
Design, Synthesis, and Repurposing of Rosmarinic Acid-β-Amino-α-Ketoamide Hybrids as Antileishmanial Agents

 ,
,  , ,
, ,  ,
,
Abstract
:
1. Introduction
2. Results and Discussion
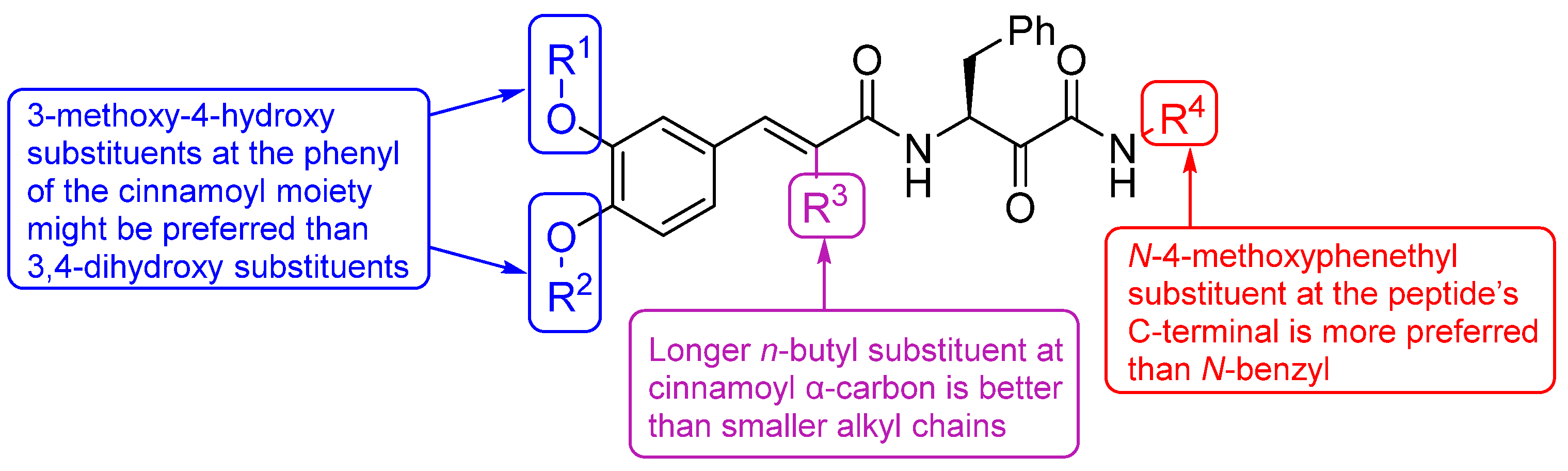
2.1. Design and Repurposing Rational
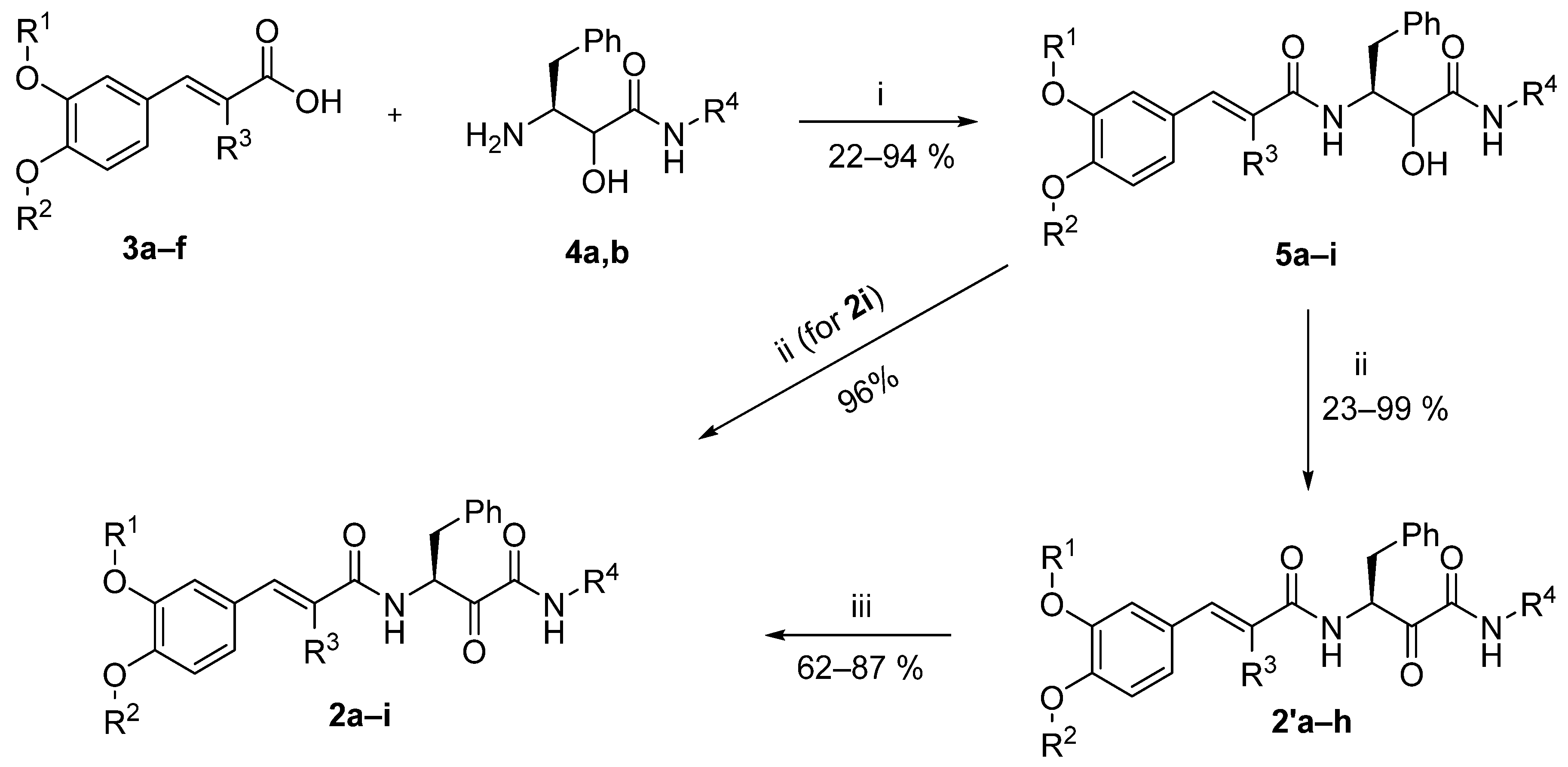
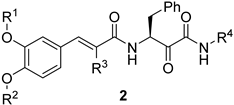
2.2. Chemistry
2.3. Biological Evaluations
2.3.1. Evaluation of Antileishmanial Activity
In Vitro L. donovani Promastigotes-Based Evaluation Model
In Vitro Potency Evaluation against L. donovani Promastigotes
Structure–Activity Relationship
In Vitro Safety Evaluation
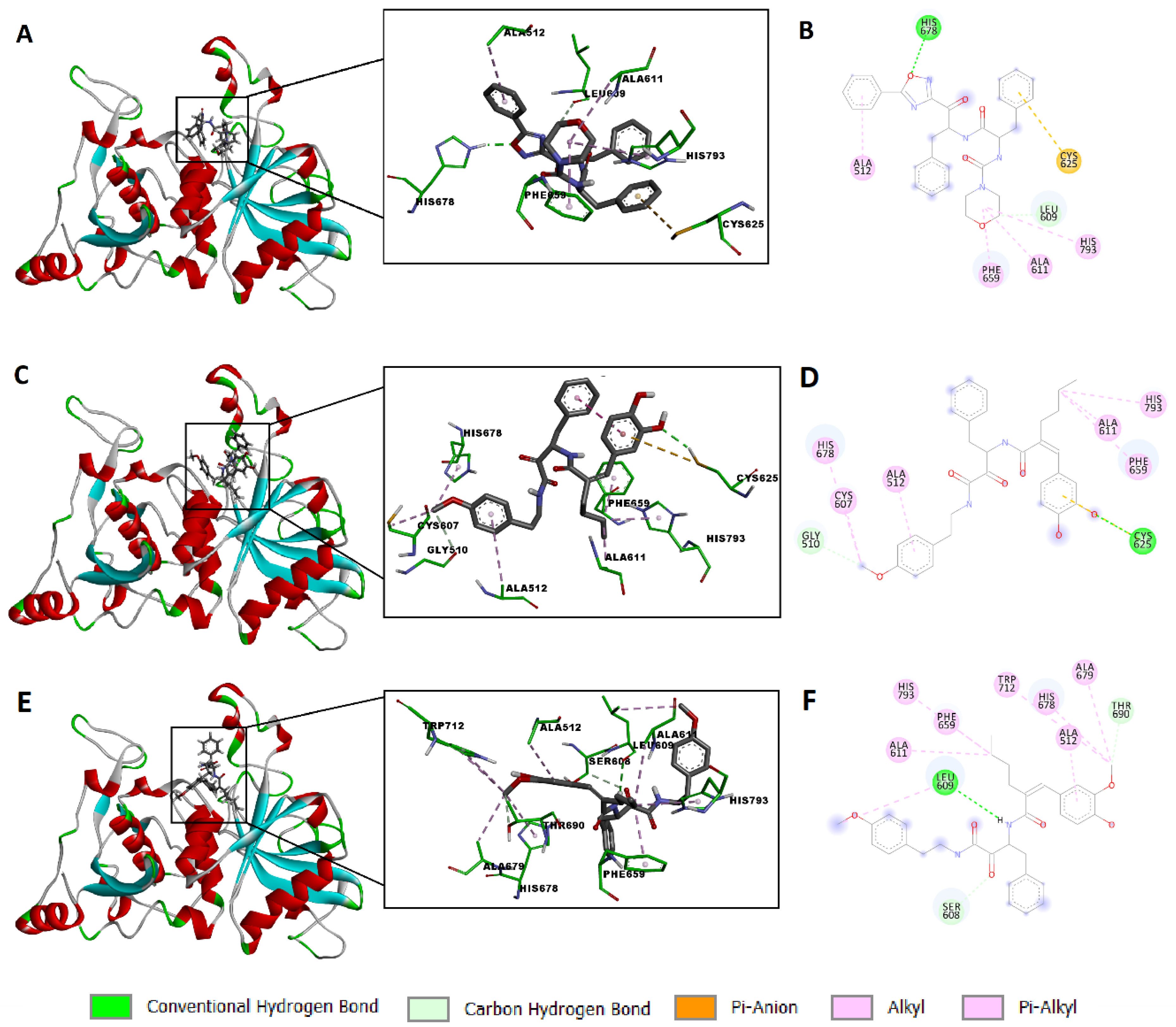
2.4. Molecular Modeling Study
3. Materials and Methods
3.1. Chemistry
3.2. Biological Evaluations
Evaluations of Antileishmanial Activity and Safety Assessment
3.3. In Silico Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bodimeade, C.; Marks, M.; Mabey, D. Neglected tropical diseases: Elimination and eradication. Clin. Med. 2019, 19, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Okwor, I.; Uzonna, J. Social and Economic Burden of Human Leishmaniasis. Am. J. Trop. Med. Hyg. 2016, 94, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Mann, S.; Frasca, K.; Scherrer, S.; Henao-Martínez, A.F.; Newman, S.; Ramanan, P.; Suarez, J.A. A Review of Leishmaniasis: Current Knowledge and Future Directions. Curr. Trop. Med. Rep. 2021, 8, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Alvar, J.; Vélez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; de Boer, M. Leishmaniasis Worldwide and Global Estimates of Its Incidence. PLoS ONE 2012, 7, e35671. [Google Scholar] [CrossRef]
- de Araújo, V.E.; Morais, M.H.; Reis, I.A.; Rabello, A.; Carneiro, M. Early clinical manifestations associated with death from visceral leishmaniasis. PLoS Negl. Trop. Dis. 2012, 6, e1511. [Google Scholar] [CrossRef]
- Tuon, F.F.; Dantas, L.R.; de Souza, R.M.; Ribeiro, V.S.T.; Amato, V.S. Liposomal drug delivery systems for the treatment of leishmaniasis. Parasitol. Res. 2022, 121, 3073–3082. [Google Scholar] [CrossRef]
- Pinheiro, A.C.; de Souza, M.V.N. Current leishmaniasis drug discovery. RSC Med. Chem. 2022, 13, 1029–1043. [Google Scholar] [CrossRef]
- Madusanka, R.K.; Silva, H.; Karunaweera, N.D. Treatment of Cutaneous Leishmaniasis and Insights into Species-Specific Responses: A Narrative Review. Infect. Dis. Ther. 2022, 11, 695–711. [Google Scholar] [CrossRef]
- de Santana, N.S.; de Oliveira de Siqueira, L.B.; do Nascimento, T.; Santos-Oliveira, R.; dos Santos Matos, A.P.; Ricci-Júnior, E. Nanoparticles for the treatment of visceral leishmaniasis: Review. J. Nanopart. Res. 2023, 25, 24. [Google Scholar] [CrossRef]
- Kumari, S.; Kumar, V.; Tiwari, R.K.; Ravidas, V.; Pandey, K.; Kumar, A. Amphotericin B: A drug of choice for Visceral Leishmaniasis. Acta Trop. 2022, 235, 106661. [Google Scholar] [CrossRef]
- Yeshaw, Y.; Tsegaye, A.T.; Nigatu, S.G. Incidence of Mortality and Its Predictors among Adult Visceral Leishmaniasis Patients at the University of Gondar Hospital: A Retrospective Cohort Study. Infect. Drug Resist. 2020, 13, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Scarpini, S.; Dondi, A.; Totaro, C.; Biagi, C.; Melchionda, F.; Zama, D.; Pierantoni, L.; Gennari, M.; Campagna, C.; Prete, A.; et al. Visceral Leishmaniasis: Epidemiology, Diagnosis, and Treatment Regimens in Different Geographical Areas with a Focus on Pediatrics. Microorganisms 2022, 10, 1887. [Google Scholar] [CrossRef] [PubMed]
- Wijnant, G.-J.; Dumetz, F.; Dirkx, L.; Bulté, D.; Cuypers, B.; Van Bocxlaer, K.; Hendrickx, S. Tackling Drug Resistance and Other Causes of Treatment Failure in Leishmaniasis. Front. Trop. Dis. 2022, 3, 837460. [Google Scholar] [CrossRef]
- Stone, S.; Newman, D.J.; Colletti, S.L.; Tan, D.S. Cheminformatic analysis of natural product-based drugs and chemical probes. Nat. Prod. Rep. 2022, 39, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J. Natural products and drug discovery. Natl. Sci. Rev. 2022, 9, nwac206. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- Young, R.J.; Flitsch, S.L.; Grigalunas, M.; Leeson, P.D.; Quinn, R.J.; Turner, N.J.; Waldmann, H. The Time and Place for Nature in Drug Discovery. JACS Au 2022, 2, 2400–2416. [Google Scholar] [CrossRef]
- Grigalunas, M.; Burhop, A.; Zinken, S.; Pahl, A.; Gally, J.-M.; Wild, N.; Mantel, Y.; Sievers, S.; Foley, D.J.; Scheel, R.; et al. Natural product fragment combination to performance-diverse pseudo-natural products. Nat. Commun. 2021, 12, 1883. [Google Scholar] [CrossRef]
- Karageorgis, G.; Foley, D.J.; Laraia, L.; Waldmann, H. Principle and design of pseudo-natural products. Nat. Chem. 2020, 12, 227–235. [Google Scholar] [CrossRef]
- Christoforow, A.; Wilke, J.; Binici, A.; Pahl, A.; Ostermann, C.; Sievers, S.; Waldmann, H. Design, Synthesis, and Phenotypic Profiling of Pyrano-Furo-Pyridone Pseudo Natural Products. Angew. Chem. Int. Ed. 2019, 58, 14715–14723. [Google Scholar] [CrossRef]
- Silva, L.P.; Santos, I.P.; Silva, D.K.C.; dos Reis, B.P.Z.C.; Meira, C.S.; Castro, M.V.B.d.S.; dos Santos Filho, J.M.; Araujo-Neto, J.H.d.; Ellena, J.A.; Silveira, R.G.d.; et al. Molecular Hybridization Strategy on the Design, Synthesis, and Structural Characterization of Ferrocene-N-acyl Hydrazones as Immunomodulatory Agents. Molecules 2022, 27, 8343. [Google Scholar] [CrossRef] [PubMed]
- Sampath Kumar, H.M.; Herrmann, L.; Tsogoeva, S.B. Structural hybridization as a facile approach to new drug candidates. Bioorg. Med. Chem. Lett. 2020, 30, 127514. [Google Scholar] [CrossRef] [PubMed]
- Fershtat, L.L.; Makhova, N.N. Molecular Hybridization Tools in the Development of Furoxan-Based NO-Donor Prodrugs. ChemMedChem 2017, 12, 622–638. [Google Scholar] [CrossRef]
- Zhang, S.; Saathoff, J.M.; He, L. Molecular Hybridization: An Emerging Tool for the Design of Novel Therapeutics for Alzheimer’s Disease. In Design of Hybrid Molecules for Drug Development; Decker, M., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 219–237. [Google Scholar]
- Bahia, S.B.B.B.; Reis, W.J.; Jardim, G.A.M.; Souto, F.T.; de Simone, C.A.; Gatto, C.C.; Menna-Barreto, R.F.S.; de Castro, S.L.; Cavalcanti, B.C.; Pessoa, C.; et al. Molecular hybridization as a powerful tool towards multitarget quinoidal systems: Synthesis, trypanocidal and antitumor activities of naphthoquinone-based 5-iodo-1,4-disubstituted-, 1,4- and 1,5-disubstituted-1,2,3-triazoles. MedChemComm 2016, 7, 1555–1563. [Google Scholar] [CrossRef]
- Ivasiv, V.; Albertini, C.; Gonçalves, E.A.; Rossi, M.; Bolognesi, L.M. Molecular Hybridization as a Tool for Designing Multitarget Drug Candidates for Complex Diseases. Curr. Top. Med. Chem. 2019, 19, 1694–1711. [Google Scholar] [CrossRef]
- Pawełczyk, A.; Sowa-Kasprzak, K.; Olender, D.; Zaprutko, L. Molecular Consortia—Various Structural and Synthetic Concepts for More Effective Therapeutics Synthesis. Int. J. Mol. Sci. 2018, 19, 1104. [Google Scholar] [CrossRef]
- Kulkarni, V.S.; Alagarsamy, V.; Solomon, V.R.; Jose, P.A.; Murugesan, S. Drug Repurposing: An Effective Tool in Modern Drug Discovery. Russ. J. Bioorg. Chem. 2023, 49, 157–166. [Google Scholar] [CrossRef]
- Jourdan, J.P.; Bureau, R.; Rochais, C.; Dallemagne, P. Drug repositioning: A brief overview. J. Pharm. Pharmacol. 2020, 72, 1145–1151. [Google Scholar] [CrossRef]
- da Silva, E.R.; Brogi, S.; Grillo, A.; Campiani, G.; Gemma, S.; Vieira, P.C.; Maquiaveli, C.d.C. Cinnamic acids derived compounds with antileishmanial activity target Leishmania amazonensis arginase. Chem. Biol. Drug Des. 2019, 93, 139–146. [Google Scholar] [CrossRef]
- Garcia, A.R.; Oliveira, D.M.P.; Claudia, F.; Amaral, A.; Jesus, J.B.; Rennó Sodero, A.C.; Souza, A.M.T.; Supuran, C.T.; Vermelho, A.B.; Rodrigues, I.A.; et al. Leishmania infantum arginase: Biochemical characterization and inhibition by naturally occurring phenolic substances. J. Enzyme Inhib. Med. Chem. 2019, 34, 1100–1109. [Google Scholar] [CrossRef]
- Abamor, E.S. Antileishmanial activities of caffeic acid phenethyl ester loaded PLGA nanoparticles against Leishmania infantum promastigotes and amastigotes in vitro. Asian Pac. J. Trop. Med. 2017, 10, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Montrieux, E.; Perera, W.H.; García, M.; Maes, L.; Cos, P.; Monzote, L. In vitro and in vivo activity of major constituents from Pluchea carolinensis against Leishmania amazonensis. Parasitol. Res. 2014, 113, 2925–2932. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudzadeh-Niknam, H.; McKerrow, J.H. Leishmania tropica: Cysteine proteases are essential for growth and pathogenicity. Exp. Parasitol. 2004, 106, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Steert, K.; Berg, M.; Mottram, J.C.; Westrop, G.D.; Coombs, G.H.; Cos, P.; Maes, L.; Joossens, J.; Van der Veken, P.; Haemers, A.; et al. α-Ketoheterocycles as Inhibitors of Leishmania mexicana Cysteine Protease CPB. ChemMedChem 2010, 5, 1734–1748. [Google Scholar] [CrossRef]
- d’Avila-Levy, C.M.; Marinho, F.A.; Santos, L.O.; Martins, J.L.; Santos, A.L.S.; Branquinha, M.H. Antileishmanial activity of MDL 28170, a potent calpain inhibitor. Int. J. Antimicrob. Agents 2006, 28, 138–142. [Google Scholar] [CrossRef]
- Marinho, F.A.; Gonçalves, K.C.S.; Oliveira, S.S.C.; Gonçalves, D.S.; Matteoli, F.P.; Seabra, S.H.; Oliveira, A.C.S.; Bellio, M.; Oliveira, S.S.; Souto-Padrón, T.; et al. The Calpain Inhibitor MDL28170 Induces the Expression of Apoptotic Markers in Leishmania amazonensis Promastigotes. PLoS ONE 2014, 9, e87659. [Google Scholar] [CrossRef]
- Marinho, F.A.; Sangenito, L.S.; Oliveira, S.S.C.; De Arruda, L.B.; D’Ávila-levy, C.M.; Santos, A.L.S.; Branquinha, M.H. The potent cell permeable calpain inhibitor MDL28170 affects the interaction of Leishmania amazonensis with macrophages and shows anti-amastigote activity. Parasitol. Int. 2017, 66, 579–583. [Google Scholar] [CrossRef]
- Casanova, M.; Gonzalez, I.J.; Sprissler, C.; Zalila, H.; Dacher, M.; Basmaciyan, L.; Späth, G.F.; Azas, N.; Fasel, N. Implication of different domains of the Leishmania major metacaspase in cell death and autophagy. Cell Death Dis. 2015, 6, e1933. [Google Scholar] [CrossRef]
- Yoo, Y.J.; Nam, D.H.; Jung, S.Y.; Jang, J.W.; Kim, H.J.; Jin, C.; Pae, A.N.; Lee, Y.S. Synthesis of cinnamoyl ketoamides as hybrid structures of antioxidants and calpain inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 2850–2854. [Google Scholar] [CrossRef]
- Kulshrestha, A.; Bhandari, V.; Mukhopadhyay, R.; Ramesh, V.; Sundar, S.; Maes, L.; Dujardin, J.C.; Roy, S.; Salotra, P. Validation of a simple resazurin-based promastigote assay for the routine monitoring of miltefosine susceptibility in clinical isolates of Leishmania donovani. Parasitol. Res. 2013, 112, 825–828. [Google Scholar] [CrossRef]
- Hassan, A.H.E.; Mahmoud, K.; Phan, T.-N.; Shaldam, M.A.; Lee, C.H.; Kim, Y.J.; Cho, S.B.; Bayoumi, W.A.; El-Sayed, S.M.; Choi, Y.; et al. Bestatin analogs-4-quinolinone hybrids as antileishmanial hits: Design, repurposing rational, synthesis, in vitro and in silico studies. Eur. J. Med. Chem. 2023, 250, 115211. [Google Scholar] [CrossRef]
- Hassan, A.H.E.; Phan, T.-N.; Choi, Y.; Moon, S.; No, J.H.; Lee, Y.S. Design, Rational Repurposing, Synthesis, In Vitro Evaluation, Homology Modeling and In Silico Study of Sulfuretin Analogs as Potential Antileishmanial Hit Compounds. Pharmaceuticals 2022, 15, 1058. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.H.E.; Phan, T.-N.; Moon, S.; Lee, C.H.; Kim, Y.J.; Cho, S.B.; El-Sayed, S.M.; Choi, Y.; No, J.H.; Lee, Y.S. Design, synthesis, and repurposing of O6-aminoalkyl-sulfuretin analogs towards discovery of potential lead compounds as antileishmanial agents. Eur. J. Med. Chem. 2023, 251, 115256. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.H.E.; Phan, T.-N.; Yoon, S.; Lee, C.J.; Jeon, H.R.; Kim, S.-H.; No, J.H.; Lee, Y.S. Pyrrolidine-based 3-deoxysphingosylphosphorylcholine analogs as possible candidates against neglected tropical diseases (NTDs): Identification of hit compounds towards development of potential treatment of Leishmania donovani. J. Enzyme Inhib. Med. Chem. 2021, 36, 1922–1930. [Google Scholar] [CrossRef]
- Phan, T.-N.; Baek, K.H.; Lee, N.; Byun, S.Y.; Shum, D.; No, J.H. In Vitro and in Vivo Activity of mTOR Kinase and PI3K Inhibitors Against Leishmania donovani and Trypanosoma brucei. Molecules 2020, 25, 1980. [Google Scholar] [CrossRef]
- Hassan, A.H.E.; Bayoumi, W.A.; El-Sayed, S.M.; Phan, T.-N.; Kim, Y.J.; Lee, C.H.; Cho, S.B.; Oh, T.; Ham, G.; Mahmoud, K.; et al. Rational repurposing, synthesis, in vitro and in silico studies of chromone-peptidyl hybrids as potential agents against Leishmania donovani. J. Enzyme. Inhib. Med. Chem. 2023, 38, 2229071. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
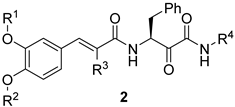
 | ||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | % Inhibition at 50 µM 1 | % Inhibition at 25 µM 1 |
| 2a | H | H | H | benzyl | 103 ± 0.4 | 97.82 ± 1.0 |
| 2b | H | H | H | 4-methoxyphenethyl | 72.83 ± 0.4 | 44.02 ± 1.2 |
| 2c | H | H | ethyl | benzyl | 101 ± 0.2 | 104 ± 0.3 |
| 2d | H | H | n-propyl | benzyl | 102 ± 0.1 | 107 ± 0.4 |
| 2e | H | H | n-propyl | 4-methoxyphenethyl | 104 ± 0.1 | 104 ± 0.2 |
| 2f | H | H | n-butyl | benzyl | 100 ± 0.3 | 104 ± 0.4 |
| 2g | H | H | n-butyl | 4-methoxyphenethyl | 103 ± 0.1 | 107 ± 0.1 |
| 2h | methyl | H | n-butyl | 4-methoxyphenethyl | 100 ± 0.3 | 103 ± 0.2 |
| 2i | methyl | methyl | n-butyl | 4-methoxyphenethyl | 94.16 ± 1.2 | 79.85 ± 1.8 |
| Erufosine | 107.6 ± 0.3 | 100 ± 1.0 | ||||
 | |||||
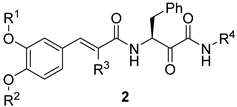
|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | IC50 (µM) 1 |
| 2a | H | H | H | benzyl | >100 |
| 2c | H | H | ethyl | benzyl | 87.3 ± 1.2 |
| 2d | H | H | n-propyl | benzyl | 45.2 ± 0.9 |
| 2e | H | H | n-propyl | 4-methoxyphenethyl | 37.6 ± 2.1 |
| 2f | H | H | n-butyl | benzyl | 26.9 ± 1.4 |
| 2g | H | H | n-butyl | 4-methoxyphenethyl | 9.5 ± 0.6 |
| 2h | methyl | H | n-butyl | 4-methoxyphenethyl | 8.8 ± 0.4 |
| Erufosine | 9.8 ± 0.7 | ||||
 | |||||
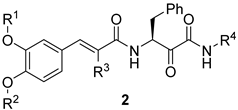
|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | THP-1 Cell CC50 (µM) |
| 2g | H | H | n-butyl | 4-methoxyphenethyl | >100 |
| 2h | methyl | H | n-butyl | 4-methoxyphenethyl | >100 |
| Erufosine | 19.3 ± 0.8 | ||||
 | ||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | LARG | LdCALP |
| 2g | H | H | n-butyl | 4-methoxyphenethyl | −7.34 | −7.80 |
| 2h | methyl | H | n-butyl | 4-methoxyphenethyl | −7.44 | −8.10 |
| Rosmarinic acid | −6.24 | — | ||||
| Compound 1 | — | −8.41 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassan, A.H.E.; Bayoumi, W.A.; El-Sayed, S.M.; Phan, T.-N.; Oh, T.; Ham, G.; Mahmoud, K.; No, J.H.; Lee, Y.S. Design, Synthesis, and Repurposing of Rosmarinic Acid-β-Amino-α-Ketoamide Hybrids as Antileishmanial Agents. Pharmaceuticals 2023, 16, 1594. https://doi.org/10.3390/ph16111594
Hassan AHE, Bayoumi WA, El-Sayed SM, Phan T-N, Oh T, Ham G, Mahmoud K, No JH, Lee YS. Design, Synthesis, and Repurposing of Rosmarinic Acid-β-Amino-α-Ketoamide Hybrids as Antileishmanial Agents. Pharmaceuticals. 2023; 16(11):1594. https://doi.org/10.3390/ph16111594
Chicago/Turabian StyleHassan, Ahmed H.E., Waleed A. Bayoumi, Selwan M. El-Sayed, Trong-Nhat Phan, Taegeun Oh, Gyeongpyo Ham, Kazem Mahmoud, Joo Hwan No, and Yong Sup Lee. 2023. "Design, Synthesis, and Repurposing of Rosmarinic Acid-β-Amino-α-Ketoamide Hybrids as Antileishmanial Agents" Pharmaceuticals 16, no. 11: 1594. https://doi.org/10.3390/ph16111594
APA StyleHassan, A. H. E., Bayoumi, W. A., El-Sayed, S. M., Phan, T.-N., Oh, T., Ham, G., Mahmoud, K., No, J. H., & Lee, Y. S. (2023). Design, Synthesis, and Repurposing of Rosmarinic Acid-β-Amino-α-Ketoamide Hybrids as Antileishmanial Agents. Pharmaceuticals, 16(11), 1594. https://doi.org/10.3390/ph16111594