
Design, Synthesis, Evaluation and Molecular Dynamics Simulation of Dengue Virus NS5-RdRp Inhibitors

Abstract
:
1. Introduction
2. Results and Discussion
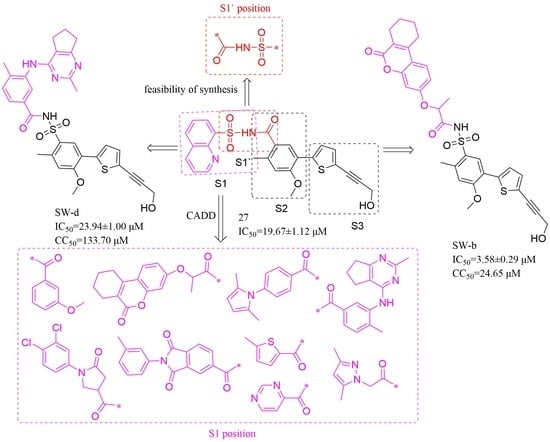
2.1. Compounds Design
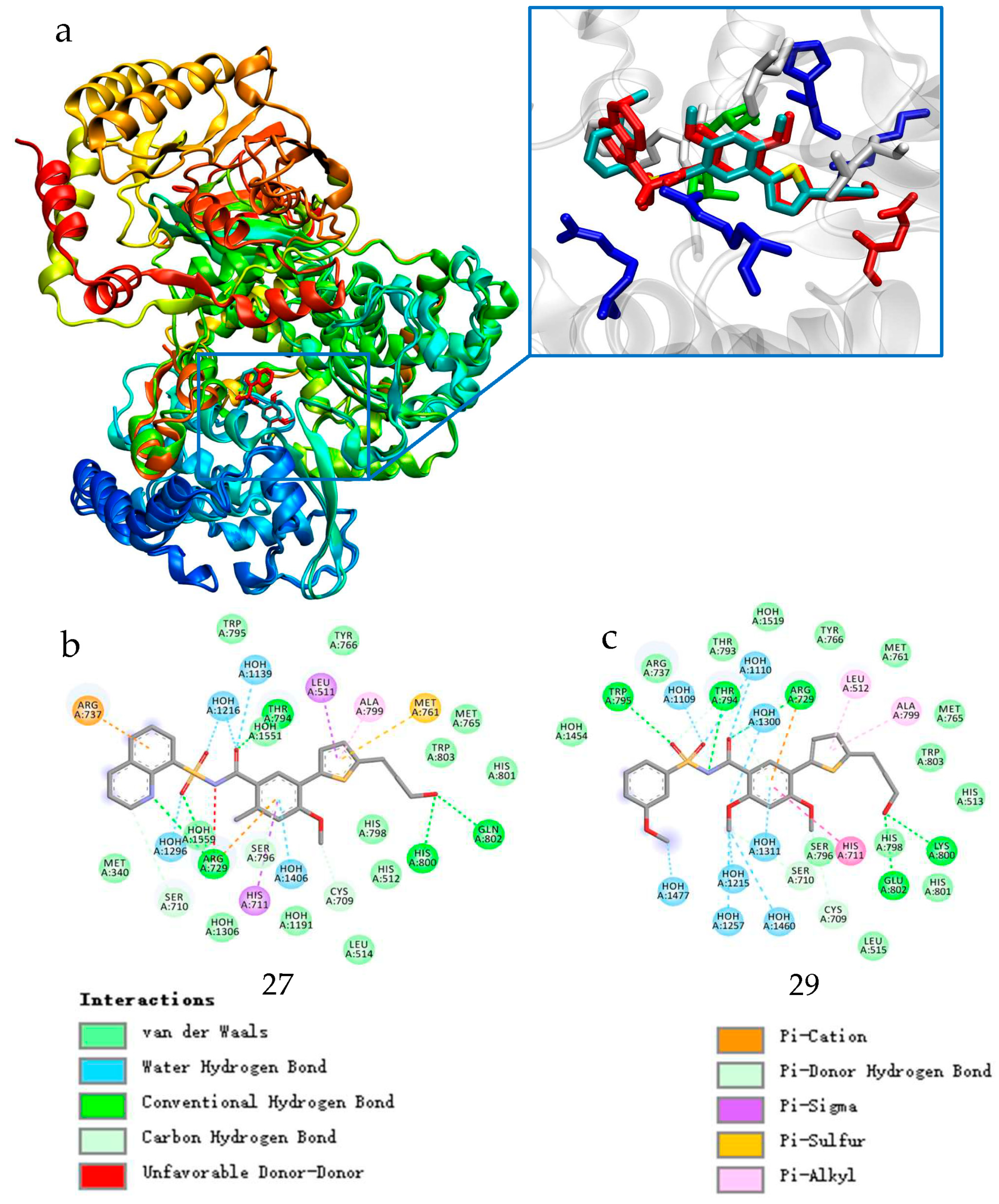
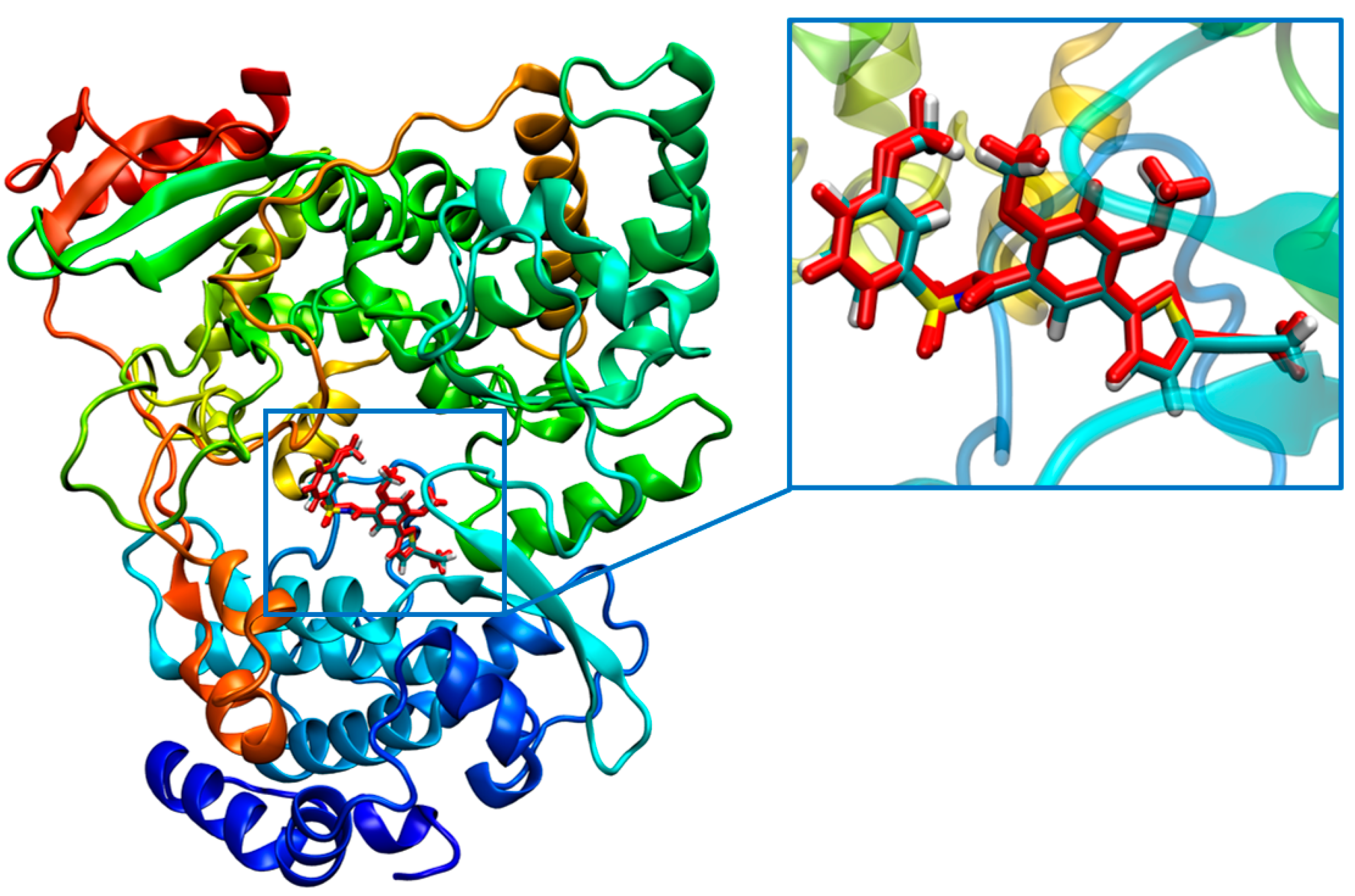
2.1.1. Binding Mode Analysis of Compounds 27 and 29
2.1.2. Construction of Virtual Compound Library
2.1.3. Virtual Screening and Selection of Target Compounds
Receptor Selection
Verification of the Docking Method Preparation
Virtual Screening, ADMET Prediction, Binding Free Energy Calculation, and Compound Selection
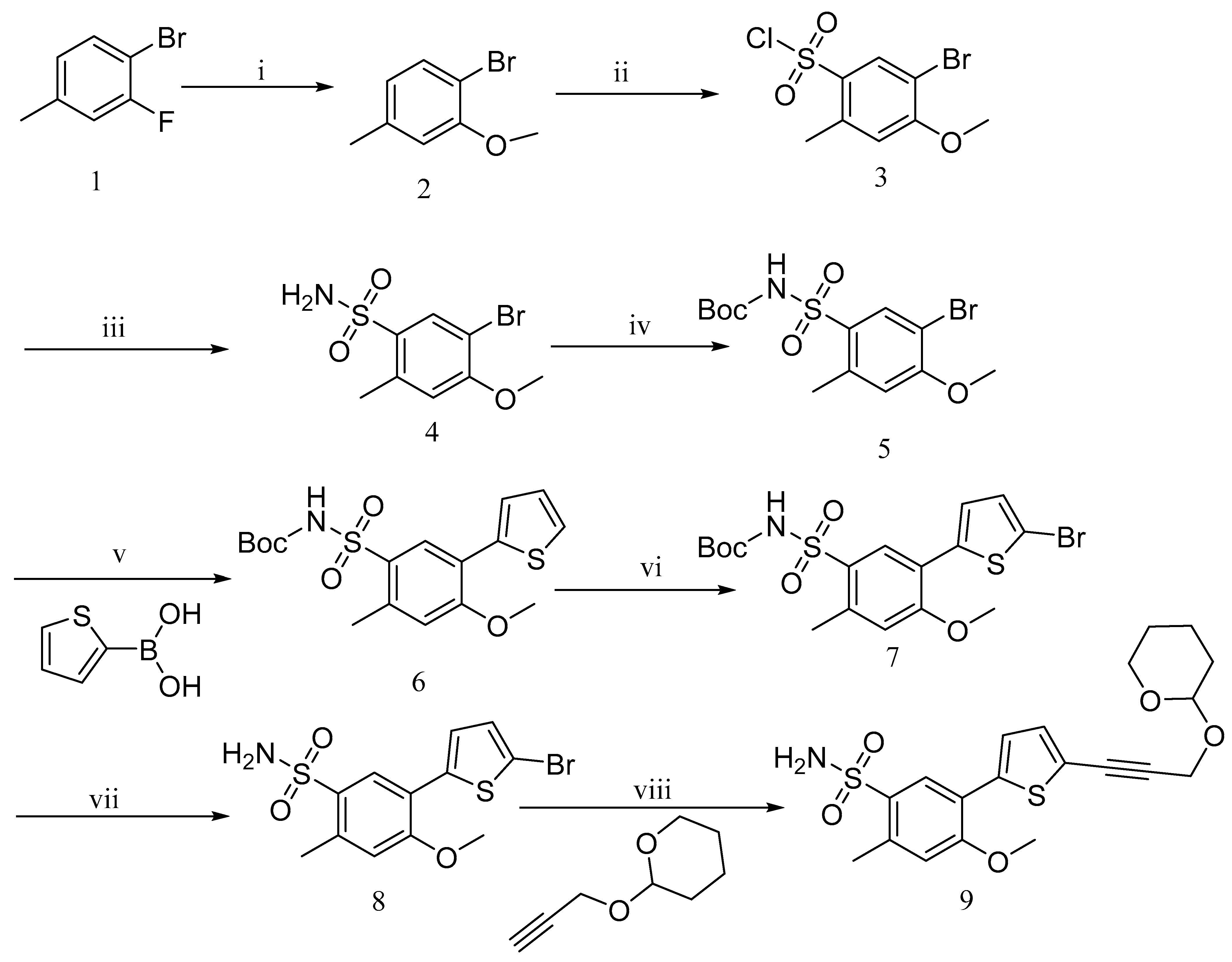
2.2. Synthesis
2.3. Biological Evaluation
2.3.1. Cytopathic Effect (CPE) Inhibition Assay and Cytotoxicity Assay
2.3.2. DENV Biochemical Enzyme Assay
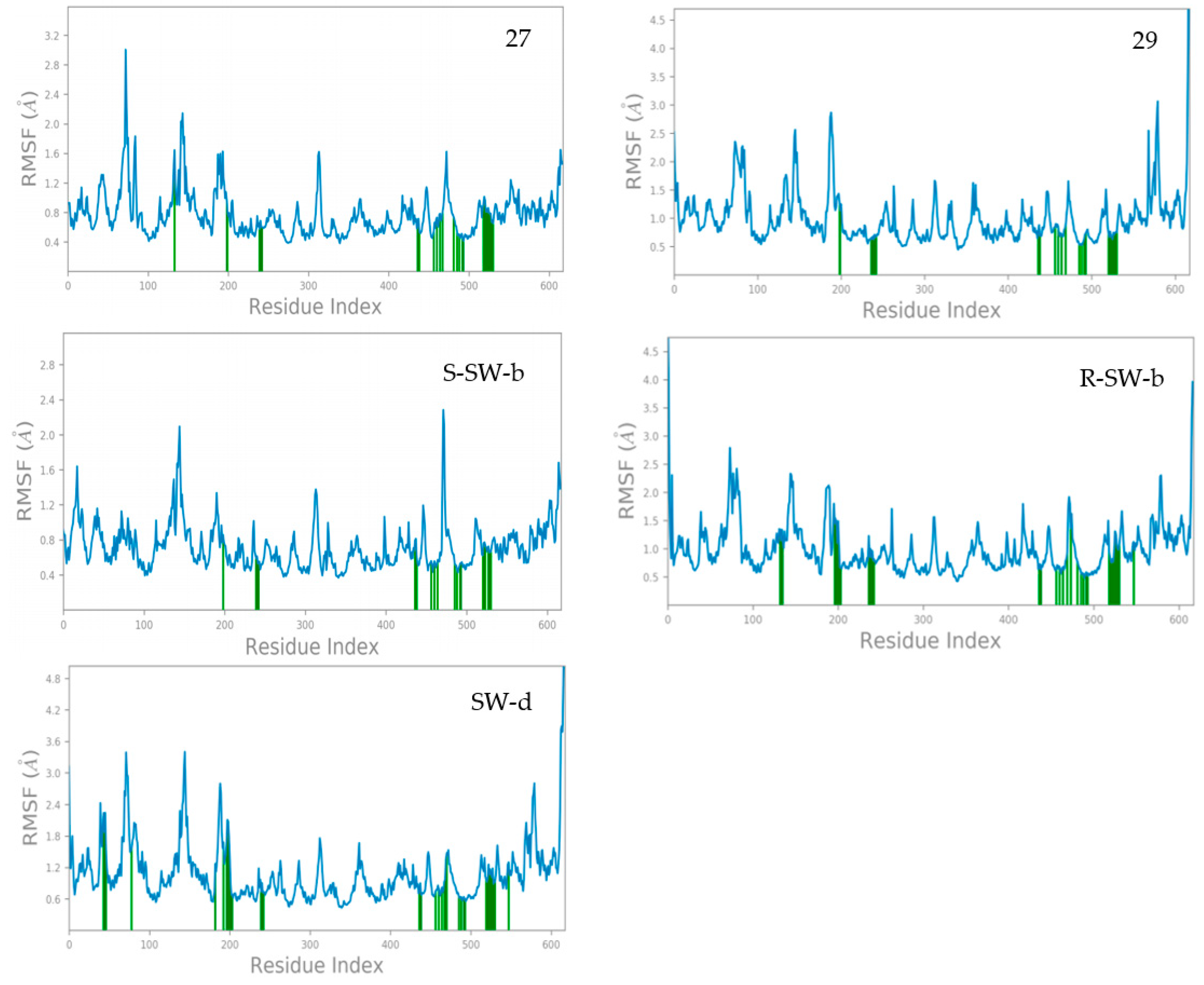
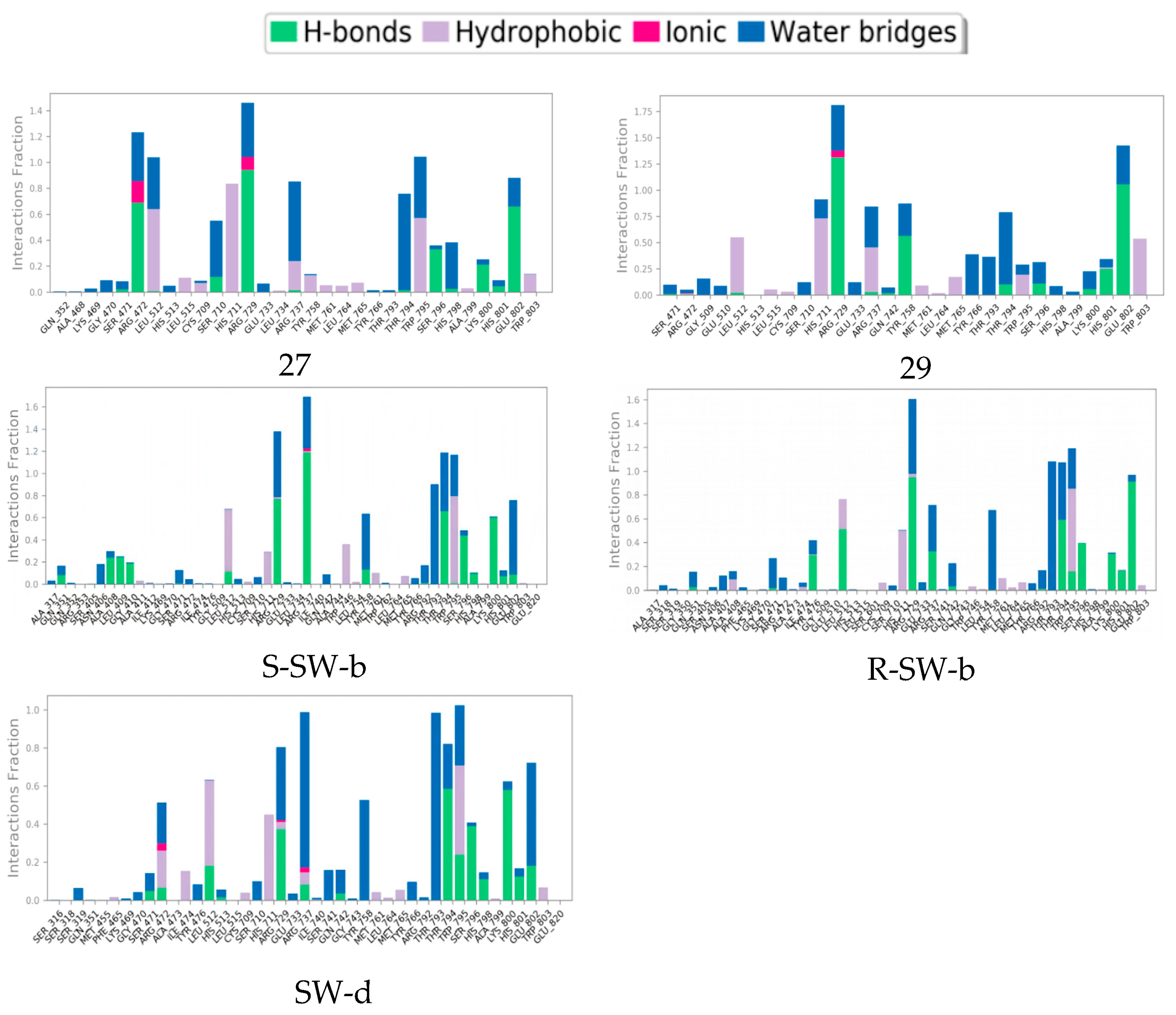
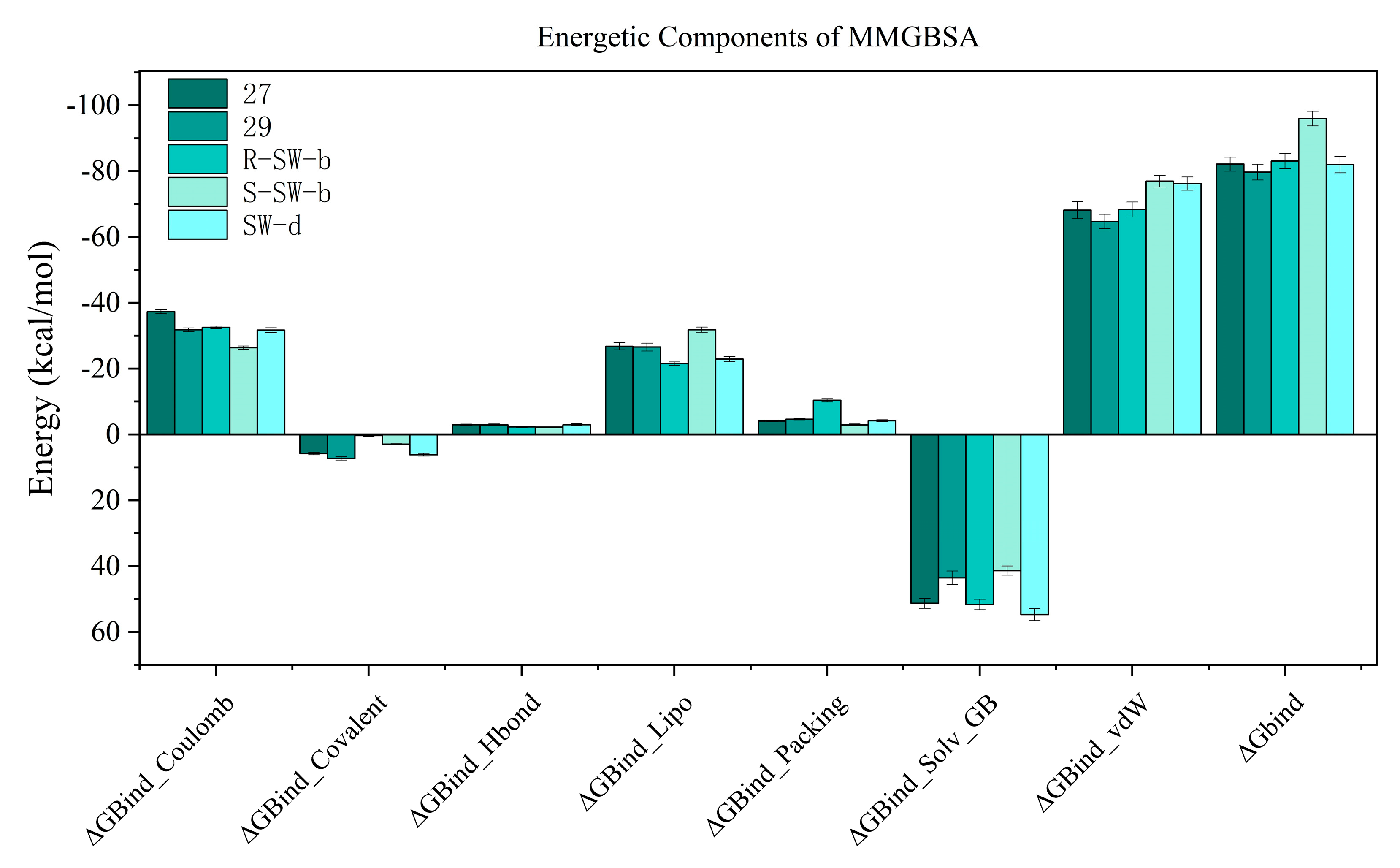
2.4. Molecular Dynamics (MD) Simulation and Analysis
3. Materials and Methods
3.1. Chemistry
General Chemistry Information
3.2. Biological Evaluation
3.2.1. Cloning, Expression, and Purification of the RdRp Domains
3.2.2. In Vitro DENV Biochemical Assay
3.2.3. Antiviral Screening and Toxicity Assays
3.2.4. DENV Biochemical Enzyme Assay
3.3. Virtual Screening Assay
3.3.1. Protein Preparation
3.3.2. Ligand Preparation
3.3.3. Docking Simulations
3.3.4. Calculation of Binding Energy
3.4. Molecular Dynamics (MD) Simulation
3.5. Thermodynamic Calculations: Binding Free Energy Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, W.H.; Urbina, A.N.; Chang, M.R.; Assavalapsakul, W.; Lu, P.L.; Chen, Y.H.; Wang, S.F. Dengue hemorrhagic fever—A systemic literature review of current perspectives on pathogenesis, prevention and control. J. Microbiol. Immunol. Infect. 2020, 53, 963–978. [Google Scholar] [CrossRef]
- Kuo, H.J.; Lee, I.K.; Liu, J.W. Analyses of clinical and laboratory characteristics of dengue adults at their hospital presentations based on the World Health Organization clinical-phase framework: Emphasizing risk of severe dengue in the elderly. J. Microbiol. Immunol. Infect. 2018, 51, 740–748. [Google Scholar] [CrossRef] [PubMed]
- Whitehorn, J.; Simmons, C.P. The pathogenesis of dengue. Vaccine 2011, 29, 7221–7228. [Google Scholar] [CrossRef] [PubMed]
- Mousson, L.; Dauga, C.; Garrigues, T.; Schaffner, F.; Vazeille, M.; Failloux, A.B. Phylogeography of Aedes (Stegomyia) aegypti (L.) and Aedes (Stegomyia) albopictus (Skuse) (Diptera: Culicidae) based on mitochondrial DNA variations. Genet. Res. 2005, 86, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Alshaikh, M.A.; Elnahary, E.K.; Elaiw, A.M. Stability of a secondary dengue viral infection model with multi-target cells. Alex. Eng. J. 2022, 61, 7075–7087. [Google Scholar] [CrossRef]
- Caillet-Saguy, C.; Lim, S.P.; Shi, P.Y. Polymerases of hepatitis C viruses and flaviviruses: Structural and mechanistic insights and drug development. Antivir. Res. 2014, 105, 8–16. [Google Scholar] [CrossRef]
- Koonin, E.V. The phylogeny of RNA-dependent RNA polymerases of positive-strand RNA viruses. J. Gen. Virol. 1991, 72, 2197–2206. [Google Scholar] [CrossRef]
- Martina, B.E.E.; Koraka, P.; Osterhaus, A.D.M.E. Dengue virus pathogenesis: An integrated view. Clin. Microbiol. Rev. 2009, 22, 564–581. [Google Scholar] [CrossRef]
- Yokokawa, F.; Nilar, S.; Noble, C.G.; Lim, S.P.; Rao, R.; Tania, S.; Wang, G.; Lee, G.; Hunziker, J.; Karuna, R.; et al. Discovery of potent non-nucleoside inhibitors of dengue viral RNA-dependent RNA polymerase from a fragment hit using structure-based drug design. J. Med. Chem. 2016, 59, 3935–3952. [Google Scholar] [CrossRef]
- Anoop, M.; Mathew, A.J.; Jayakumar, B.; Issac, A.; Nair, S.; Abraham, R.; Anupriya, M.G.; Sreekumar, E. Complete genome sequencing and evolutionary analysis of dengue virus serotype 1 isolates from an outbreak in Kerala, South India. Virus Genes 2012, 45, 1–13. [Google Scholar] [CrossRef]
- Qian, W.; Xue, J.X.; Xu, J.; Li, F.; Zhou, G.F.; Wang, F.; Luo, R.H.; Liu, J.; Zheng, Y.T.; Zhou, G.C. Design, synthesis, discovery and SAR of the fused tricyclic derivatives of indoline and imidazolidinone against DENV replication and infection. Bioorg. Chem. 2022, 120, 105639. [Google Scholar] [CrossRef] [PubMed]
- Songprakhon, P.; Thaingtamtanha, T.; Limjindaporn, T. Peptides targeting dengue viral nonstructural protein 1 inhibit dengue virus production. Sci. Rep. 2020, 10, 12933. [Google Scholar] [CrossRef]
- Hadinegoro, S.R.; Arredondo-García, J.L.; Capeding, M.R.; Deseda, C.; Chotpitayasunondh, T.; Dietze, R.; Hj Muhammad Ismail, H.I.; Reynales, H.; Limkittikul, K.; Rivera-Medina, D.M.; et al. Efficacy and long-term safety of a dengue vaccine in regions of endemic disease. N. Engl. J. Med. 2015, 373, 1195–1206. [Google Scholar] [CrossRef]
- Sridhar, S.; Luedtke, A.; Langevin, E.; Zhu, M.; Bonaparte, M.; Machabert, T.; Savarino, S.; Zambrano, B.; Moureau, A.; Khromava, A.; et al. Effect of dengue serostatus on dengue vaccine safety and efficacy. N. Engl. J. Med. 2018, 379, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.H.; Tsai, Y.T.; Wang, S.F.; Wang, W.H.; Chen, Y.H. Dengue vaccine: An update. Expert Rev. Anti. Infect. Ther. 2021, 19, 1495–1502. [Google Scholar] [CrossRef]
- Wilder-Smith, A. Dengue vaccine development by the year 2020: Challenges and prospects. Curr. Opin. Virol. 2020, 43, 71–78. [Google Scholar] [CrossRef]
- Siriphanitchakorn, T.; Kini, R.M.; Ooi, E.E.; Choy, M.M. Revisiting dengue virus-mosquito interactions: Molecular insights into viral fitness. J. Gen. Virol. 2021, 102, 001693. [Google Scholar] [CrossRef] [PubMed]
- Campos, R.K.; Garcia-Blanco, M.A.; Bradrick, S.S. Roles of pro-viral host factors in mosquito-borne flavivirus infections. Curr. Top. Microbiol. Immunol. 2018, 419, 43–67. [Google Scholar]
- Krishnan, M.N.; Garcia-Blanco, M.A. Targeting host factors to treat West Nile and dengue viral infections. Viruses 2014, 6, 683–708. [Google Scholar] [CrossRef]
- Egloff, M.P.; Decroly, E.; Malet, H.; Selisko, B.; Benarroch, D.; Ferron, F.; Canard, B. Structural and functional analysis of methylation and 5’-RNA sequence requirements of short capped RNAs by the methyltransferase domain of dengue virus NS5. J. Mol. Biol. 2007, 372, 723–736. [Google Scholar] [CrossRef]
- Issur, M.; Geiss, B.J.; Bougie, I.; Picard-Jean, F.; Despins, S.; Mayette, J.; Hobdey, S.E.; Bisaillon, M. The flavivirus NS5 protein is a true RNA guanylyltransferase that catalyzes a two-step reaction to form the RNA cap structure. RNA 2009, 15, 2340–2350. [Google Scholar] [CrossRef] [PubMed]
- Acosta, E.G.; Kumar, A.; Bartenschlager, R. Revisiting dengue virus-host cell interaction: New insights into molecular and cellular virology. Adv. Virus Res. 2014, 88, 1–109. [Google Scholar] [PubMed]
- Selisko, B.; Wang, C.; Harris, E.; Canard, B. Regulation of Flavivirus RNA synthesis and replication. Curr. Opin. Virol. 2014, 9, 74–83. [Google Scholar] [CrossRef]
- Bollati, M.; Milani, M.; Mastrangelo, E.; Ricagno, S.; Tedeschi, G.; Nonnis, S.; Decroly, E.; Selisko, B.; De Lamballerie, X.; Coutard, B.; et al. Recognition of RNA Cap in the wesselsbron virus NS5 methyltransferase domain: Implications for RNA-capping mechanisms in flavivirus. J. Mol. Biol. 2009, 385, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.P.; Noble, C.G.; Shi, P.Y. The dengue virus NS5 protein as a target for drug discovery. Antivir. Res. 2015, 119, 57–67. [Google Scholar] [CrossRef]
- Koonin, E.V. Computer-assisted identification of a putative methyltransferase domain in NS5 protein of flaviviruses and λ2 protein of reovirus. J. Gen. Virol. 1993, 74, 733–740. [Google Scholar] [CrossRef]
- Xu, H.T.; Colby-Germinario, S.P.; Hassounah, S.; Quashie, P.K.; Han, Y.; Oliveira, M.; Stranix, B.R.; Wain-berg, M.A. Identification of a pyridoxine-derived small-molecule inhibitor targeting dengue virus RNA-dependent RNA polymerase. Antimicrob Agents Chemother. 2015, 60, 600–608. [Google Scholar] [CrossRef]
- Cannalire, R.; Tarantino, D.; Astolfi, A.; Barreca, M.L.; Sabatini, S.; Massari, S.; Tabarrini, O.; Milani, M.; Querat, G.; Mastrangelo, E.; et al. Functionalized 2,1-benzothiazine 2,2-dioxides as new inhibitors of Dengue NS5 RNA-dependent RNA polymerase. Eur. J. Med. Chem. 2018, 143, 1667–1676. [Google Scholar] [CrossRef]
- Noble, C.G.; Lim, S.P.; Chen, Y.L.; Liew, C.W.; Yap, L.; Lescar, J.; Shi, P.Y. Conformational flexibility of the dengue virus RNA-dependent RNA polymerase revealed by a complex with an inhibitor. J. Virol. 2013, 87, 5291–5295. [Google Scholar] [CrossRef]
- Qadir, A.; Riaz, M.; Saeed, M.; Shahzad-Ul-Hussan, S. Potential targets for therapeutic intervention and structure based vaccine design against Zika virus. Eur. J. Med. Chem. 2018, 156, 444–460. [Google Scholar] [CrossRef]
- Manvar, D.; Kucukguzel, I.; Erensoy, G.; Tatar, E.; Deryabasogullari, G.; Reddy, H.; Talele, T.T.; Cevik, O.; Kaushik-Basu, N. Discovery of conjugated thiazolidinone-thiadiazole scaffold as anti-dengue virus polymerase inhibitors. Biochem. Biophys. Res. Commun. 2016, 469, 743–747. [Google Scholar] [CrossRef] [PubMed]
- Anusuya, S.; Velmurugan, D.; Gromiha, M.M. Identification of dengue viral RNA-dependent RNA polymerase inhibitor using computational fragment-based approaches and molecular dynamics study. J. Biomol. Struct. Dyn. 2016, 34, 1512–1532. [Google Scholar] [CrossRef]
- Lim, S.P.; Noble, C.G.; Seh, C.C.; Soh, T.S.; El Sahili, A.; Chan, G.K.; Lescar, J.; Arora, R.; Benson, T.; Nilar, S.; et al. Potent allosteric dengue virus NS5 polymerase inhibitors: Mechanism of action and resistance profiling. PLoS Pathog. 2016, 12, 1005737. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.L.; Xu, T.; Chen, Y.L.; Malet, H.; Egloff, M.P.; Canard, B.; Vasudevan, S.G.; Lescar, J. Crystal structure of the dengue virus RNA-dependent RNA polymerase catalytic domain at 1.85-angstrom resolution. J. Virol. 2007, 81, 4753–4765. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Kalnmals, C.A. Stereoselective synthesis of exocyclic tetrasubstituted vinyl halides via ru-catalyzed halotropic cycloisomerization of 1,6-haloenynes. Org. Lett. 2017, 19, 2346–2349. [Google Scholar] [CrossRef] [PubMed]
- Schröder, F.; Tugny, C.; Salanouve, E.; Clavier, H.; Giordano, L.; Moraleda, D.; Gimbert, Y.; Mouriès-Mansuy, V.; Goddard, J.-P.; Fensterbank, L. Secondary phosphine oxide–gold(i) complexes and their first application in catalysis. Organometallics 2014, 33, 4051–4056. [Google Scholar] [CrossRef]
- Wucher, P.; Goldbach, V.; Mecking, S. Electronic influences in phosphinesulfonato palladium(II) polymerization catalysts. Organometallics 2013, 32, 4516–4522. [Google Scholar] [CrossRef]
- Alexandre, F.-R.; Brandt, G.; Caillet, C.; Chaves, D.; Convard, T.; Derock, M.; Gloux, D.; Griffon, Y.; Lallos, L.; Leroy, F.; et al. Synthesis and antiviral evaluation of a novel series of homoserine-based inhibitors of the hepatitis C virus NS3/4A serine protease. Bioorg. Med. Chem. Lett. 2015, 25, 3984–3991. [Google Scholar] [CrossRef]
- Nguyen, S.S.; Ferreira, A.J.; Long, Z.G.; Heiss, T.K.; Dorn, R.S.; Row, R.D.; Prescher, J.A. Butenolide synthesis from functionalized cyclopropenones. Org. Lett. 2019, 21, 8695–8699. [Google Scholar] [CrossRef]
- Xie, L.; Takeuchi, Y.; Cosentino, L.M.; Mcphail, A.T.; Lee, K.-H. Anti-AIDS agents. 42. synthesis and anti-HIV activity of disubstituted (3′R,4′R)-3′,4′-di-O-(S)-camphanoyl-(+)-cis-khellactone analogues. J. Med. Chem. 2001, 44, 664–671. [Google Scholar] [CrossRef]
- Mohsenzadeh, F.; Darabi, H.R.; Alivand, M.; Aghapoor, K.; Balavar, Y. Naturally occurring organic acids for organocatalytic synthesis of pyrroles via Paal–Knorr reaction. Res Chem. Intermed. 2020, 46, 5255–5262. [Google Scholar] [CrossRef]
- Gangjee, A.; Zhao, Y.; Raghavan, S.; Rohena, C.C.; Mooberry, S.L.; Hamel, E. Structure–activity relationship and in vitro and in vivo evaluation of the potent cytotoxic anti-microtubule agent N-(4-methoxyphenyl)-N,2,6-trimethyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-aminium chloride and its analogues as antitumor agents. J. Med. Chem. 2013, 56, 6829–6844. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.; Shi, Y.; Chen, J.; He, Z.; Xu, Z.; Zhao, Z.; Zhu, W.; Li, H.; Xu, Y.; Li, B.; et al. The discovery of new plant activators and scaffolds with potential induced systemic resistance: From jasmonic acid to pyrrolidone. MedChemComm 2016, 7, 1849–1857. [Google Scholar] [CrossRef]
- Mansoori, Y.; Atghia, S.V.; Sanaei, S.S.; Zamanloo, M.R.; Imanzadeh, G.; Eskandari, H. New, organo-soluble, thermally stable aromatic polyimides and poly(amide-imide) based on 2-[5-(3,5-dinitrophenyl)-1,3, 4-oxadiazole-2-yl]pyridine. Polym. Int. 2012, 61, 1213–1220. [Google Scholar] [CrossRef]
- Tang, L.; Ma, M.; Zhang, Q.; Luo, H.; Wang, T.; Chai, Y. Metal-free synthesis of pyrazoles from 1,3-diarylpropenes and hydrazines via multiple inter-/intramolecular C–H aminations. Adv. Synth. Catal. 2017, 359, 2610–2620. [Google Scholar] [CrossRef]
- Gunaseelan, S.; Arunkumar, M.; Aravind, M.K.; Gayathri, S.; Rajkeerthana, S.; Mohankumar, V.; Ashok-kumar, B.; Varalakshmi, P. Probing marine brown macroalgal phlorotannins as antiviral candidate against SARS-CoV-2: Molecular docking and dynamics simulation approach. Mol. Divers. 2022, 26, 3205–3224. [Google Scholar] [CrossRef]
- Qi, C.; Zhang, R.; Liu, F.; Zheng, T.; Wu, W. Molecular mechanism of interactions between inhibitory tripeptide GEF and angiotensin-converting enzyme in aqueous solutions by molecular dynamic simulations. J. Mol. Liq. 2018, 249, 389–396. [Google Scholar] [CrossRef]
- Medina, F.; Medina, J.F.; Colon, C.; Vergne, E.; Santiago, G.A.; Munoz-Jordan, J.L. Dengue virus: Isolation, propagation, quantification, and storage. Curr. Protoc. Microbiol. 2012, 27, 15D.2.1–15D.2.24. [Google Scholar] [CrossRef]
- Gong, E.Y.; Clynhens, M.; Ivens, T.; Lory, P.; Simmen, K.; Kraus, G. Cell-based antiviral assays for screening and profiling inhibitors against dengue virus. Methods Mol. Biol. 2013, 1030, 185–194. [Google Scholar]
- Farias, K.J.S.; Machado, P.R.L.; Da Fonseca, B.A.L. Chloroquine inhibits dengue virus type 2 replication in vero cells but not in C6/36 cells. Sci. World J. 2013, 2013, 282734. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Entry No. | R | Docking Score | QPPCaco | Aqueous Solubility Level | ADMET Absorption Level | MMGBSA ∆G Bind (kcal/mol) |
| SW-a |  | −10.687 | 108.021 | 2 | 0 | −89.28 |
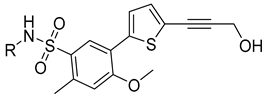
| S-SW-b | 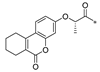 | −11.660 | 84.455 | 3 | 2 | −115.26 |
| R-SW-b |  | −12.094 | 115.112 | 4 | 1 | −108.82 |
| SW-c | 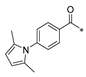 | −11.352 | 114.214 | 3 | 1 | −98.68 |
| SW-d |  | −13.083 | 138.478 | 4 | 2 | −98.02 |
| SW-e | 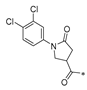 | −12.564 | 123.874 | 3 | 0 | −98.76 |
| SW-f |  | −11.158 | 90.057 | 3 | 1 | −99.24 |
| SW-g |  | −11.157 | 130.258 | 4 | 2 | −87.40 |
| SW-h | 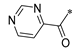 | −10.655 | 187.064 | 3 | 0 | −81.71 |
| SW-i | 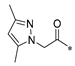 | −11.415 | 150.241 | 14 | 2 | −93.36 |
| 27 | - | −12.295 | 179.421 | 3 | 1 | −92.13 |
| 29 | - | −11.995 | 273.700 | 3 | 0 | −88.56 |
| Entry No. | a DENV-NGC-IC50 (μM) | b CC50 (μM) | SI (Selectivity Index) |
|---|---|---|---|
| SW-a | 57.60 ± 2.37 | ND | - |
| SW-b | 3.58 ± 0.29 | 24.65 | 6.89 |
| SW-c | 41.49 ± 1.37 | >200 | - |
| SW-d | 23.94 ± 1.00 | 133.70 | 5.58 |
| SW-e | >200 | 69.86 | - |
| SW-f | 53.79 ± 4.53 | >200 | >3.72 |
| SW-g | >200 | ND | - |
| SW-h | >200 | ND | - |
| SW-i | >200 | ND | - |
| BCX4430 | 41.59 ± 2.95 | 100 | 2.40 |
| NITD008 | 15.45 ± 5.54 | 20 | 1.29 |
| 27 | 19.67 ± 1.12 | ND | - |
| Entry No. | a RdRp-IC50 (μM) |
|---|---|
| SW-b | 11.54 ± 1.30 |
| SW-d | 13.54 ± 0.32 |
| 3′-dATP | 30.09 ± 8.26 |
| BCX4430 | ND |
| NITD008 | ND |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zong, K.; Li, W.; Xu, Y.; Zhao, X.; Cao, R.; Yan, H.; Li, X. Design, Synthesis, Evaluation and Molecular Dynamics Simulation of Dengue Virus NS5-RdRp Inhibitors. Pharmaceuticals 2023, 16, 1625. https://doi.org/10.3390/ph16111625
Zong K, Li W, Xu Y, Zhao X, Cao R, Yan H, Li X. Design, Synthesis, Evaluation and Molecular Dynamics Simulation of Dengue Virus NS5-RdRp Inhibitors. Pharmaceuticals. 2023; 16(11):1625. https://doi.org/10.3390/ph16111625
Chicago/Turabian StyleZong, Keli, Wei Li, Yijie Xu, Xu Zhao, Ruiyuan Cao, Hong Yan, and Xingzhou Li. 2023. "Design, Synthesis, Evaluation and Molecular Dynamics Simulation of Dengue Virus NS5-RdRp Inhibitors" Pharmaceuticals 16, no. 11: 1625. https://doi.org/10.3390/ph16111625
APA StyleZong, K., Li, W., Xu, Y., Zhao, X., Cao, R., Yan, H., & Li, X. (2023). Design, Synthesis, Evaluation and Molecular Dynamics Simulation of Dengue Virus NS5-RdRp Inhibitors. Pharmaceuticals, 16(11), 1625. https://doi.org/10.3390/ph16111625