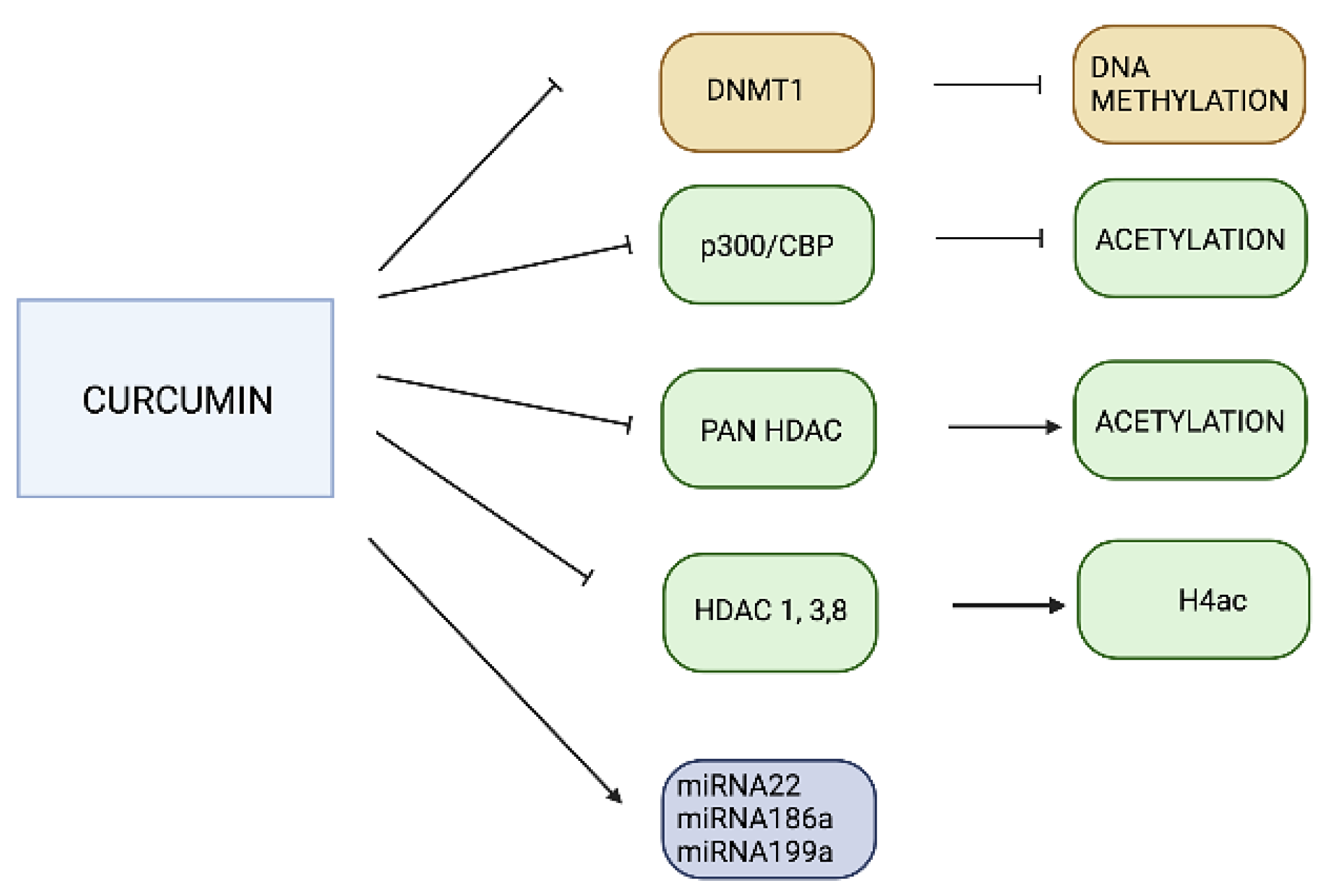
3.2. Curcumin
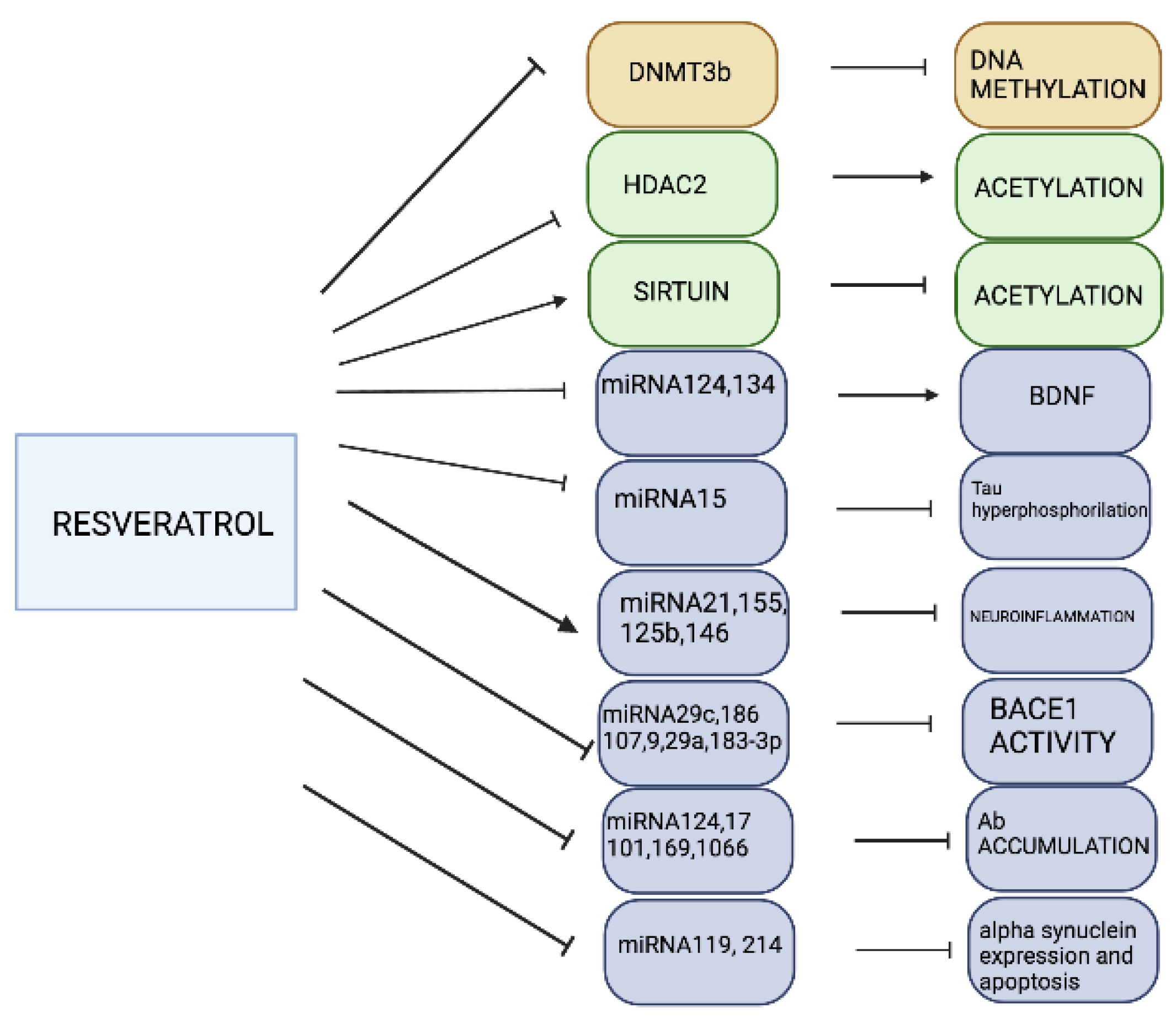
3.3. Resveratrol
3.4. Epigallocatechin-3-Gallate (EGCG)
3.5. AtreMorine
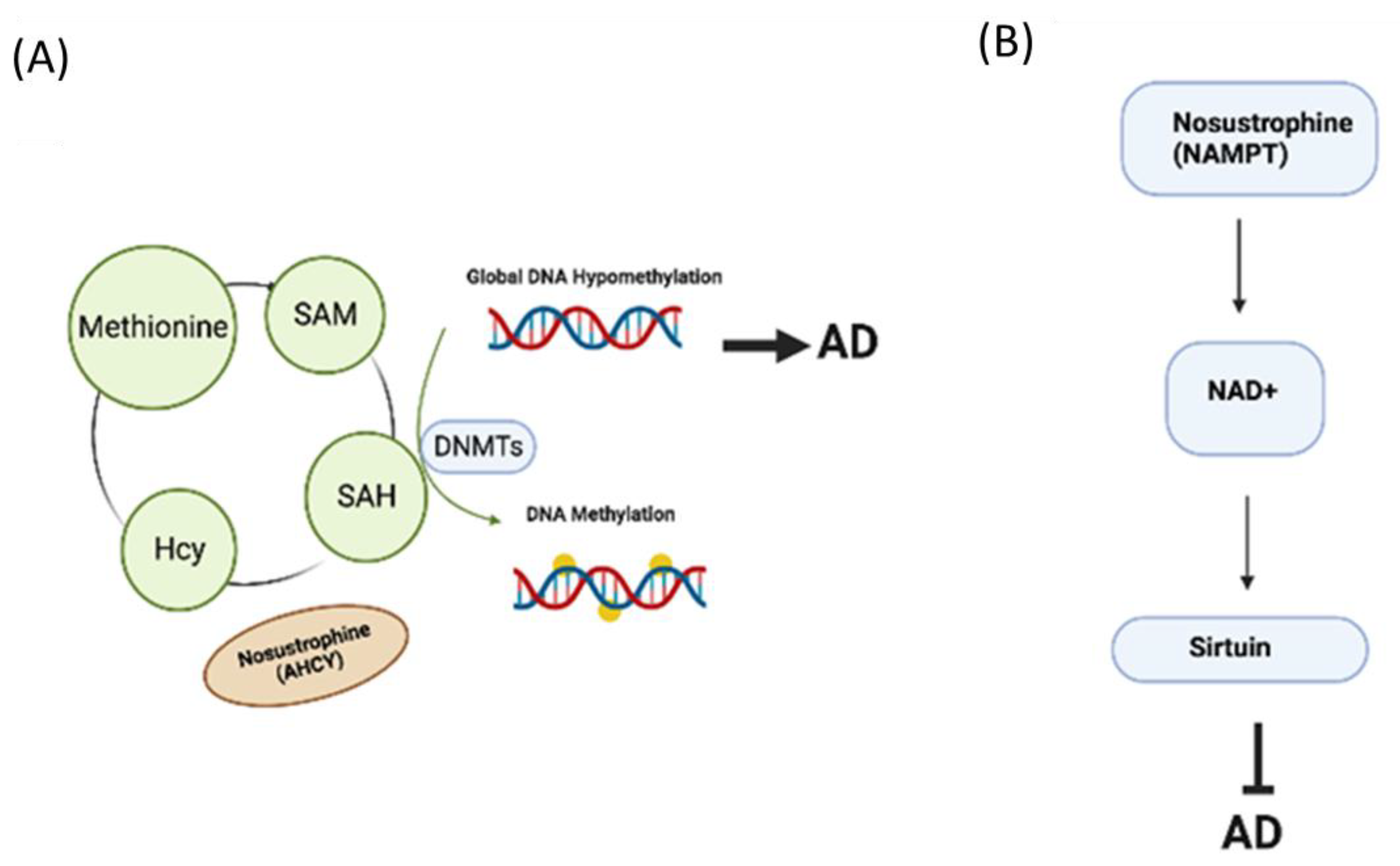
3.6. Nosustrophine
4. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Patents
References
- Alzheimer’s Association. 2020 Alzheimer’s disease facts and figures. Alzheimers Dement. 2020, 16, 391–460. [Google Scholar] [CrossRef]
- GDB. 2019 Dementia Forecasting Collaborators. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the Global Burden of Disease Study. Lancet Public Health 2022, 7, e105–e125. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Teijido, O.; Carril, J.C. Can cloud-based tools accelerate Alzheimer’s disease drug discovery? Expert Opin. Drug. Discov. 2016, 11, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R. Pharmacogenomics of Alzheimer’s and Parkinson’s Diseases. Neurosci. Lett. 2018, 726, 133807. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Férnandez-Novoa, L.; Lombardi, V.; Kubota, Y.; Takeda, M. Molecular genetics of Alzheimer’s disease and ageing. Methods Find Exp. Clin. Pharm. 2005, 27, 1673. [Google Scholar]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Pang, S.; Ho, P.; Liu, H.-F.; Leung, C.-T.; Chang, E.; Ramsden, D.; Ho, S.-L. The interplay of aging, genetics and environmental factors in the pathogenesis of Parkinson’s disease. Transl Neurodeg. 2019, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.; Schmidt, M.; Lee, V.; Trojanowski, J.; Goedert, M. Alpha-synuclein in Lewi bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Urdinguio, R.; Sanchez-Mut, J.; Esteller, M. Epigenetic mechanisms in neurological diseases: Genes, syndromes, and therapies. Lancet Neurol. 2009, 8, 1056–1072. [Google Scholar] [CrossRef]
- Sweatt, J.D. The emerging field of neuroepigenetics. Neuron 2013, 80, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Tellado, I.; Cacabelos, P. The epigenetic machinery in the life cycle and pharmacoepigenetics. Pharmacoepigenetics 2019, 10, 1–100. [Google Scholar]
- Delgado-Morales, R.; Esteller, M. Opening up the DNA methylome of dementia. Mol. Psychiatry 2017, 22, 485–496. [Google Scholar] [CrossRef]
- Maloney, B.; Lahiri, D. Epigenetics of dementia: Understanding the disease as a transformation rather than a state. Lancet Neurol. 2016, 15, 760–774. [Google Scholar] [CrossRef]
- Bird, A. The essentials of DNA methylation. Cell 1992, 70, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Gräff, J.; Mansuy, I. Epigenetic codes in cognition and behaviour. Behav. Brain Res. 2008, 1, 70–87. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Cross, S.; Bird, A. Gene silencing by methyl-CpG-binding proteins. Novartis Found. Symp. 1998, 214, 6–16. [Google Scholar] [PubMed]
- Nan, X.; Campoy, F.; Bird, A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 1997, 88, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Su, Y.; Shin, J.; Shin, J.; Li, H.; Xie, B.; Zhong, C.; Hu, S.; Le, T.; Fan, G.; et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci. 2014, 17, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Hermann, A.; Goya, R.; Jeltsch, A. The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J. Biol. Chem. 2004, 279, 48350–48359. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Xie, S.; Li, E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat. Genet. 1998, 19, 219–220. [Google Scholar] [CrossRef]
- Growher, H.; Liebert, K.; Hermann, A.; Xu, G.; Jeltsch, A. Mechanism of stimulation of catalytic activity of Dnmt3A and Dnmt3B DNA-(cytosine-C5)-methyltransferases by Dnmt3L. J. Biol. Chem. 2005, 80, 13341–13348. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Su, Y.; Zhong, C.; Ming, G.-l.; Song, H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 2011, 145, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E.; Ott, M. 50 years of protein acetylation: From gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 2015, 16, 258–264. [Google Scholar] [CrossRef]
- Carafa, V.; Rotili, D.; Forgione, M.; Cuomo, F.; Serretiello, E.; Hailu, G.S.; Jarho, E.; Lahtela-Kakkonen, M.; Mai, A.; Altucci, L. Sirtuin functions and modulation: From chemistry to the clinic. Clin. Epigenet. 2016, 8, 61. [Google Scholar] [CrossRef] [PubMed]
- Grbowska, W.; Sikora, E.; Bielak-Zmijewska, A. Sirtuins, a promising target in slowing down the ageing process. Biogerontology 2017, 18, 447–476. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA biogenesis, mechanisms of action and circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef]
- Condrat, C.E.; Thompson, D.C.; Barbu, M.G.; Bugnar, O.L.; Boboc, A.; Cretoiu, D.; Suciu, N.; Cretoiu, S.M.; Voinea, S.C. miRNAs as biomarkers in disease:latest findings regarding their role in diagnosis and prognosis. Cells 2020, 9, 276. [Google Scholar] [CrossRef]
- Roy, B.; Lee, E.; Li, T.; Rampersaud, M. Role of miRNAs in Neurodegeneration: From disease cause to tools of biomarker discovery and therapeutics. Genes 2022, 13, 425. [Google Scholar] [CrossRef]
- Chakraborty, C.; Sharma, A.; Sharma, G.; Lee, S.-S. Therapeutic advances of miRNAs: A preclinical and clinical update. J. Adv. Res. 2021, 28, 127–138. [Google Scholar] [CrossRef]
- El Omari, N.; Bakrim, S.; Bakha, M.; Lorenzo, J.M.; Rebezov, M.; Shariati, M.A.; Aboulaghras, S.; Balahbib, A.; Khayrullin, M.; Bouyahya, A. Natural bioactive compounds targeting epigenetic pathways in cancer: A review on alkaloids, terpenoids, quinones and isothiocyanates. Nutrients 2021, 13, 3714. [Google Scholar] [CrossRef]
- Bautista-Garcia, P.; González-López, L.; González-Esparza, B.; Del Castillo-Rosas, C. Effect of bioactive nutriments in health and disease: The role of epigenetic modifications. Funct. Food 2017, 7, 123. [Google Scholar]
- Carrera, I.; Martínez, O.; Cacabelos, R. Neuroprotection with natural antioxidants and nutraceuticals in the context of brain cell degeenration: The epigenetic connection. Curr. Top. Med. Chem. 2019, 19, 2999–3011. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.-Y.; Aromolaran, K.A.; Zukin, R.S. The emerging field of epigenetics in neurodegeneration and neuroprotection. Nat. Rev. Neurosci. 2017, 18, 347–361. [Google Scholar] [CrossRef]
- Berson, A.; Nativio, R.; Berger, S.; Bonini, N. Epigenetic regulation in Neurodegeenrative Diseases. Trends Neurosci. 2018, 9, 587–598. [Google Scholar] [CrossRef]
- Teijido, O.; Cacabelos, R. Pharmacoepigenomic interventions as novel potential treatments for alzheimer’s and parkinson’s diseases. Int. J. Mol. Sci. 2018, 19, 3199. [Google Scholar] [CrossRef]
- Cacabelos, R.; Torrellas, C. Epigenetics of Aging and alzheimer’s disease: Implications for pharmacogenomics and drug response. Int. J. Mol. Sci. 2015, 16, 30483–30543. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Takae, H.; Miyake, K. Epigenetic mechanisms and therapeutic perspectives for neurodevelopmental disorders. Pharmaceuticals 2012, 5, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Iglesias, O.; Naidoo, V.; Cacabelos, N.; Cacabelos, R. Epigenetic Biomarkers as Diagnostic Tools for Neurodegenerative Disorders. Int. J. Mol. Sci. 2022, 23, 13. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Iglesias, O.; Carera, I.; Carril, J.C.; Fernández-Novoa, L.; Cacabelos, N.; Cacabelos, R. DNA methylation in neurodegenerative and cerebrovascular disorders. Int. J. Mol. Sci. 2020, 21, 2220. [Google Scholar] [CrossRef]
- Martínez-Iglesias, O.; Naidoo, V.; Carrera, I.; Cacabelos, R. Epigenetic studies in the male APP/BIN1/COPS5 triple-transgenic mouse model of Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 2446. [Google Scholar] [CrossRef]
- Kaur, G.; Rathod, S.; Ghoneim, M.; Alshehri, S.; Ahmad, J.; Mishra, A.; Alhakamy, N.A. DNA methylation: A promising approach in management of Alzheiemr’s Disease and other neurodegenerative disorders. Biology 2022, 11, 90. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Romero, C.; Hur, J.; Bender, D.E.; Delaney, C.E.; Cataldo, M.D.; Smith, A.L.; Yung, R.; Ruden, D.M.; Callaghan, B.C.; Feldman, E.L. Identification of epigenetically altered genes in sporadic amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e52672. [Google Scholar] [CrossRef] [PubMed]
- Nicolia, V.; Fuso, A.; Cavallaro, R.A.; Di Luzio, A.; Scarpa, S. B vitamin deficiency promotes tau phosphorylation through regulation of GSK3beta and PP2A. J. Alzheimers Dis. 2010, 19, 95–907. [Google Scholar] [CrossRef] [PubMed]
- Mastroeni, D.; Grover, A.; Delvaux, E.; Whiteside, C.; Coleman, P.D.; Rogers, J. Epigenetic mechanisms in alzheimer’s disease. Neurobiol. Aging 2011, 32, 1161–1180. [Google Scholar] [PubMed]
- Teijido, O.; Cacabelos, R. Interrogating the Epigenome to Unveil the Secrets of Neurodegeneration: Promising Epigenetic Therapies. J. Genome Med. Pharm. 2016, 1, 95–150. [Google Scholar]
- Sanchez-Mut, J.V.; Aso, E.; Panayotis, N.; Lott, I.; Dierssen, M.; Rabano, A.; Urdinguio, R.G.; Fernandez, A.F.; Astudillo, A.; Martin-Subero, J.I.; et al. DNA methylation map of mouse and human brain identifies target genes in Alzheimer’s disease. Brain 2013, 136, 3018–3027. [Google Scholar] [CrossRef]
- Pérez, R.F.; Alba-Linares, J.J.; Tejedor, J.R.; Fernández, A.F.; Calero, M.; Román-Domínguez, A.; Borrás, C.; Viña, J.; Ávila, J.; Medina, M.; et al. Blood DNA methylation patterns in older adults with evolving dementia. J. Gerontol. A Biol. Sci. Med. Sci. 2022, 77, 1743–1749. [Google Scholar] [CrossRef]
- Cilla, C.; Stoccoro, A. Epigenetic peripheral biomarkers for early diagnosis of Alzheimer’s Disease. Genes 2022, 13, 1308. [Google Scholar]
- Matsumoto, L.; Takuma, H.; Tamaoka, A.; Kurisaki, H.; Date, H.; Tsuji, S.; Iwata, A. CpG demethylation enhances alpha-synuclein expression and affects the pathogenesis of Parkinson’s disease. PLoS ONE 2010, 5, e15522. [Google Scholar] [CrossRef]
- Martínez-Iglesias, O.; Cacabelos, R. Epigenetic treatment of neurodegenerative disorders. Histone Modif. Ther. 2020, 20, 311–335. [Google Scholar]
- Lu, X.; Wang, L.; Yu, C.; Yu, D.; Yu, G. Histone Acetylation Modifiers in the Pathogenesis of Alzheimer’s Disease. Front. Cell. Neurosci. 2015, 9, 226. [Google Scholar] [CrossRef]
- Kontopoulos, E.; Parvin, J.D.; Feany, M.B. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum. Mol. Genet. 2006, 15, 3012–3023. [Google Scholar] [CrossRef] [PubMed]
- Guhathakurta, S.; Kim, J.; Adams, L.; Basu, S.; Song, M.K.; Adler, E.; Je, G.; Fiadeiro, M.B.; Kim, Y.S. Targeted attenuation of elevated histone marks at SNCA alleviates α-synuclein in Parkinson’s disease. EMBO Mol. Med. 2021, 13, 2. [Google Scholar] [CrossRef]
- Nicholas, A.P.; Lubin, F.D.; Hallett, P.J.; Vattem, P.; Ravenscroft, P.; Bezard, E.; Zhou, S.; Fox, S.H.; Brotchie, J.M.; Sweatt, J.D.; et al. Striatal histone modifications in models of levodopa-induced dyskinesia. J. Neurochem. 2008, 106, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gu, Z.; Lin, S.; Chen, L.; Dzreyan, V.; Eid, M.; Demyanenko, S.; He, B. Histone Deacetylases as Epigenetic Targets for Treating Parkinson’s Disease. Brain Sci. 2022, 12, 672. [Google Scholar] [CrossRef] [PubMed]
- St Laurent, R.; O’Brien, L.M.; Ahmad, S.T. Sodium butyrate improves locomotor impairment and early mortality in a rotenoneinduced drosophila model of parkinson’s disease. Neuroscience 2013, 246, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Sinclair, D.A. Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. 2010, 5, 253–295. [Google Scholar] [CrossRef] [PubMed]
- Julien, C.; Tremblay, C.; Emond, V.; Lebbadi, M.; Salem, N., Jr.; Bennett, D.A.; Calon, F. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2009, 68, 48–58. [Google Scholar] [CrossRef]
- Basavarajappa, B.; Subbanna, S. Histone Methylation Regulation in Neurodegenerative Disorders. Int. J. Mol. Sci. 2021, 22, 4654. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A. Targeting histone-modifications in Alzheimer’s disease. What is the evidence that this is a promising therapeutic avenue? Neuropharmacology 2014, 80, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Li, T.; Li, J.; Yang, J.; Liu, H.; Zhang, X.J.; Le, W. Jmjd3 is essential for the epigenetic modulation of microglia phenotypes in the immune pathogenesis of Parkinson’s disease. Cell Death Differ. 2014, 21, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Sadlon, A.; Takousis, P.; Alexopoulos, P.; Evangelou, E.; Prokopenko, I.; Perneczky, R. miRNAs Identify Shared Pathways in Alzheimer’s and Parkinson’s Diseases. Trends Mol. Med. 2019, 25, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Sheinerman, K.S.; Umansky, S.R. Circulating cell-free microRNA as biomarkers for screening, diagnosis and monitoring of neurodegenerative diseases and other neurologic pathologies. Front. Cell. Neurosci. 2013, 7, 150. [Google Scholar] [CrossRef] [PubMed]
- Sheinerman, K.S.; Tsivinsky, V.G.; Abdullah, L.; Crawford, F.; Umansky, S.R. Plasma microRNA biomarkers for detection of mild cognitive impairment: Biomarker validation study. Aging Cell 2013, 5, 925–938. [Google Scholar] [CrossRef]
- Siedlecki-Wullich, D.; Català-Solsona, J.; Fábregas, C.; Hernández, I.; Clarimon, J.; Lleó, A.; Boada, M.; Saura, C.A.; Rodríguez-Álvarez, J.; Miñano-Molina, A.J. Altered microRNAs related to synaptic function as potential plasma biomarkers for Alzheimer’s disease. Alzheimer’s Res. Ther. 2019, 11, 46. [Google Scholar] [CrossRef]
- Cacabelos, R. Epigenomic networking in Drug Development: From Pathogenic Mechanisms to Pharmacogenomics. Drug. Dev. Res. 2014, 75, 348–365. [Google Scholar] [CrossRef]
- Golla, U. Emergence of nutraceuticals as the alternative medications for pharmaceuticals. Int. J. Complement. Altern. Med. 2018, 11, 155–158. [Google Scholar] [CrossRef]
- Aggarwal, R.; Jha, M.; Shrivastava, A.; Jha, A. Natural compounds:role in reversal of epigenetic changes. Biochemistry 2015, 80, 972–989. [Google Scholar]
- Huang, D.; Cui, L.; Ahmed, S.; Zainab, F.; Wu, Q.; Wang, X.; Yuan, Z. An overview of epigenetic and natural nutrition products targeting DNA methyltransferase, histone deacetylases and microRNAs. Food Chem. Toxicol. 2019, 123, 574–594. [Google Scholar] [CrossRef]
- Alissa, E.M.; Ferns, G.A. Functional Foods and Nutraceuticals in the Primary Prevention of Cardiovascular Diseases. J. Nutr. Metab. 2012, 2012, 569486. [Google Scholar] [CrossRef]
- Kordiak, J.; Bielec, F.; Jabłoński, S.; Pastuszak-Lewandoska, D. Role of Beta-Carotene in Lung Cancer Primary Chemoprevention: A Systematic Review with Meta-Analysis and Meta-Regression. Nutrients 2022, 14, 1361. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhou, Y.; Yang, J.; Li, H.; Zhang, H.; Zheng, P. Curcumin induces apoptotic cell death and protective autophagy in human gastric cancer cells. Oncol. Rep. 2017, 37, 3459–3466. [Google Scholar] [CrossRef] [PubMed]
- FAO. Human vitamin and mineral requirements. In Report of a Joint FAO/WHO Expert Consultation; World Health Organization and Food and Agriculture Organization of the United Nations FAO: Rome, Italy, 2015; Chapter 7 (Vitamin C). [Google Scholar]
- Alshahrani, F.; Aljohani, N. Vitamin D: Deficiency, sufficiency and toxicity. Nutrients 2013, 5, 3605–3616. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.R., III; Pastor-Barriuso, R.; Dalal, D.; Riemersma, R.A.; Appel, L.J.; Guallar, E. Meta-analysis: High-dosage vitamin E supplementation may increase all-cause mortality. Ann. Intern. Med. 2005, 142, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Bast, A.; Haenen, G.R. The toxicity of antioxidants and their metabolites. Environ. Toxicol. Pharmacol. 2002, 11, 251–258. [Google Scholar] [CrossRef]
- Mohajeri, M.H.; Troesch, B.; Weber, P. Inadequate supply of vitamins and DHA in the elderly: Implications for brain aging and Alzheimer-type dementia. Nutrition 2015, 31, 261–275. [Google Scholar] [CrossRef]
- Athanasopoulos, D.; Karagiannis, G.; Tsolaki, M. Recent findings in alzheimer disease and nutrition focusing on epigenetics. Adv. Nutr. 2016, 7, 917–927. [Google Scholar] [CrossRef]
- Martin, S.L.; Hardy, T.M.; Tollefsbol, T.O. Medicinal chemistry of the epigenetic diet and caloric restriction. Curr. Med. Chem. 2013, 20, 4050–4059. [Google Scholar] [CrossRef]
- Dauncey, M.J. Genomic and epigenomic insights into nutrition and brain disorders. Nutrients 2013, 5, 887–914. [Google Scholar] [CrossRef]
- Monti, N.; Cavallaro, R.A.; Stoccoro, A.; Nicolia, V.; Scarpa, S.; Kovacs, G.G.; Fiorenza, M.T.; Lucarelli, M.; Aronica, E.; Ferrer, I.; et al. CpG and non-CpG Presenilin1 methylation pattern in course of neurodevelopment and neurodegeneration is associated with gene expression in human and murine brain. Epigenetics 2020, 15, 781–799. [Google Scholar] [CrossRef]
- Lin, H.C.; Hsieh, H.-M.; Chen, Y.-H.; Hu, M.-L. S-Adenosylhomocysteine increases β-amyloid formation in BV-2 microglial cells by increased expressions of β-amyloid precursor protein and presenilin 1 and by hypomethylation of these gene promoters. NeuroToxicology 2009, 30, 622–627. [Google Scholar] [CrossRef]
- Aisen, P.S.; Schneider, L.S.; Sano, M.; Diaz-Arrastia, R.; Van Dyck, C.H.; Weiner, M.F.; Bottiglieri, T.; Jin, S.; Stokes, K.T.; Thomas, R.G.; et al. High-dose B vitamin supplementation and cognitive decline in Alzheimer disease: A randomized controlled trial. JAMA 2008, 300, 1774–1783. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Liu, Y.H.; Zhang, C.E.; Wang, Q.; Wei, Z.; Mousseau, D.D.; Wang, J.Z.; Tian, Q.; Liu, G.P. Folate/vitamin-B12 prevents chronic hyperhomocysteinemia-induced tau hyperphosphorylation and memory deficits in aged rats. J. Alzheimers Dis. 2011, 27, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, A.; Sapehia, D.; Thakur, S.; Mohanraj, P.; Bagga, R.; Kaur, J. Effect of imbalance in folate and vitamin B12 in maternal/parental diet on global methylation and regulatory miRNAs. Sci. Rep. 2019, 9, 17602. [Google Scholar] [CrossRef]
- Ghoshal, K.; Li, X.; Datta, J.; Bai, S.; Pogribny, I.; Pogribny, M.; Huang, Y.; Young, D.; Jacob, S.T. A folate- and methyl-deficient diet alters the expression of DNA methyltransferases and methyl CpG binding proteins involved in epigenetic gene silencing in livers of F344 rats. J. Nutr. 2006, 136, 1522–1527. [Google Scholar] [CrossRef]
- Cavalcante Da Silva, V.; Fernandes, L.; Agamme, A.; Haseyama, E.; Muniz, M.T.; Almeida, V. Maternal Vitamin B Deficiency and Epigenetic Changes of Genes Involved in the Alzheimer’ s Disease Pathogenesis. Biol. Med. 2017, 9, 3. [Google Scholar] [CrossRef]
- Shen, L. Associations between B Vitamins and Parkinson’s Disease. Nutrients 2015, 7, 7197–7208. [Google Scholar] [CrossRef]
- Reynolds, E. Vitamin B12, folic acid and the nervous system. Lancet Neurol. 2006, 5, 949–960. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Shea, T.B. Folate and homocysteine metabolism in neural plasticity and neurodegenerative disorders. Trends Neurosci. 2003, 26, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, J.; Tang, M.-X.; Miller, J.; Green, R.; Mayeux, R. Relation of higher folate intake to lower risk of Alzheimer’s Disease in the elederly. Arch. Neurol. 2007, 64, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Rolstein, A.; Kodesh, A.; Goldberg, Y.; Reichenberg, A.; Levine, S. Serum folate deficiency and the risks of dementia and all-cause mortality: A national study of old age. Evid. Based Ment. Health 2022, 25, 63–68. [Google Scholar] [CrossRef]
- Clarke, R.; Smith, A.D.; Jobst, K.A.; Refsum, H.; Sutton, L.; Ueland, P.M. Folate, vitamin B12 and serum total homocysteine levels in corfirmed Alzheimer Disease. Arch. Neurol. 1998, 55, 1449–1455. [Google Scholar] [CrossRef]
- Chen, H.; Liu, S.; Ji, L.; Wu, T.; Ji, Y.; Zhou, Y.; Zheng, M.; Zhang, M.; Xu, W.; Huang, G. Folic acid supplementation mitigates Alzheimer’s disease by reducing inflammation: A randomized controlled trial. Mediat. Inflamm. 2016, 2016, 5912146. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Wu, T.; Zhao, J.; Song, A.; Liu, H.; Xu, W.; Huang, G. Folic acid supplementation improves cognitive function by reducing the levels of peripheral inflammatory cytokines in elderly Chinese subbjects with MCI. Sci. Rep. 2016, 6, 37486. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bao, G.; Liu, D.; Yang, Y.; Li, X.; Cai, G.; Liu, Y.; Wu, Y. The association between folate and Alzheimer’s disease: A systematic review and meta-analysis. Front. Neurosci. 2021, 15, 661198. [Google Scholar] [CrossRef]
- Dong, B.; Wu, R. Plasma homocysteibe, folate and vitamin B12 levels in parkinson’s disease in China: A meta-analysis. Clin. Neurol. Neurosurg. 2020, 188, 105587. [Google Scholar] [CrossRef] [PubMed]
- de Lau, L.; Koudstaal, P.J.; Witteman, J.; Hofman, A.; Breteler, M. Dietary folate, vitamin B12, and vitamin B6 and the risk of Parkinson’s disease. Neurology 2006, 67, 315–318. [Google Scholar] [CrossRef] [PubMed]
- McCarter, S.; Stang, C.; Turcano, P.; Mielke, M.; Ali, F.; Bower, J.; Savica, R. Higher vitamin B12 level at Parkisnon’s disease diagnosis is associated with lower risk of future dementia. Park. Relat. Disord. 2020, 73, 19–22. [Google Scholar] [CrossRef] [PubMed]
- McCarter, S.; Teigen, L.; McCarter, A.; Benarroch, E.; St Lois, E.; Savica, R. Low vitamin B12 and Parkinson Disease. Mayo Clin. Proc. 2019, 94, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Betch, M.; Harandi-Zadeh, S.; Shen, K.; Lubecka, K.; Kitts, D.; O’Hagan, H.; Stefanska, B. Dietary antioxidants remodel DNA methylation patterns in chronic disease. Br. J. Pharm. 2020, 177, 1382–1408. [Google Scholar] [CrossRef]
- Park, L.K.; Friso, S.; Choi, S.-W. Nutritional influences on epigenetics and age-related disease. Proc. Nutr. Soc. 2012, 71, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Joven, J.; Micol, V.; Segura-Carretero, A.; Alonso-Villaverde, V.; Menéndez, J. Bioactive food components platforms: Polyphenols and the modulation of gene expression pathways: Can we eat or way out of the danger of chronic disease? Crit. Rev. Food. Sci. Nutr. 2014, 54, 985–1001. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Castegna, A.; Drake, J.; Scapagnini, G.; Calabrese, V. Vitamin E and Neurodegenrative Disorders associated with oxidative stress. Nutr. Neurosci. 2002, 5, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Ulatowski, L.; Mannor, D. Vitamin E and neurodegeneration. Neurobiol. Dis. 2015, 84, 78–83. [Google Scholar] [CrossRef]
- Camarena, V.; Wang, G. The epigenetic role of vitamin C in health and disease. Cell Mol. Life Sci. 2016, 73, 1645–1658. [Google Scholar] [CrossRef]
- Kuttan, R.; Bhanumathy, P.; Nirmala, K.; George, M.C. Potential anticancer activity of turmeric (Curcuma longa). Cancer Lett. 1985, 29, 197–202. [Google Scholar] [CrossRef]
- Nelson, K.M.; Dahlin, J.L.; Bisson, J.; Graham, J.; Pauli, G.F.; Walters, M.A. The essential medicinal chemistry of curcumin: Miniperspective. J. Med. Chem. 2017, 60, 1620–1637. [Google Scholar] [CrossRef]
- Beltzig, L.; Frumkina, A.; Schwarzenbach, C.; Kaina, B. Cytotoxic, Genotoxic and Senolytic Potential of Native and Micellar Curcumin. Nutrients 2021, 13, 2385. [Google Scholar] [CrossRef]
- Oon, S.; Nallappan, M.; tee, T.; Shohaimi, S.; Kassim, N.; Sa’ariwijaya M and Cheah, Y. Xanthorrhizol: A review of its pharmacological activities and anticancer properties. Cancer Cell Int. 2015, 15, 10. [Google Scholar] [CrossRef]
- Kocaadam, B.; ¸Sanlier, N. Curcumin, an active component of turmeric (Curcuma longa), and its effects on health. Crit. Rev. Food Sci. Nutr. 2017, 57, 2889–2895. [Google Scholar] [CrossRef]
- Lao, C.D.; Ruffin, M.T., 4th; Normolle, D.; Heath, D.D.; Murray, S.I.; Bailey, J.M.; Boggs, M.E.; Crowell, J.; Rock, C.L.; Brenner, D.E. Dose escalation of a curcuminoid formulation. BMC Complement. Altern. Med. 2006, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Chainani-Wu, N. Safety and anti-inflammatory activity of curcumin: A component of tumeric (Curcuma longa). J. Altern. Complement. Med. 2003, 9, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, N.; Aggarwal, B.B.; Newman, R.A.; Wolff, R.A.; Kunnumakkara, A.B.; Abbruzzese, J.L.; Ng, C.S.; Badmaev, V.; Kurzrock, R. Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 4491–4499. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.A.; McLelland, H.R.; Hill, K.A.; Ireson, C.R.; Euden, S.A.; Manson, M.M.; Pirmohamed, M.; Marnett, L.J.; Gescher, A.J.; Steward, W.P. Pharmacodynamic and pharmacokinetic study of oral Curcuma extract in patients with colorectal cancer. Clin. Cancer Res. Off. J. 2001, 7, 1894–1900. [Google Scholar]
- Ryan, J.L.; Heckler, C.E.; Ling, M.; Katz, A.; Williams, J.P.; Pentland, A.P.; Morrow, G.R. Curcumin for radiation dermatitis: A randomized, double-blind, placebo-controlled clinical trial of thirty breast cancer patients. Radiat. Res. 2013, 180, 34–43. [Google Scholar] [CrossRef]
- Kurien, B.T.; Singh, A.; Matsumoto, H.; Scofield, R.H. Improving the solubility and pharmacological efficacy of curcumin by heat treatment. Assay. Drug. Dev. Technol. 2007, 5, 567–576. [Google Scholar] [CrossRef]
- Song, Y.; Sonawane, N.; Salinas, D.; Qian, L.; Pedemonte, N.; Galietta, L.J.; Verkman, A. Evidence against the rescue of defective ΔF508-CFTR cellular processing by curcumin in cell culture and mouse models. J. Biol. Chem. 2004, 279, 40629–40633. [Google Scholar] [CrossRef]
- European Medicines Agency. Assessment Report on Curcuma xanthorrhiza Roxb. (C. xanthorrhiza D. dietrich), Rhizome. 2014 (EMA/HMPC/604598/2012). Available online: https://www.ema.europa.eu/en/documents/herbal-report/draft-assessment-report-curcuma-xanthorrhiza-roxb-rhizoma_en.pdf (accessed on 8 January 2023).
- Liu, W.; Zhai, Y.; Heng, X.; Che, F.Y.; Chen, W.; Sun, D.; Zhai, G. Oral bioavailability of curcumin: Problems and advancements. J. Drug Target. 2016, 24, 694–702. [Google Scholar] [CrossRef]
- Cuomo, J.; Appendino, G.; Dern, A.S.; Schneider, E.; McKinnon, T.P.; Brown, M.J.; Togni, S.; Dixon, B.M. Comparative absorption of a standardized curcuminoid mixture and its lecithin formulation. J. Nat. Prod. 2011, 74, 664–669. [Google Scholar] [CrossRef]
- Gota, V.S.; Maru, G.B.; Soni, T.G.; Gandhi, T.R.; Kochar, N.; Agarwal, M.G. Safety and pharmacokinetics of a solid lipid curcumin particle formulation in osteosarcoma patients and healthy volunteers. J. Agric. Food Chem. 2010, 58, 2095–2099. [Google Scholar] [CrossRef]
- Dadhaniya, P.; Patel, C.; Muchhara, J.; Bhadja, N.; Mathuria, N.; Vachhani, K.; Soni, M.G. Safety assessment of a solid lipid curcumin particle preparation: Acute and subchronic toxicity studies. Food. Chem. Toxicol. 2011, 49, 1834–1842. [Google Scholar] [CrossRef] [PubMed]
- Shoba, G.; Joy, D.; Joseph, T.; Majeed, M.; Rajendran, R.; Srinivas, P.S. Influence of piperine on the pharmacokinetics of curcumin in animals and human volunteers. Planta Med. 1998, 64, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, T.; Sunagawa, Y.; Katanassaka, Y.; Hiraon, S.; Namiki, M.; Watanabe, Y.; Suzuki, H.; Doi, O.; Suzuki, K.; Yamauchi, M.; et al. Drinkable preparation of Theracurmin exhibits high absorption efficiency—A single-dose, double-blind, 4-way crossover study. Biol. Pharm. Bull. 2013, 36, 708–1714. [Google Scholar] [CrossRef]
- Maiti, P.; Dunbar, G.L. Use of curcumin, a natural polyphenol for targeting molecular pathways in treating age-related neurodegenerative diseases. Int. J. Mol. Sci. 2018, 19, 1637. [Google Scholar] [CrossRef]
- Bahramsoltani, R.; Rahimi, R.; Farzaei, M. Pharmacokinetic interactions of curcuminoids with conventional drugs: A review. J. Ethnopharmacol. 2017, 14, 1–12. [Google Scholar] [CrossRef]
- Hassan, F.U.; Rehman, M.S.U.; Khan, M.S.; Ali, M.A.; Javed, A.; Nawaz, A.; Yang, C. Curcumin as an alternative epigenetic modulator: Mechanism of action and potential effects. Front. Genet. 2019, 10, 514. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Gupta, S.C.; Park, B.; Goel, A.; Aggarwal, B.B. Epigenetic changes induced by curcumin and other natural compounds. Genes Nutr. 2011, 6, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Suresh, D.; Srinivasan, K. Tissue distribution & elimination of capsaicin, piperine & curcumin following oral intake in rats. Indian J. Med. Res. 2010, 131, 682–691. [Google Scholar]
- Begum, A.N.; Jones, M.R.; Lim, G.P.; Morihara, T.; Kim, P.; Heath, D.D.; Rock, C.L.; Pruitt, M.A.; Yang, F.; Hudspeth, B.; et al. Curcumin structure-function, bioavailability, and efficacy in models of neuroinflammation and Alzheimer’s disease. J. Pharm. Exp. 2008, 326, 196–208. [Google Scholar] [CrossRef]
- Mythri, R.B.; Bharath, M.S. Curcumin: A potential neuroprotective agent in Parkinson’s disease. Curr. Pharm. Des. 2012, 18, 91–99. [Google Scholar] [CrossRef]
- Witkin, J.M.; Li, X. Curcumin, an active constiuent of the ancient medicinal herb Curcuma longa L.: Some uses and the establishment and biological basis of medical efficacy. CNS Neurol. Disord. Drug Targets 2013, 12, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanyam, K.; Varier, R.A.; Altaf, M.; Swaminathan, V.; Siddappa, N.B.; Ranga, U.; Kundu, T.K. Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J. Biol. Chem. 2004, 279, 51163–51171. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; McClure, D.; Jimenez, L.A.; Megson, I.L.; Rahman, I. Curcumin induces glutathione biosynthesis and inhibits NFkappaB activation and interleukin-8 release in alveolar epithelial cells: Mechanism of free radical scavenging activity. Antioxid. Redox Signal. 2005, 7, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Li, Q.; Chang, R.; Yang, D.; Song, Z.; Guo, Q.; Huang, C. Curcumin alleviates neuropathic pain by inhibiting p300/CBP histone acetyltransferase activity-regulated expression of BDNF and cox-2 in a rat model. PLoS ONE 2014, 9, e91303. [Google Scholar] [CrossRef]
- Ogiwara, H.; Ui, A.; Shiotani, B.; Zou, L.; Yasui, A.; Kohno, T. Curcumin suppresses multiple DNA damage response pathways and has potency as a sensitizer to PARP inhibitor. Carcinogenesis 2013, 34, 2486–2497. [Google Scholar] [CrossRef] [PubMed]
- Bora-Tatar, G.; Dayangaç-Erden, D.; Demir, A.S.; Dalkara, S.; Yelekçi, K.; Erdem-Yurter, H. Molecular modifications on carboxylic acid derivatives as potent histone deacetylase inhibitors: Activity and docking studies. Bioorg. Med. Chem. 2009, 17, 52195228. [Google Scholar] [CrossRef]
- Liu, H.L.; Chen, Y.; Cui, G.H.; Zhou, J.F. Curcumin, a potent antitumor reagent, is a novel histone deacetylase inhibitor regulating BNHL cell line Raji proliferation. Acta. Pharmacol. Sin. 2005, 26, 603–609. [Google Scholar] [CrossRef]
- Chiu, S.; Woodbury-Fariña, M.A.; Shad, M.U.; Husni, M.; Copen, J.; Bureau, Y.; Cernovsky, Z.; Hou, J.J.; Raheb, H.; Terpstra, K.; et al. The role of nutrient-based epigenetic changes in buffering against stress, aging, and Alzheimer’s disease. Psychiatr. Clin. N. Am. 2014, 37, 591–623. [Google Scholar] [CrossRef]
- Lu, X.; Deng, Y.; Yu, D.; Cao, H.; Wang, L.; Liu, L.; Yu, C.; Zhang, Y.; Guo, X.; Yu, G. Histone acetyl-transferase p300 mediates histone acetylation of PS1 and BACE1 in a cellular model of Alzheimer’s disease. PLoS ONE 2014, 9, e103067. [Google Scholar]
- Meng, J.; Li, Y.; Camarillo, C.; Yao, Y.; Zhang, Y.; Xu, C.; Jiang, L. The antitumor histone deacetylase inhibitor SAHA and the natural flavonoid curcumin exhibit synergistic neuroprotection against amyloid-beta toxicity. PLoS ONE 2014, 9, e85570. [Google Scholar]
- Adwan, L.; Zawia, N. Epigenetics: A no vel therapeutic approach for the treatment of Alzheimer’s disease. Pharm. Ther. 2013, 139, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Mrcu, M.G.; Jung, Y.-J.; Lee, S.; Chung, E.-J.; Lee, M.J.; Trepel, J.; Neckers, L. Curcumin is an inhibitor of p300 histone acetyltransferase. Med. Chem. 2006, 2, 169–174. [Google Scholar]
- Wei, W.; Wang, Z.-Y.; Ma, L.-N.; Zhang, T.-T.; Cao, Y.; Li, H. MicroRNAs in Alzheiemr’s Disease: Function and potential applications as diagnostic biomarkers. Front. Mol. Neurosci. 2020, 13, 160. [Google Scholar] [CrossRef]
- Sun, A.Y.; Wang, Q.; Simonyi, A.; Sun, G.Y. Resveratrol as a therapeutic agent for Neurodegenerative Diseases. Mol. Neurobiol. 2010, 41, 375–383. [Google Scholar] [CrossRef]
- Zhang, L.X.; Li, C.X.; Kakar, M.U.; Khan, M.S.; Wu, P.F.; Amir, R.M.; Dai, D.F.; Naveed, M.; Li, Q.Y.; Saeed, M.; et al. Resveratrol (RV): A pharmacological review and call for further research. Biomed Pharmacother. 2021, 143, 112164. [Google Scholar] [CrossRef] [PubMed]
- Vang, N.O.; Ahmad, C.A.; Baile, J.A.; Baur, K.; Brown, A.; Csiszar, D.K.; Das, D.; Delmas, C.; Gottfried, H.Y.; Lin, Q.Y.; et al. What is new for an old molecule? Systematic review and recommendations on the use of resveratrol. PLoS ONE 2011, 6, 19881. [Google Scholar] [CrossRef]
- Patel, K.R.; Scott, E.; Brown, V.A.; Gescher, A.J.; Steward, W.P.; Brown, K. Clinical trials of resveratrol. Ann. N. Y. Acad. Sci. 2011, 1215, 161–169. [Google Scholar] [CrossRef]
- Cicero, A.; Ruscica, M.; Banach, M. Resveratrol and cognitive decline: A clinician perspective. Arc. Med. Sci. 2019, 15, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.R.; Appel, C.L. Polyphenols as dietary supplements: A double-edged sword. Nutr. Diet. Suppl. 2010, 2, 1–12. [Google Scholar] [CrossRef]
- Walle, T.; Hsieh, F.; DeLegge, M.; Oatis, J.E., Jr.; Walle, U.K. High absortion but very low bioavailability of oral resveratrol in humans. Drug Metab. Dispos. 2004, 32, 1377–1382. [Google Scholar] [CrossRef]
- Almeida, L.; Vaz-da-Silva, M.; Falcão, A.; Soares, E.; Costa, R.; Loureiro, A.I.; Fernandes-Lopes, C.; Rocha, J.F.; Nunes, T.; Wright, L.; et al. Pharmacokinetic and safety profile of trans-resveratrol in a rising multiple-dose study in healthy volunteers. Mol. Nutr. Food Res. 2009, 53 (Suppl 1), S7–S15. [Google Scholar] [CrossRef] [PubMed]
- Delmas, D.; Aires, V.; Limagne, E.; Dutartre, P.; Mazue, F.; Ghiringhelli, F.; Latruffe, N. Transport, stability, and biological activity of resveratrol. Ann. N. Y. Acad. Sci. 2011, 1215, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Sang, S. Metabolism and pharmacokinetics of resveratrol and pterostilbene. Biofactors 2018, 44, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Lancon, A.; Delmas, D.; Osman, H.; Thenot, J.P.; Jannin, B.; Latruffe, N. Human hepatic cell uptake of resveratrol: Involvement of both passive diffusion and carrier-mediated process. Biochem. Biophys. Res. Commun. 2004, 316, 1132–1137. [Google Scholar] [CrossRef]
- Baur, J.A.; Sinclair, D.A. Therapeutic potential of resveratrol: The in vivo evidence. Nat. Rev. Drug Discov. 2006, 5, 493–506. [Google Scholar] [CrossRef]
- Arbo, B.D.; André-Miral, C.; Nasre-Nasser, R.G.; Schimith, L.E.; Santos, M.G.; Costa-Silva, D.; Muccillo-Baisch, A.L.; Hort, M.A. Resveratrol Derivatives as Potential Treatments for Alzheimer’s and Parkinson’s Disease. Front. Aging Neurosci. 2020, 20, 103. [Google Scholar] [CrossRef]
- Arbo, B.D.; André-Miral, C.; Nasre-Nasser, R.G.; Schimith, L.E.; Santos, M.G.; Costa-Silva, D.; Muccillo-Baisch, A.L.; Hort, M.A. Comparision of piceid and resveratrol in antioxidation and antiproliferation activities in vitro. PLoS ONE 2013, 8, e54505. [Google Scholar]
- Chen, J.; Zhou, Y.; Mueller-Steiner, S.; Chen, L.F.; Kwon, H.; Yi, S.; Mucke, L.; Gan, L. SIRT1 protects against microglia-dependent amyloid-beta toxicity through inhibiting NF-kappaB signaling. J. Biol. Chem. 2005, 280, 40364–40374. [Google Scholar] [CrossRef]
- Feng, X.; Liang, N.; Zhu, D.; Gao, Q.; Peng, L.; Dong, H.; Yue, Q.; Liu, H.; Bao, L.; Zhang, J.; et al. Resveratrol Inhibits β-Amyloid-Induced neuronal apoptosis through regulation of SIRT1-ROCK1 signaling pathway. PLoS ONE 2013, 8, e59888. [Google Scholar] [CrossRef]
- Zhang, J.; Feng, X.; Wu, J.; Xu, H.; Li, G.; Zhu, D.; Yue, Q.; Liu, H.; Zhang, Y.; Sun, D.; et al. Neuroprotective effects of resveratrol on damages of mouse cortical neurons induced by β-amyloid through activation of SIRT1/Akt1 pathway. Biofactors 2013, 40, 258–267. [Google Scholar] [CrossRef]
- Granzotto, A.; Zatta, P. Resveratrol acts not through antiaggregative pathways but mainly via its scavenging properties against Aβ and Aβ-metal complexes toxicity. PLoS ONE 2011, 6, e21565. [Google Scholar] [CrossRef] [PubMed]
- Tosatti, J.; Fialho da Silva, A.; Caramelli, P.; Braga Gomes, K. Effects of Resveratrol supplementation on the cognitive function of patients with Alzheimer’s Disease: A systematic review of randomized controlled assays. Drugs Aging 2022, 39, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Tellone, E.; Galtieri, A.; Russo, A.; Giardina, B.; Ficarra, S. Resveratrol: A Focus on Several Neurodegenerative Diseases. Oxid. Med. Cell Longev. 2015, 2015, 392169. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Xu, L.; Qu, C.; Sun, H.; Zhang, J. Resveratrol prevents cognitive deficits induced by chronic unpredictable mild stress: Sirt1/miR-134 signalling pathway regulates CREB/BDNF expression in hippocampus in vivo and in vitro. Behav. Brain. Res. 2018, 349, 1–7. [Google Scholar] [CrossRef]
- Zhao, Y.-N.; Li, W.-F.; Li, F.; Zhang, Z.; Dai, Y.-D.; Xu, A.L.; Qi, C.; Gao, J.M.; Gao, J. Resveratrol improves learning and memory in normally aged mice through microRNA-CREB pathway. Biochem. Biophys. Res. Comm. 2013, 435, 597–602. [Google Scholar] [CrossRef]
- Kou, X.; Chen, N. Resveratrol as a natural autophagy regulator for prevention and treatment of Alzheimer’s Disease. Nutrients 2017, 9, 927. [Google Scholar] [CrossRef]
- Ferretta, A.; Gaballo, A.; Tanzarella, P.; Piccoli, C.; Capitanio, N.; Nico, B.; Annese, T.; Di Paola, M.; Dell’Aquila, C.; De Mari, M.; et al. Effect of resveratrol on mitochondrial function: Implications in parkin-associated familiar Parkinson’s disease. Biochim. Biophys. Acta 2014, 1842, 902–915. [Google Scholar] [CrossRef]
- Donmez, G.; Arun, A.; Chung, C.Y.; McLean, P.J.; Lindquist, S.; Guarente, L. SIRT1 protects against alpha-synuclein aggregation by activating molecular chaperones. J. Neurosci. 2012, 32, 124–132. [Google Scholar] [CrossRef]
- Hardy, T.M.; Tollefsbol, T. Epigenetic diet: Impact on the epigenome and cancer. Epigenomics 2012, 3, 503–518. [Google Scholar] [CrossRef]
- Feng, Y.; Liu, T.; Dong, S.Y.; Guo, Y.J.; Jankovic, J.; Xu, H.; Wu, Y.C. Rotenone affects p53 transcriptional activity and apoptosis via targeting SIRT1 and H3K9 acetylation in SH-SY5Y cells. J. Neurochem. 2015, 134, 668–676. [Google Scholar] [CrossRef]
- Xia, D.; Sui, R.; Zhang, Z. Administration of resveratrol improved parkinson’s disease-like phenotype by suppresing apoptosis of neurons via modulating the MALAT1/miR-129/SNCA signaling pathway. J. Cell Biochem. 2019, 120, 4942–4951. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-H.; Zhang, J.-L.; Duan, Y.-L.; Zhang, Q.-S.; Li, G.-F.; Zheng, D.-L. MicroRNA-214 participates in the neuroprotective effect of Resveratrol via inhibiting a-synuclein expression in MPTP-induced Parkinson’s disease mouse. Biomed Pharm. 2015, 74, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Fujiki, H.; Sueoka, E.; Watanabe, T.; Suganuma, M. Primary cancer prevention by green tea, and tertiary cancer prevention by the combination of green tea catechins and anticancer compounds. J. Cancer Prev. 2015, 20, 1–4. [Google Scholar] [CrossRef]
- Blumberg, J.; Bolling, B.W.; Chen, C.O.; Xiao, H. Review and perspective on the composition and safety of green tea extracts. Eur. J. Nutr. Food Saf. 2015, 5, 1–31. [Google Scholar]
- Teschke, R.; Zhang, L.; Melzer, L.; Schulze, J.; Eickhoff, A. Green tea extract and the risk of drug-induced liver injury. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1663–1676. [Google Scholar] [CrossRef]
- Yates, A.A.; Erdman, J.W., Jr.; Shao, A.; Dolan, L.C.; Griffiths, J.C. Bioactive nutrients-Time for tolerable upper intake levels to address safety. Regul. Toxicol. Pharmacol. RTP 2017, 84, 94–101. [Google Scholar]
- EFSA ANS Panel (EFSA Panel on Food Additives and Nutrient Sources added to Food); Younes, M.; Aggett, P.; Aguilar, F.; Crebelli, R.; Dusemund, B.; Filipič, M.; Frutos, M.J.; Galtier, P.; Gott, D.; et al. Scientific Opinion on the safety of green tea catechins. EFSA J. 2018, 16, 5239. [Google Scholar]
- Hu, J.; Webster, D.; Cao, J.; Shao, A. The safety of green tea and green tea extract consumption in adults-Results of a systematic review. Regul. Toxicol. Pharmacol. RTP 2018, 95, 412–433. [Google Scholar]
- Mähler, A.; Mandel, S.; Lorenz, M.; Ruegg, U.; Wanker, E.E.; Boschmann, M.; Paul, F. Epigallocatechin-3-gallate: A useful, effective and safe clinical approach for targeted prevention and individualised treatment of neurological diseases? EPMA J. 2013, 4, 5. [Google Scholar]
- Cerbin-Koczorowska, M.; Waszyk-Nowaczyk, M.; Bakun, P.; Goslinski, T.; Koczorowski, T. Current View on Green Tea Catechins Formulations, Their Interactions with Selected Drugs, and Prospective Applications for Various Health Conditions. Appl. Sci. 2021, 11, 4905. [Google Scholar] [CrossRef]
- Lee, M.-J.; Maliakal, P.; Chen, L.; Meng, X.; Bondoc, F.Y.; Prabhu, S.; Lambert, G.; Mohr, S.; Yang, C.S. Pharmacokinetics of tea catechins after ingestion of green tea and (−)-epigallocatechin-3-gallate by humans: Formation of different metabolites and individual variability. Cancer Epidemiol. Biomark. Prev. 2002, 11, 1025–1032. [Google Scholar]
- Mereles, D.; Hunstein, W. Epigallocatechin-3-gallate (EGCG) for clinical trials: More pitfalls than promises? Int. J. Mol. Sci. 2011, 12, 5592–5603. [Google Scholar] [PubMed]
- Zwolak, I. Epigallocatechin Gallate for Management of Heavy Metal-Induced Oxidative Stress: Mechanisms of Action, Efficacy, and Concerns. Int. J. Mol. Sci. 2021, 22, 4027. [Google Scholar]
- Swezey, R.R.; Aldridge, D.E.; LeValley, S.E.; Crowell, J.A.; Hara, Y.; Green, C.E. Absorption, tissue distribution and elimination of 4-[H-3]-epigallocatechin gallate in beagle dogs. Int. J. Toxicol. 2003, 22, 187–193. [Google Scholar] [CrossRef]
- Lambert, J.D.; Sang, S.; Yang, C.S. Biotransformation of green tea polyphenols and the biological activities of those metabolites. Mol. Pharm. 2007, 4, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Ullmann, U.; Haller, J.; Decourt, J.P.; Girault, N.; Girault, J.; Richard-Caudron, A.S.; Pineau, B.; Weber, P. A single ascending dose study of epigallocatechin gallate in healthy volunteers. J. Int. Med. Res. 2003, 31, 88–101. [Google Scholar] [CrossRef]
- Elbling, L.; Herbacek, I.; Weiss, R.M.; Jantschitsch, C.; Micksche, M.; Gerner, C.; Pangratz, H.; Grusch, M.; Knasmüller, S.; Berger, W. Hydrogen peroxide mediates EGCG-induced antioxidant protection in human keratinocytes. Free Radic. Biol. Med. 2010, 49, 1444–1452. [Google Scholar] [CrossRef]
- Satoh, M.; Takemura, Y.; Hamada, H.; Sekido, Y.; Kubota, S. EGCG induces human mesothelioma cell death by inducing reactive oxygen species and autophagy. Cancer Cell Int. 2013, 13, 19. [Google Scholar] [CrossRef]
- Fujimura, Y.; Kumazoe, M.; Tachibana, H. 67-kDa Laminin Receptor-Mediated Cellular Sensing System of Green Tea Polyphenol EGCG and Functional Food Pairing. Molecules 2022, 27, 5130. [Google Scholar] [CrossRef]
- Xu, M.J.; Liu, B.J.; Wang, C.L.; Wang, G.H.; Tian, Y.; Wang, S.H.; Li, J.; Li, P.Y.; Zhang, R.H.; Wei, D.; et al. Epigallocatechin-3-gallate inhibits TLR4 signaling through the 67-kDa laminin receptor and effectively alleviates acute lung injury induced by H9N2 swine influenza virus. Int. Immunopharmacol. 2017, 52, 24–33. [Google Scholar] [CrossRef]
- Kim, H.S.; Quon, M.J.; Kim, J.A. New insights into the mechanisms of polyphenols beyond antioxidant properties; lessons from the green tea polyphenol, epigallocatechin 3-gallate. Redox Biol. 2014, 2, 187–195. [Google Scholar] [PubMed]
- Ide, K.; Yamada, H.; Takuma, N.; Park, M.; Wakamiya, N.; Nakase, J.; Ukawa, Y.; Sagesaka, Y.M. Green tea consumption affects cognitive dysfunction in the elderly: A pilot study. Nutrients 2014, 6, 4032–4042. [Google Scholar] [CrossRef]
- Gu, Y.J.; He, C.H.; Li, S.; Zhang, S.Y.; Duan, S.Y.; Sun, H.P.; Shen, Y.P.; Xu, Y.; Yin, J.Y.; Pan, C.W. Tea consumption is associated with cognitive impairment in older Chinese adults. Aging Ment. Health 2018, 22, 1232–1238. [Google Scholar]
- Lee, J.W.; Lee, Y.K.; Ban, J.O.; Ha, T.Y.; Yun, Y.P.; Han, S.B.; Oh, K.W.; Hong, J.T. Green tea (-)-epigallocatechin-3-gallate inhibits beta-amyloid-induced cognitive dysfunction through modification of secretase activity via inhibition of ERK and NF-kappaB pathways in mice. J. Nutr. 2009, 139, 1987–1993. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, Y.; Quan, Z.; Wong, W.; Guo, J.; Zhang, R.; Yang, Q.; Dai, R.; McGeer, P.L.; Qing, H. Epigallocatechin Gallate (EGCG) Inhibits Alpha-Synuclein Aggregation: A Potential Agent for Parkinson’s Disease. Neurochem. Res. 2016, 41, 2788–2796. [Google Scholar]
- Tanaka, K.; Miyake, Y.; Fukushima, W.; Sasaki, S.; Kiyohara, C.; Tsuboi, Y.; Yamada, T.; Oeda, T.; Miki, T.; Kawamura, N.; et al. Intake of Japanese and Chinese teas reduces risk of Parkinson’s disease. Park. Relat. Disord. 2011, 17, 446–450. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. US National Institute of Health. Available online: https://clinicaltrials.gov/ (accessed on 8 January 2023).
- Chang, X.; Romg, C.; Chen, Y.; Yang, C.; Hu, Q. Epigallocatechin-3-gallate attenuates cognitive deterioration in Alzheimer’s disease model mice by upregulating neprylisin expression. Exp. Cell Res. 2015, 334, 136–145. [Google Scholar]
- Choudhury, S.; Balasubramanian, S.; Chin Chew, Y.; Han, B.; Marquez, V.; Eckert, R. (-)-Epigallocatechin-3-gallate and DZNep reduce polycomb protein level via a proteasome-dependent mechanism in skin cancer cells. Carcinogenesis 2011, 32, 1525–1532. [Google Scholar]
- Corzo, L.; Fernández-Novoa, L.; Carrera, I.; Martínez, O.; Rodríguez, S.; Alejo, R.; Cacabelos, R. Nutrition, Health, and Disease: Role of Selected Marine and Vegetal Nutraceuticals. Nutrients 2020, 12, 747. [Google Scholar] [CrossRef]
- Zhao, W.-Q.; Latinwo, L.; Liu, X.; Lee, E.S.; Lamango, N.; Charlton, C.G. L-Dopa Upregulates the Expression and Activities of Methionine Adenosyl Transferase and Catechol-O-Methyltransferase. Exp. Neurol. 2001, 171, 127–138. [Google Scholar] [CrossRef]
- Zoccolella, S.; dell’Aquila, C.; Specchio, L.; Logroscino, G.; Lamberti, P. Elevated Homocysteine Levels in Parkinson’s Disease: Is there Anything Besides L-Dopa Treatment? Curr. Med. Chem. 2010, 17, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, I.; Kaut, O.; Khazneh, H.; deBoni, L.; Ahmad, A.; Berg, D.; Klein, C.; Fröhlich, H.; Wüllner, U. L-dopa increases α-synuclein DNA methylation in Parkinson’s disease patients in vivo and in vitro. Mov. Disord. 2015, 30, 1794–1801. [Google Scholar] [PubMed]
- Bottiglieri, T.; Arning, E.; Wasek, B.; Nunbhakdi-Craig, N.; Sontag, J.-M.; Sontag, E. Acute Administration of l-Dopa Induces Changes in Methylation Metabolites, Reduced Protein Phosphatase 2A Methylation, and Hyperphosphorylation of Tau Protein in Mouse Brain. J. Neurosci. 2012, 32, 9173–9181. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.; Parada, E.; González-Lafuente, L.; Farré-Alins, V.; Ramos, E.; Cacabelos, R.; Egea, J. Neuroprotective effects of E-PodoFavalin-15999 (Atremorine®). CNS Neurosci. Ther. 2017, 23, 450–452. [Google Scholar] [CrossRef]
- Carrera, I.; Fernandez-Novoa, L.; Sampedro, C.; Cacabelos, R. Neuroprotective Effect of Atremorine in an Experimental Model of Parkinson’s Disease. Curr. Pharm. Des. 2017, 23, 2673–2684. [Google Scholar] [CrossRef]
- Cacabelos, R.; Fernández-Novoa, L.; Alejo, R.; Corzo, L.; Rodríguez, S.; Alcaraz, M.; Nebril, L.; Cacabelos, P.; Fraile, C.; Carrera, I.; et al. E-PodoFavalin-15999 (Atremorine®)-induced neurotransmitter and hormonal response in Parkinson’s Disease. J. Exp. Res. Pharmacol. 2016, 1, 1–12. [Google Scholar]
- Cacabelos, R.; Fernández-Novoa, L.; Alejo, R.; Corzo, L.; Alcaraz, M.; Nebril, L.; Cacabelos, P.; Fraile, C.; Carrera, I.; Carril, J.C. E-PodoFavalin-15999 (Atremorine®)-induced dopamine response in Parkinson’s Disease: Pharmacogenetics-related effects. J. Genom. Med. Pharm. 2016, 1, 1–26. [Google Scholar]
- Martínez-Iglesias, O.; Naidoo, V.; Carril, J.C.; Carrera, I.; Corzo, L.; Rodriguez, S.; Alejo, R.; Cacabelos, N.; Cacabelos, R. AtreMorine treatment regulates DNA methylation in Neurodegenerative Disorders: Epigenetic and pharmacogenetic studies. Curr. Pharm. Pers. Med. 2020, 17, 159–171. [Google Scholar]
- Cacabelos, R. Parkinson’s Disease: From Pathogenesis to Pharmacogenomics. Int. J. Mol. Sci. 2017, 18, 551. [Google Scholar] [CrossRef]
- Cacabelos, R.; Carrera, I.; Martínez, O.; Alejo, R.; Fernández-Novoa, L.; Cacabelos, P.; Corzo, L.; Rodríguez, S.; Alcaraz, M.; Nebril, L.; et al. Atremorine in Parkinson’s disease: From dopaminergic neuroprotection to pharmacogenomics. Med. Res. Rev. 2021, 41, 2841–2886. [Google Scholar]
- Martínez-Iglesias, O.; Naidoo, V.; Carrera, I.; Corzo, L. Cacabelos Nosustrophine: An epinutraceutical bioproduct with effects on DNA methylation, Histone acetylation and Sirtuin expression in Alzheimer’s Disease. Pharmaceutics 2022, 14, 2447. [Google Scholar] [PubMed]
- Dominguez, L.; Barbagallo, M. Nutritional prevention of cognitive decline and dementia. Acta Biomed. 2018, 89, 276–290. [Google Scholar] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNA Methylation | Histone Modifications | miRNA Regulation | |
|---|---|---|---|
| Vitamins A, C, and E | YES | YES | NO |
| Vitamin B12 | YES | NO | NO |
| Curcumin | YES | YES | YES |
| Resveratrol | YES | YES | YES |
| AtreMorine | YES | NO | NO |
| Nosustrophine | YES | YES | NO |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Iglesias, O.; Naidoo, V.; Carrera, I.; Corzo, L.; Cacabelos, R. Natural Bioactive Products as Epigenetic Modulators for Treating Neurodegenerative Disorders. Pharmaceuticals 2023, 16, 216. https://doi.org/10.3390/ph16020216
Martínez-Iglesias O, Naidoo V, Carrera I, Corzo L, Cacabelos R. Natural Bioactive Products as Epigenetic Modulators for Treating Neurodegenerative Disorders. Pharmaceuticals. 2023; 16(2):216. https://doi.org/10.3390/ph16020216
Chicago/Turabian StyleMartínez-Iglesias, Olaia, Vinogran Naidoo, Iván Carrera, Lola Corzo, and Ramón Cacabelos. 2023. "Natural Bioactive Products as Epigenetic Modulators for Treating Neurodegenerative Disorders" Pharmaceuticals 16, no. 2: 216. https://doi.org/10.3390/ph16020216
APA StyleMartínez-Iglesias, O., Naidoo, V., Carrera, I., Corzo, L., & Cacabelos, R. (2023). Natural Bioactive Products as Epigenetic Modulators for Treating Neurodegenerative Disorders. Pharmaceuticals, 16(2), 216. https://doi.org/10.3390/ph16020216