
HSPB5 Inhibition by NCI-41356 Reduces Experimental Lung Fibrosis by Blocking TGF-β1 Signaling

 , ,
, ,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
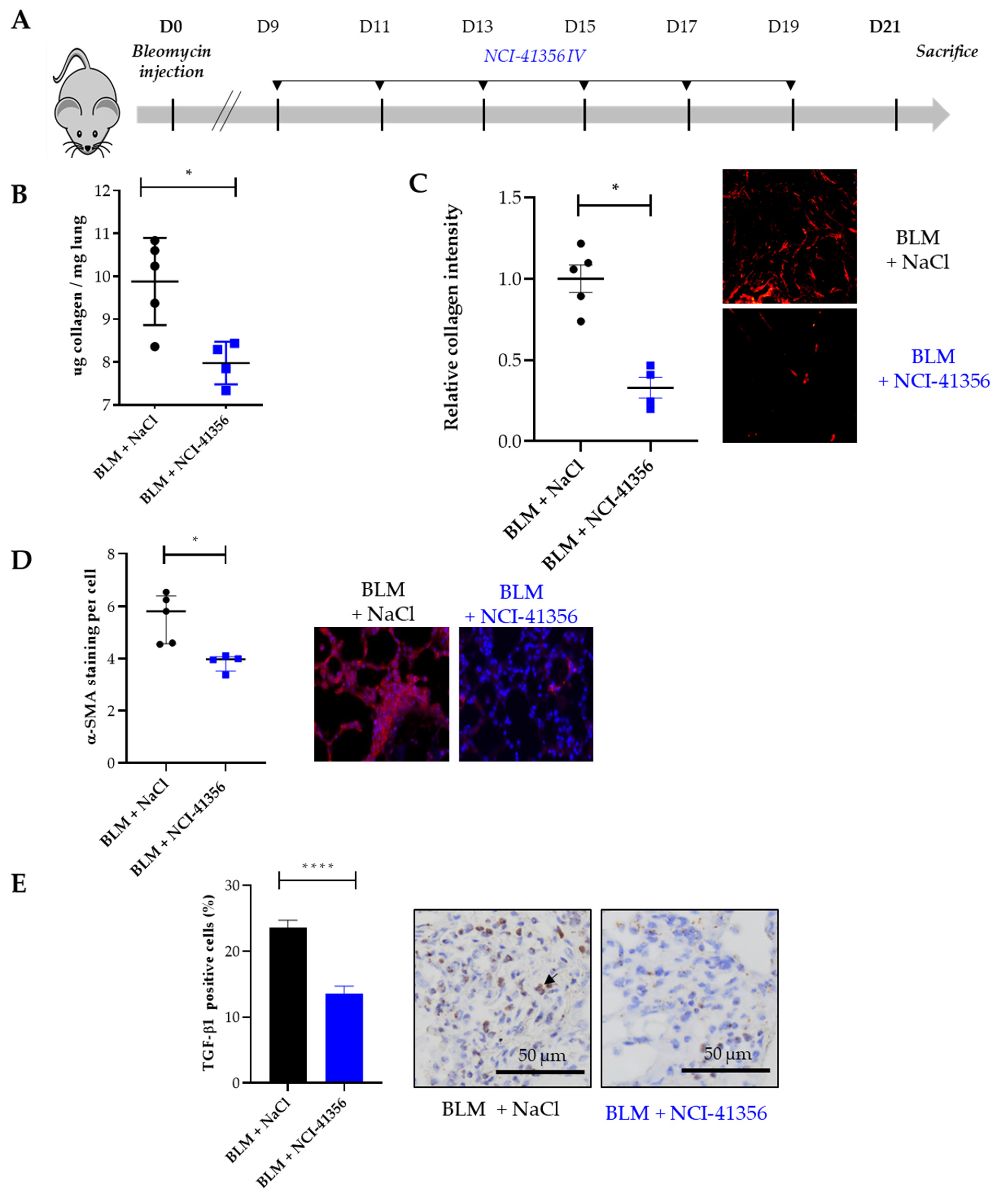
2.1. Systemic Intravenous Administration of NCI-41356 Protects from Bleomycin (BLM) Induced Lung Fibrosis
2.2. Local Intra-Tracheal Administration of NCI-41356 Protects from BLM-Induced Lung Fibrosis
2.3. NCI-41356 Inhibits HSPB5 Activity and Prevents HSPB5/SMAD4 Interaction
3. Discussion
4. Materials and Methods
4.1. Animal Procedures
4.2. Collagen Quantification
4.3. Ashcroft Scoring
4.4. Duolink Proximity-Ligation Assay (PLA)
4.5. Immunofluorescence
4.6. RNAscope Assay
4.7. RNAscope Quantification
4.8. Protein Interaction Studies
4.9. Cell Fractionation and Western Blot Analysis
4.10. Quantitative PCR Analysis
4.11. Anti-Aggregation Activity of HSPB5
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Raghu, G.; Remy-Jardin, M.; Richeldi, L.; Thomson, C.C.; Inoue, Y.; Johkoh, T.; Kreuter, M.; Lynch, D.A.; Maher, T.M.; Martinez, F.J.; et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2022, 205, e18–e47. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef] [PubMed]
- Maher, T.M.; Bendstrup, E.; Dron, L.; Langley, J.; Smith, G.; Khalid, J.M.; Patel, H.; Kreuter, M. Global Incidence and Prevalence of Idiopathic Pulmonary Fibrosis. Respir. Res. 2021, 22, 197. [Google Scholar] [CrossRef] [PubMed]
- Burgy, O.; Bellaye, P.-S.; Beltramo, G.; Goirand, F.; Bonniaud, P. Pathogenesis of Fibrosis in Interstitial Lung Disease. Curr. Opin. Pulm. Med. 2020, 26, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Bonniaud, P.; Kolb, M.; Galt, T.; Robertson, J.; Robbins, C.; Stampfli, M.; Lavery, C.; Margetts, P.J.; Roberts, A.B.; Gauldie, J. Smad3 Null Mice Develop Airspace Enlargement and Are Resistant to TGF-Beta-Mediated Pulmonary Fibrosis. J. Immunol. 2004, 173, 2099–2108. [Google Scholar] [CrossRef]
- Bellaye, P.-S.; Burgy, O.; Causse, S.; Garrido, C.; Bonniaud, P. Heat Shock Proteins in Fibrosis and Wound Healing: Good or Evil? Pharmacol. Ther. 2014, 143, 119–132. [Google Scholar] [CrossRef]
- Bellaye, P.-S.; Wettstein, G.; Burgy, O.; Besnard, V.; Joannes, A.; Colas, J.; Causse, S.; Marchal-Somme, J.; Fabre, A.; Crestani, B.; et al. The Small Heat-Shock Protein AB-Crystallin Is Essential for the Nuclear Localization of Smad4: Impact on Pulmonary Fibrosis. J. Pathol. 2014, 232, 458–472. [Google Scholar] [CrossRef]
- Bellaye, P.-S.; Burgy, O.; Colas, J.; Fabre, A.; Marchal-Somme, J.; Crestani, B.; Kolb, M.; Camus, P.; Garrido, C.; Bonniaud, P. Antifibrotic Role of AB-Crystallin Inhibition in Pleural and Subpleural Fibrosis. Am. J. Respir. Cell Mol. Biol. 2015, 52, 244–252. [Google Scholar] [CrossRef]
- Chen, Z.; Ruan, Q.; Han, S.; Xi, L.; Jiang, W.; Jiang, H.; Ostrov, D.A.; Cai, J. Discovery of Structure-Based Small Molecular Inhibitor of AB-Crystallin against Basal-like/Triple-Negative Breast Cancer Development in Vitro and in Vivo. Breast Cancer Res. Treat. 2014, 145, 45–59. [Google Scholar] [CrossRef]
- Vancheri, C.; Failla, M.; Crimi, N.; Raghu, G. Idiopathic Pulmonary Fibrosis: A Disease with Similarities and Links to Cancer Biology. Eur. Respir. J. 2010, 35, 496–504. [Google Scholar] [CrossRef]
- Jego, G.; Hazoumé, A.; Seigneuric, R.; Garrido, C. Targeting Heat Shock Proteins in Cancer. Cancer Lett. 2013, 332, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Tanguy, J.; Pommerolle, L.; Garrido, C.; Kolb, M.; Bonniaud, P.; Goirand, F.; Bellaye, P.-S. Extracellular Heat Shock Proteins as Therapeutic Targets and Biomarkers in Fibrosing Interstitial Lung Diseases. Int. J. Mol. Sci. 2021, 22, 9316. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Burns, T.F. Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approach. Int. J. Mol. Sci. 2017, 18, 1978. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.; Carter, T.R.; Cohen, M.S.; Blagg, B.S.J. Old and New Approaches to Target the Hsp90 Chaperone. Curr. Cancer Drug Targets 2020, 20, 253–270. [Google Scholar] [CrossRef] [PubMed]
- Boutanquoi, P.-M.; Burgy, O.; Beltramo, G.; Bellaye, P.-S.; Dondaine, L.; Marcion, G.; Pommerolle, L.; Vadel, A.; Spanjaard, M.; Demidov, O.; et al. TRIM33 Prevents Pulmonary Fibrosis by Impairing TGF-Β1 Signalling. Eur. Respir. J. 2020, 55, 1901346. [Google Scholar] [CrossRef]
- Teixeira, A.F.; ten Dijke, P.; Zhu, H.-J. On-Target Anti-TGF-β Therapies Are Not Succeeding in Clinical Cancer Treatments: What Are Remaining Challenges? Front. Cell Dev. Biol. 2020, 8, 605. [Google Scholar] [CrossRef]
- Burgy, O.; Crestani, B.; Bonniaud, P. Targeting the Nasty Nestin to Shoot Lung Fibrosis. Eur. Respir. J. 2022, 59, 2103146. [Google Scholar] [CrossRef] [PubMed]
- Barratt, S.L.; Blythe, T.; Ourradi, K.; Jarrett, C.; Welsh, G.I.; Bates, D.O.; Millar, A.B. Effects of Hypoxia and Hyperoxia on the Differential Expression of VEGF-A Isoforms and Receptors in Idiopathic Pulmonary Fibrosis (IPF). Respir. Res. 2018, 19, 9. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, K.; Ochiana, S.O.; Dunphy, M.P.; Gerecitano, J.F.; Corben, A.D.; Peter, R.I.; Janjigian, Y.Y.; Gomes-DaGama, E.M.; Koren, J.; Modi, S.; et al. Heat Shock Protein 90 Inhibitors in the Treatment of Cancer: Current Status and Future Directions. Expert Opin. Investig. Drugs 2014, 23, 611–628. [Google Scholar] [CrossRef]
- Colak, S.; Ten Dijke, P. Targeting TGF-β Signaling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef]
- Zoz, D.F.; Lawson, W.E.; Blackwell, T.S. Idiopathic Pulmonary Fibrosis: A Disorder of Epithelial Cell Dysfunction. Am. J. Med. Sci. 2011, 341, 435–438. [Google Scholar] [CrossRef] [PubMed]
- Planté-Bordeneuve, T.; Pilette, C.; Froidure, A. The Epithelial-Immune Crosstalk in Pulmonary Fibrosis. Front. Immunol. 2021, 12, 631235. [Google Scholar] [CrossRef] [PubMed]
- Hirani, N.; MacKinnon, A.C.; Nicol, L.; Ford, P.; Schambye, H.; Pedersen, A.; Nilsson, U.J.; Leffler, H.; Sethi, T.; Tantawi, S.; et al. Target Inhibition of Galectin-3 by Inhaled TD139 in Patients with Idiopathic Pulmonary Fibrosis. Eur. Respir. J. 2021, 57, 2002559. [Google Scholar] [CrossRef] [PubMed]
- Kaminskas, L.M.; Landersdorfer, C.B.; Bischof, R.J.; Leong, N.; Ibrahim, J.; Davies, A.N.; Pham, S.; Beck, S.; Montgomery, A.B.; Surber, M.W. Aerosol Pirfenidone Pharmacokinetics after Inhaled Delivery in Sheep: A Viable Approach to Treating Idiopathic Pulmonary Fibrosis. Pharm. Res. 2019, 37, 3. [Google Scholar] [CrossRef]
- Surber, M.W.; Beck, S.; Pham, S.; Marsden, A.T.; Gandi, S.K.; Baily, J.; McElroy, M.C. Inhaled Nintedanib Is Well-Tolerated and Delivers Key Pharmacokinetic Parameters Required to Treat Bleomycin-Induced Pulmonary Fibrosis. Pulm. Pharmacol. Ther. 2020, 63, 101938. [Google Scholar] [CrossRef]
- Cipolla, D. Will Pulmonary Drug Delivery for Systemic Application Ever Fulfill Its Rich Promise? Expert Opin. Drug Deliv. 2016, 13, 1337–1340. [Google Scholar] [CrossRef]
- Decologne, N.; Wettstein, G.; Kolb, M.; Margetts, P.; Garrido, C.; Camus, P.; Bonniaud, P. Bleomycin Induces Pleural and Subpleural Fibrosis in the Presence of Carbon Particles. Eur. Respir. J. 2010, 35, 176–185. [Google Scholar] [CrossRef]
- Decologne, N.; Kolb, M.; Margetts, P.J.; Menetrier, F.; Artur, Y.; Garrido, C.; Gauldie, J.; Camus, P.; Bonniaud, P. TGF-Beta1 Induces Progressive Pleural Scarring and Subpleural Fibrosis. J. Immunol. 2007, 179, 6043–6051. [Google Scholar] [CrossRef]
- Hübner, R.-H.; Gitter, W.; El Mokhtari, N.E.; Mathiak, M.; Both, M.; Bolte, H.; Freitag-Wolf, S.; Bewig, B. Standardized Quantification of Pulmonary Fibrosis in Histological Samples. Biotechniques 2008, 44, 507–517. [Google Scholar] [CrossRef]
- Wang, A.; Fogel, A.L.; Murphy, M.J.; Panse, G.; McGeary, M.K.; McNiff, J.M.; Bosenberg, M.; Vesely, M.D.; Cohen, J.M.; Ko, C.J.; et al. Cytokine RNA In Situ Hybridization Permits Individualized Molecular Phenotyping in Biopsies of Psoriasis and Atopic Dermatitis. JID Innov. 2021, 1, 100021. [Google Scholar] [CrossRef]
- Oh, H.J.; Easton, D.; Murawski, M.; Kaneko, Y.; Subjeck, J.R. The Chaperoning Activity of Hsp110. Identification of Functional Domains by Use of Targeted Deletions. J. Biol. Chem. 1999, 274, 15712–15718. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanguy, J.; Boutanquoi, P.-M.; Burgy, O.; Dondaine, L.; Beltramo, G.; Uyanik, B.; Garrido, C.; Bonniaud, P.; Bellaye, P.-S.; Goirand, F. HSPB5 Inhibition by NCI-41356 Reduces Experimental Lung Fibrosis by Blocking TGF-β1 Signaling. Pharmaceuticals 2023, 16, 177. https://doi.org/10.3390/ph16020177
Tanguy J, Boutanquoi P-M, Burgy O, Dondaine L, Beltramo G, Uyanik B, Garrido C, Bonniaud P, Bellaye P-S, Goirand F. HSPB5 Inhibition by NCI-41356 Reduces Experimental Lung Fibrosis by Blocking TGF-β1 Signaling. Pharmaceuticals. 2023; 16(2):177. https://doi.org/10.3390/ph16020177
Chicago/Turabian StyleTanguy, Julie, Pierre-Marie Boutanquoi, Olivier Burgy, Lucile Dondaine, Guillaume Beltramo, Burhan Uyanik, Carmen Garrido, Philippe Bonniaud, Pierre-Simon Bellaye, and Françoise Goirand. 2023. "HSPB5 Inhibition by NCI-41356 Reduces Experimental Lung Fibrosis by Blocking TGF-β1 Signaling" Pharmaceuticals 16, no. 2: 177. https://doi.org/10.3390/ph16020177
APA StyleTanguy, J., Boutanquoi, P.-M., Burgy, O., Dondaine, L., Beltramo, G., Uyanik, B., Garrido, C., Bonniaud, P., Bellaye, P.-S., & Goirand, F. (2023). HSPB5 Inhibition by NCI-41356 Reduces Experimental Lung Fibrosis by Blocking TGF-β1 Signaling. Pharmaceuticals, 16(2), 177. https://doi.org/10.3390/ph16020177