


Analgesic Activity of 5-Acetamido-2-Hydroxy Benzoic Acid Derivatives and an In-Vivo and In-Silico Analysis of Their Target Interactions

 ,
, 
 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Results
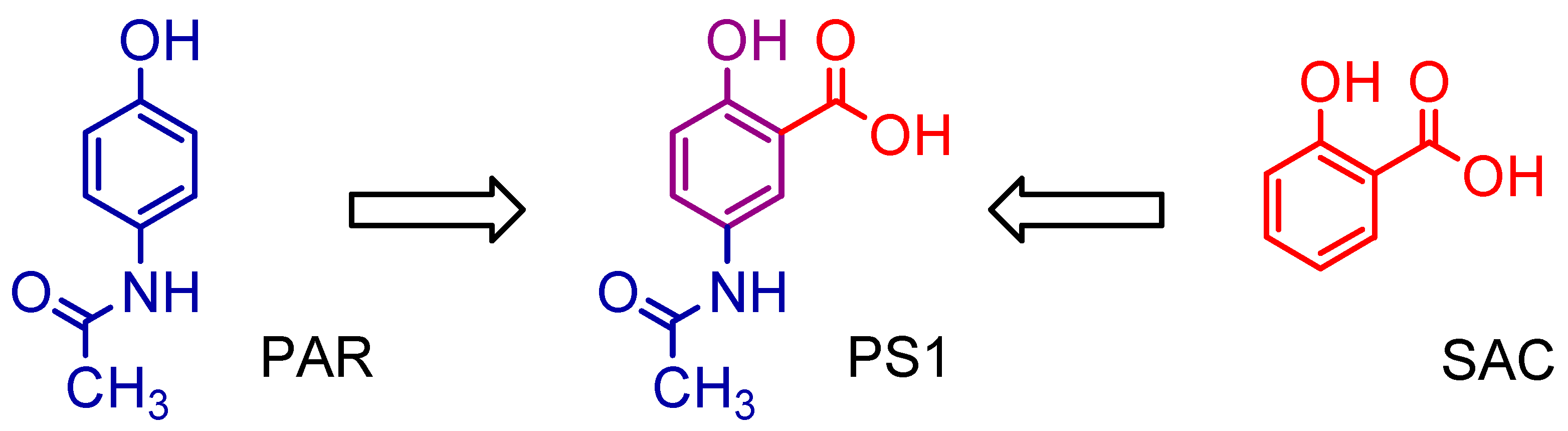
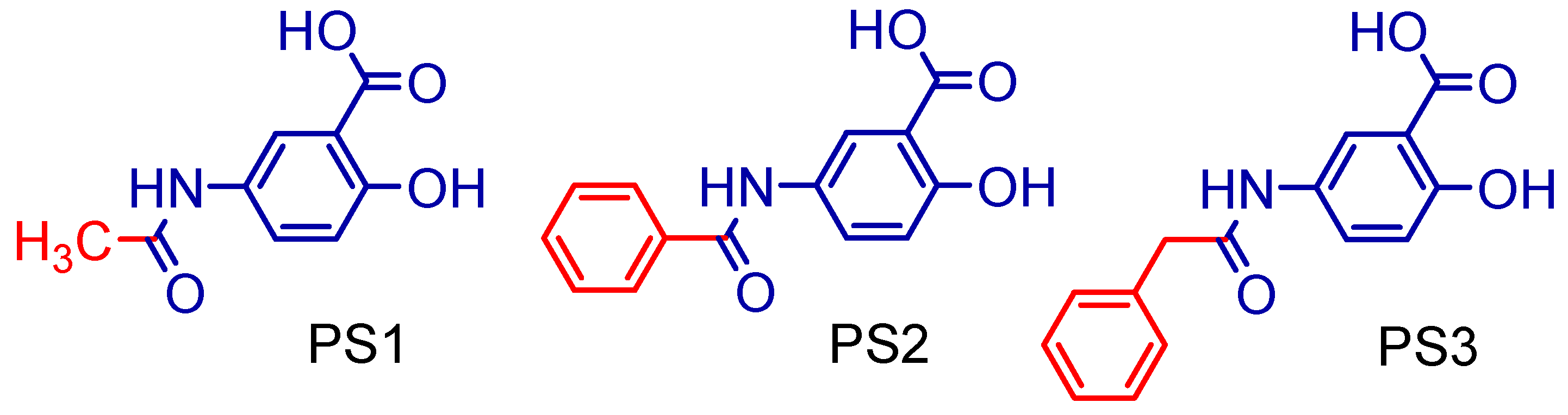
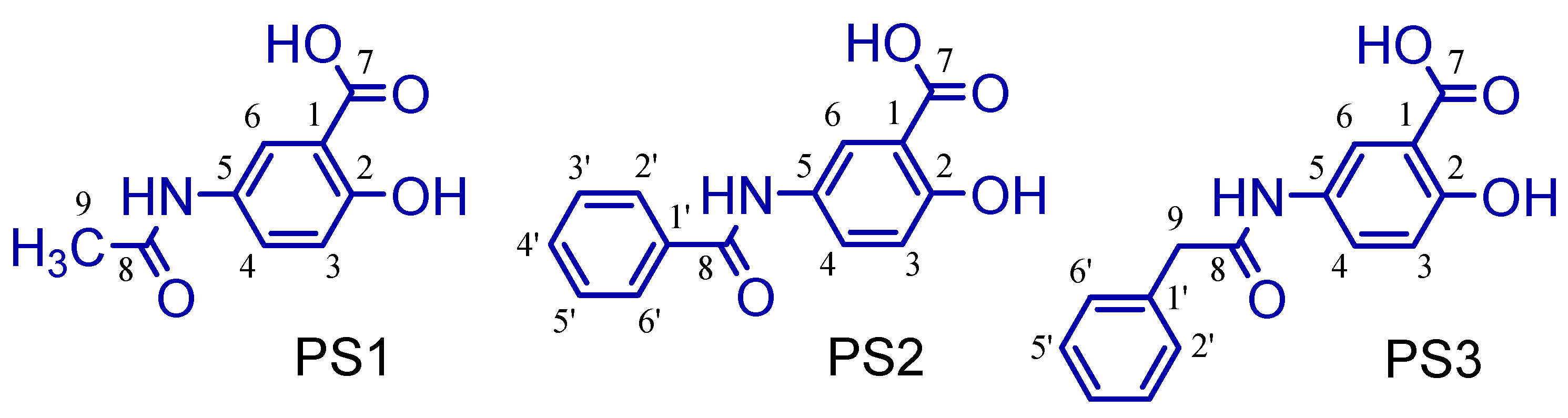
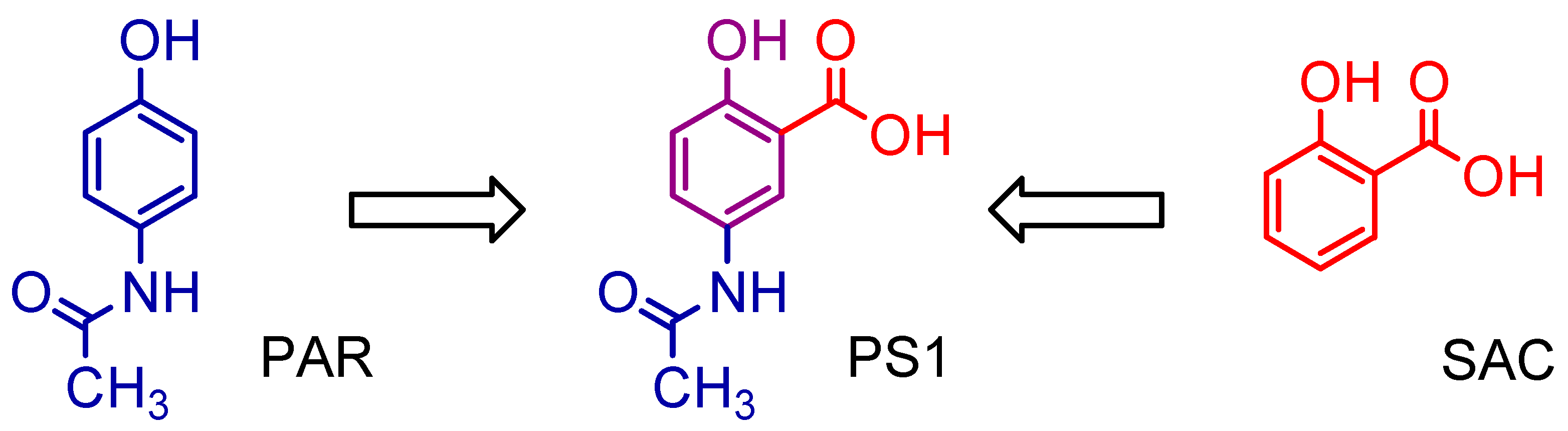
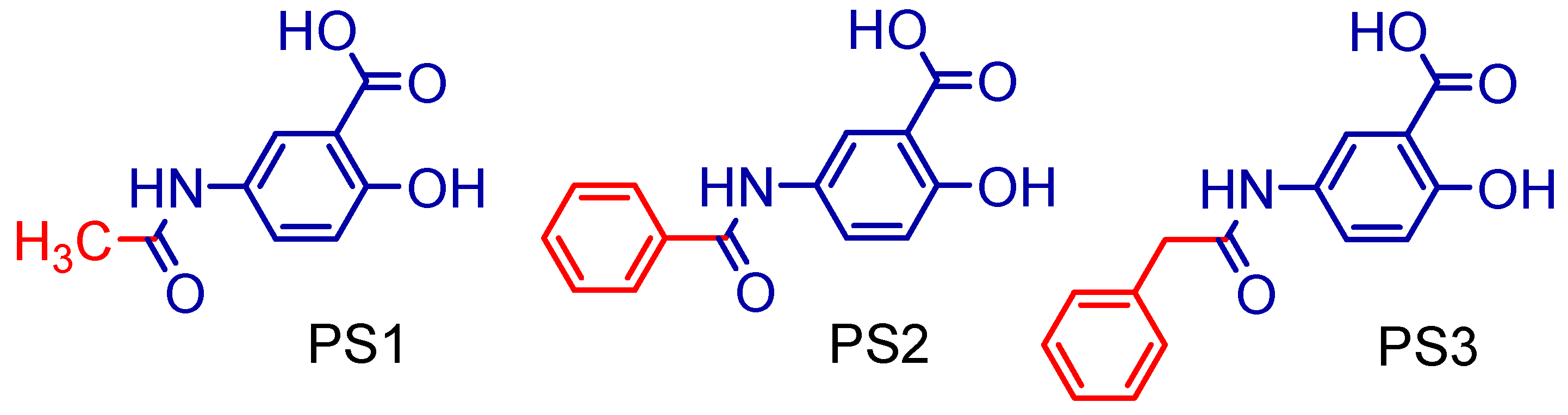
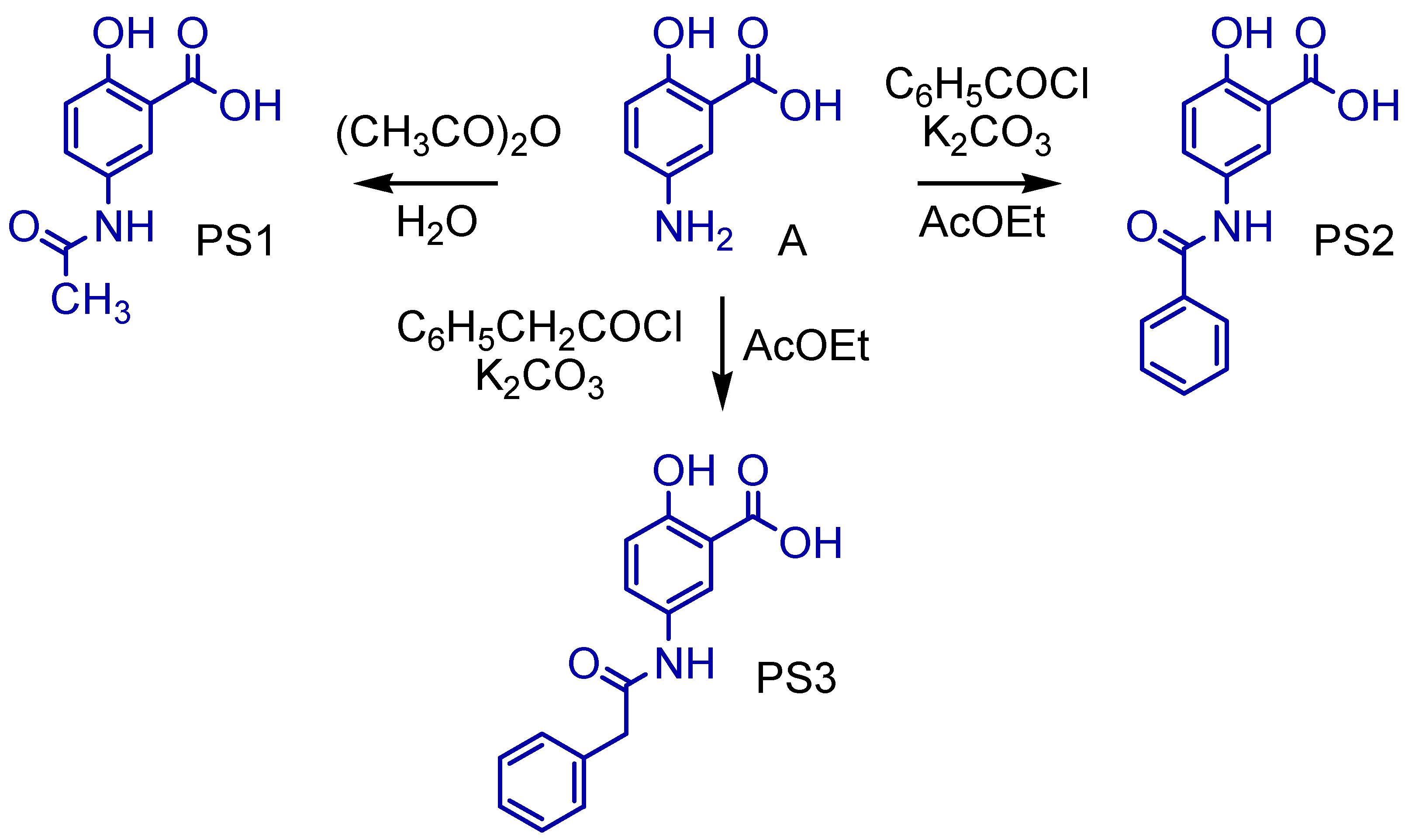
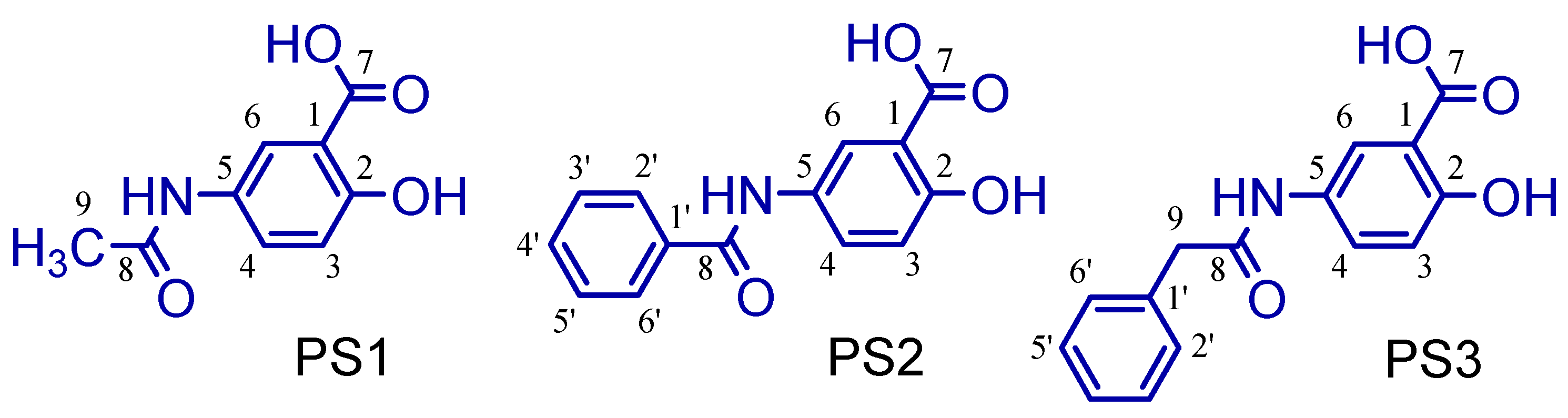
2.1. Synthesis of 5-Acetamido-2-Hydroxy Benzoic Acid and Derivatives
2.2. In-Silico Study of Oral Bioavailability, Bioactivity, ADME and Toxicity Risk Assessment
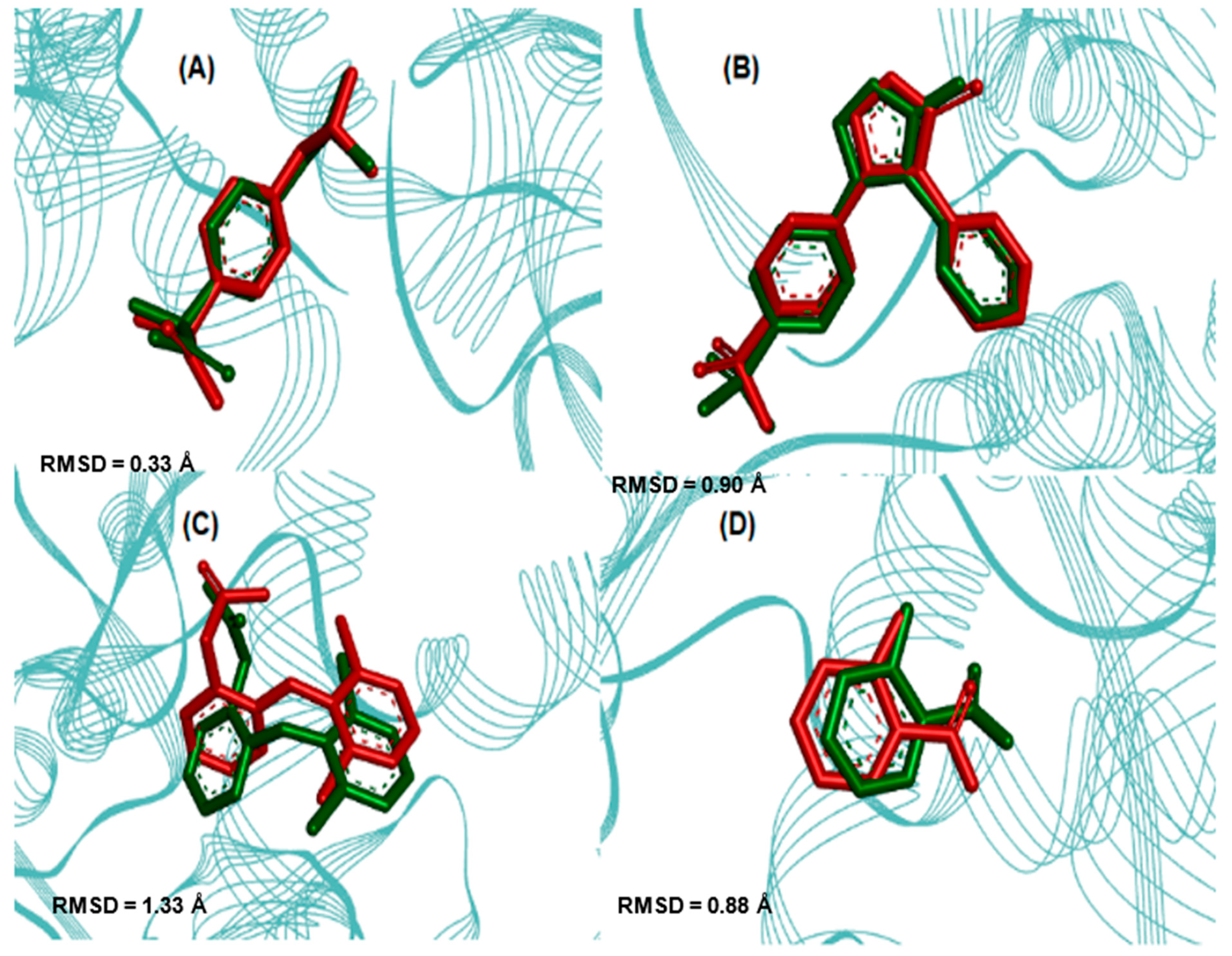
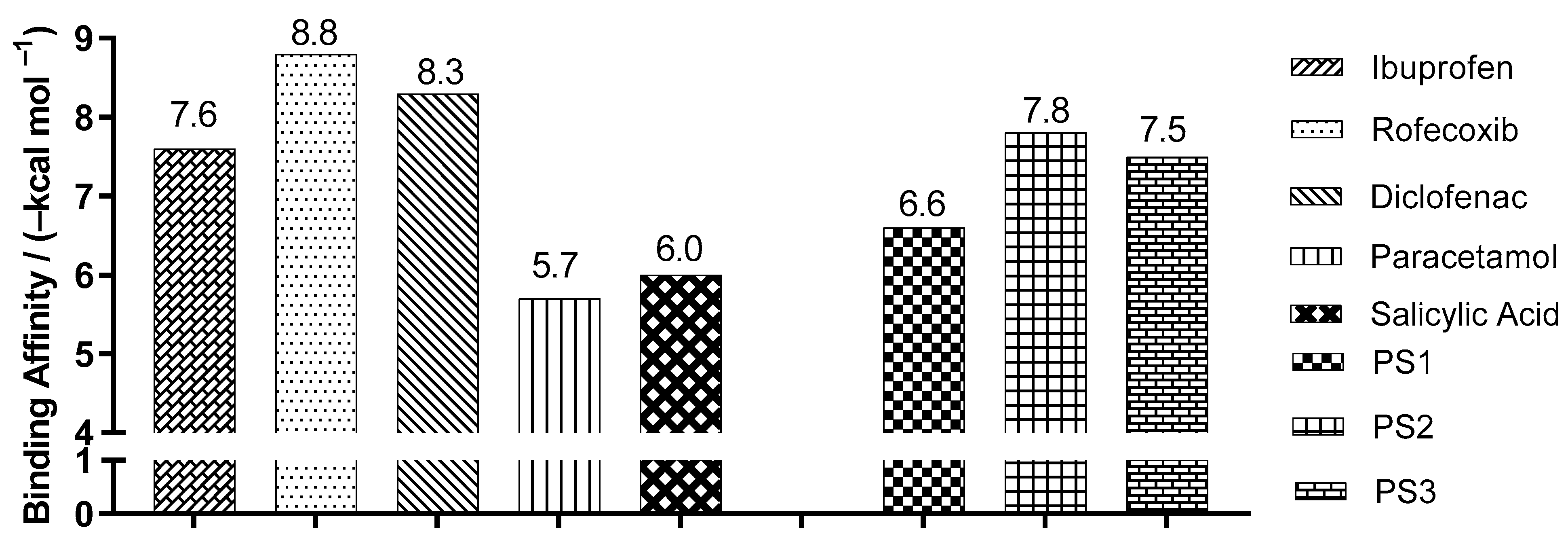
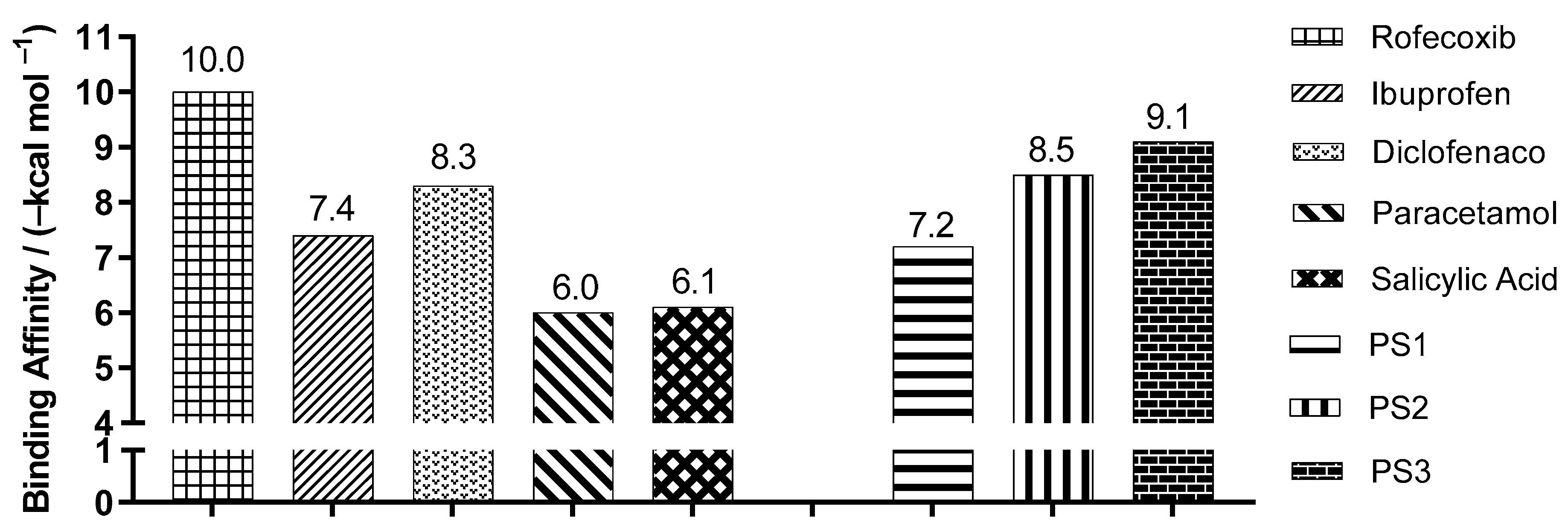
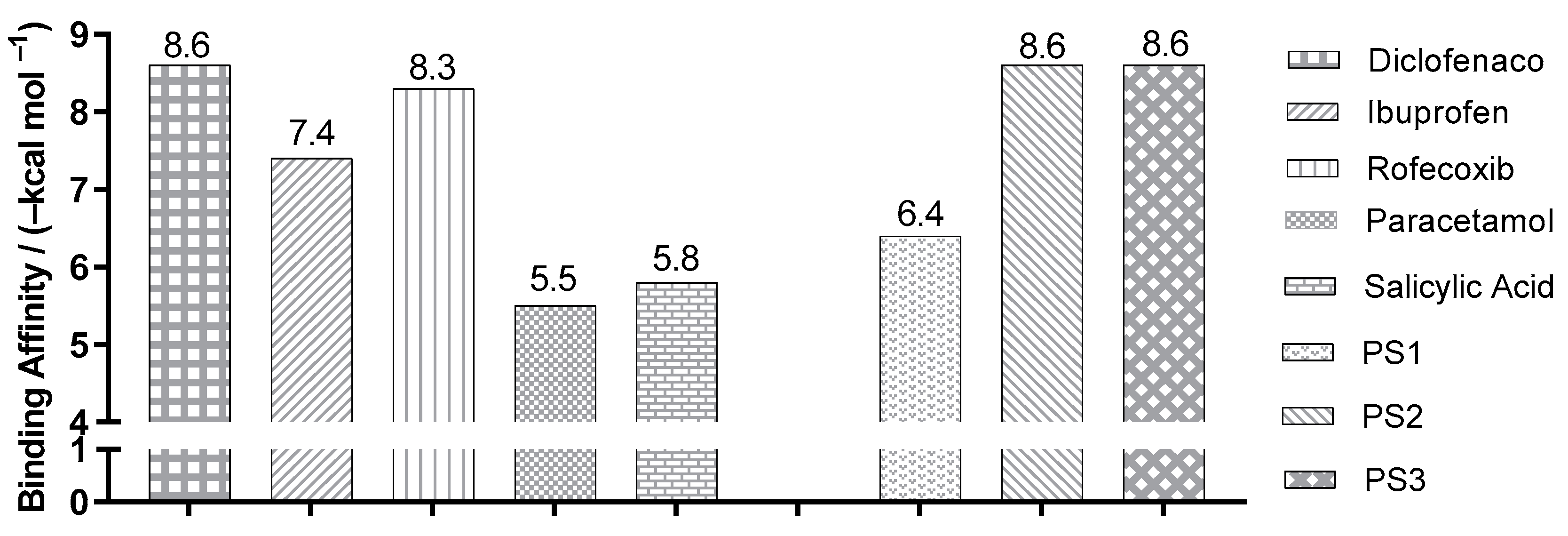
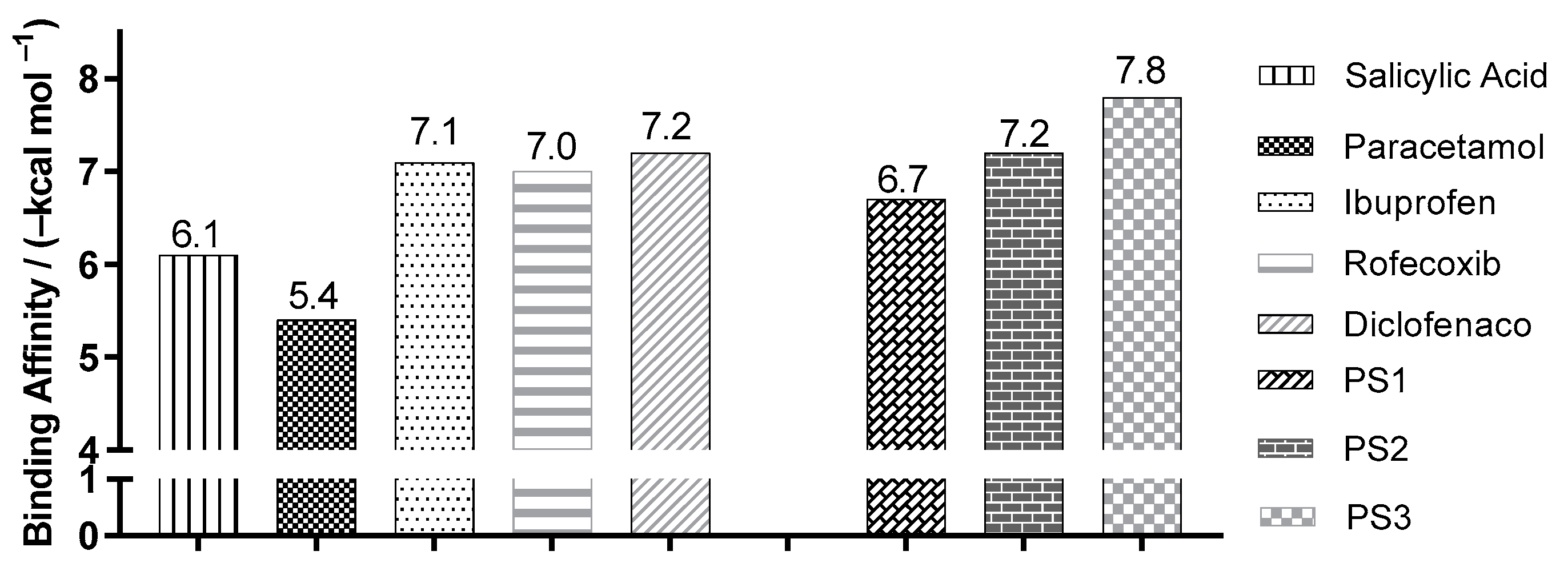
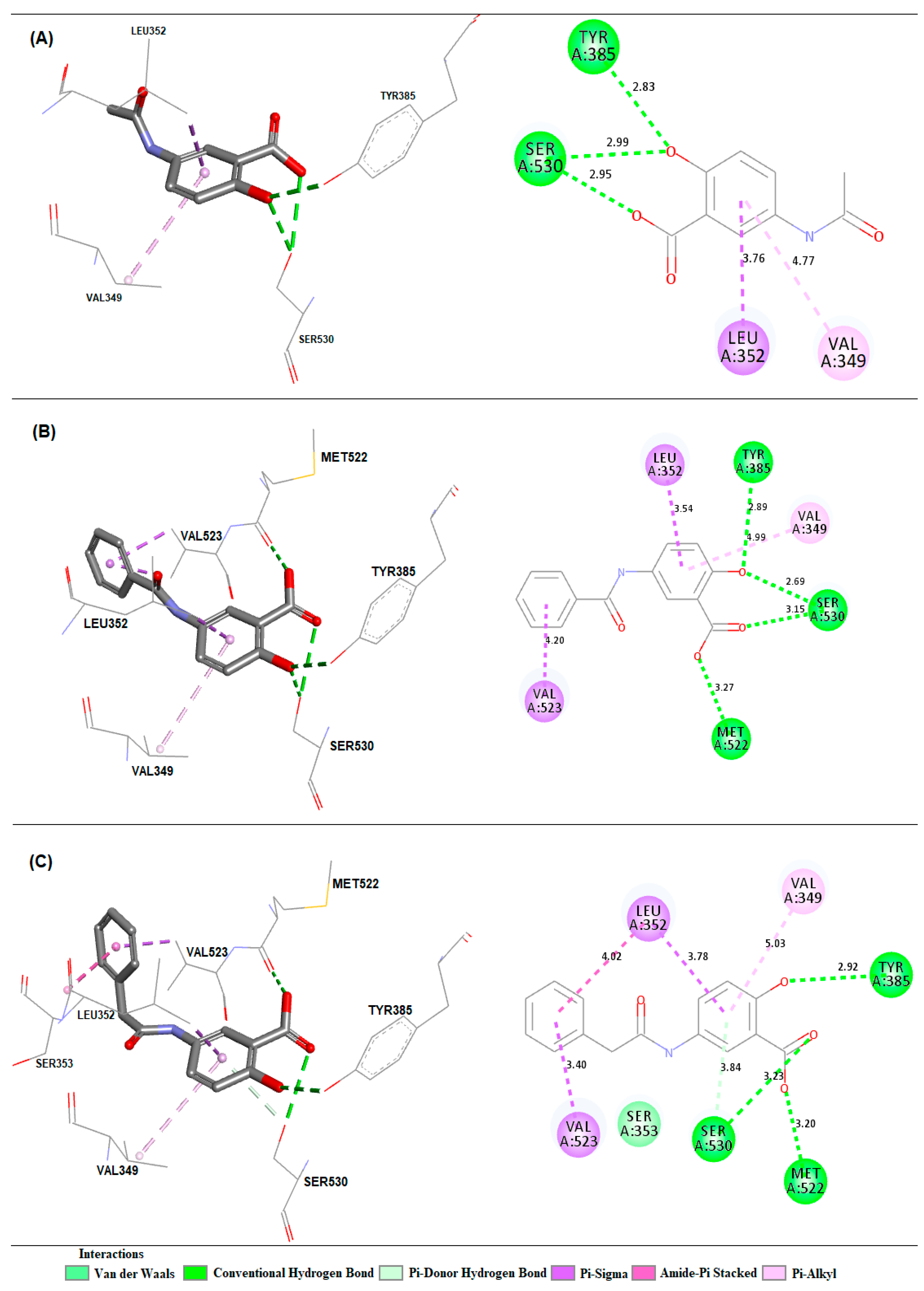
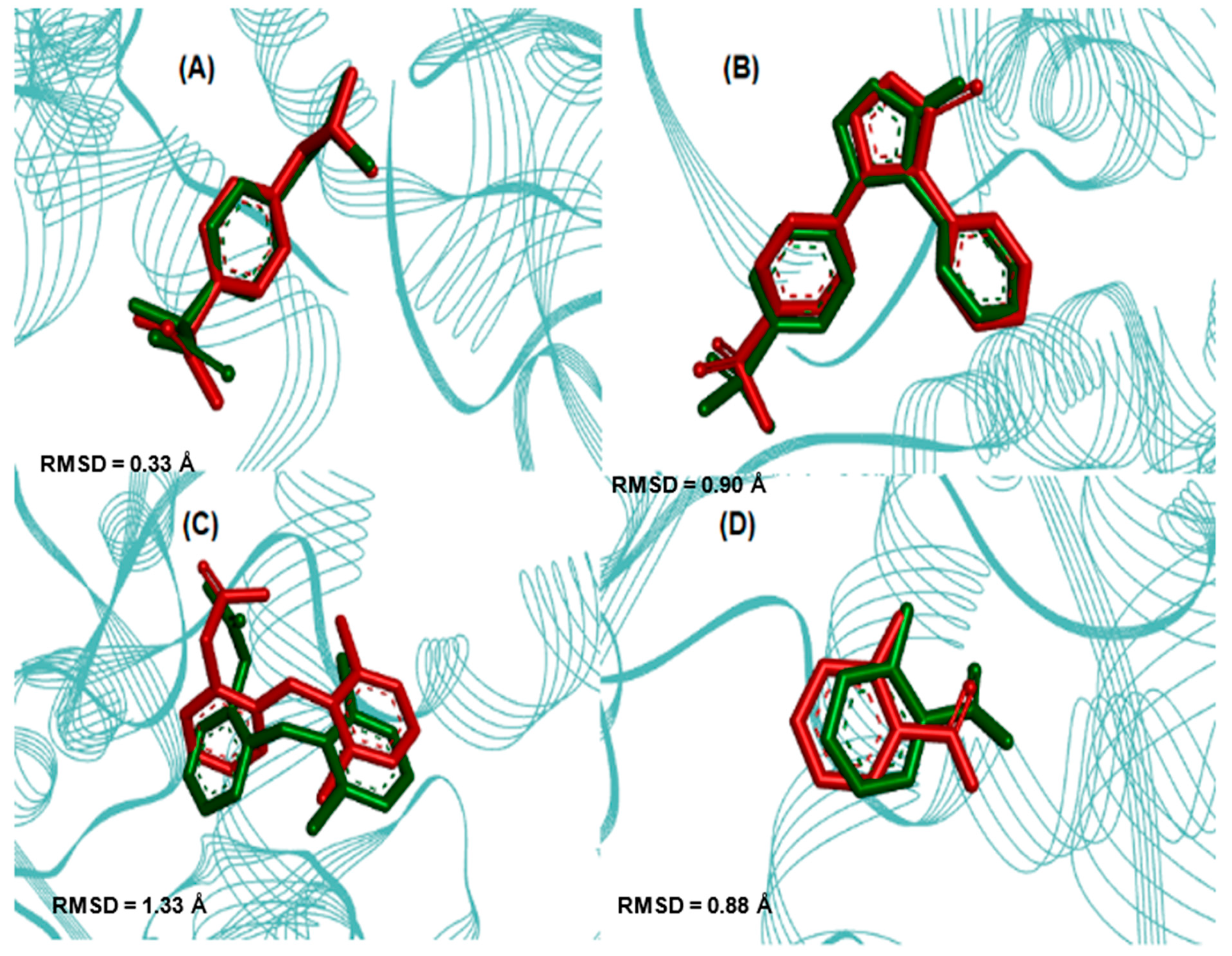
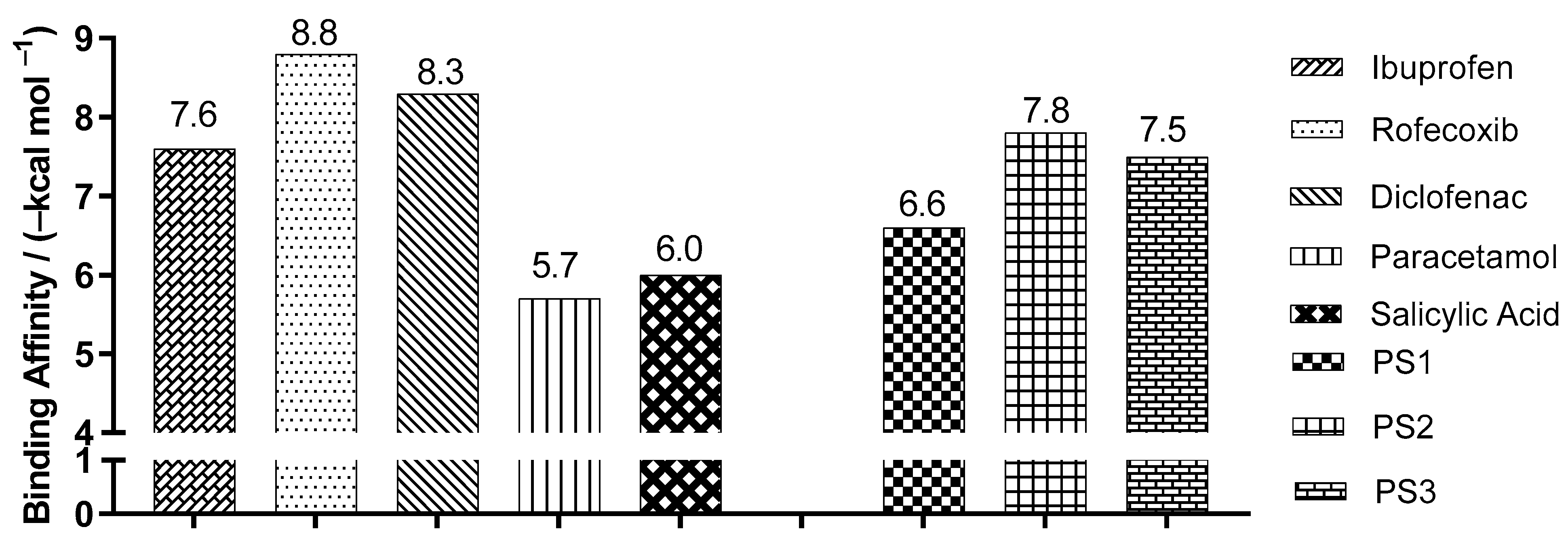
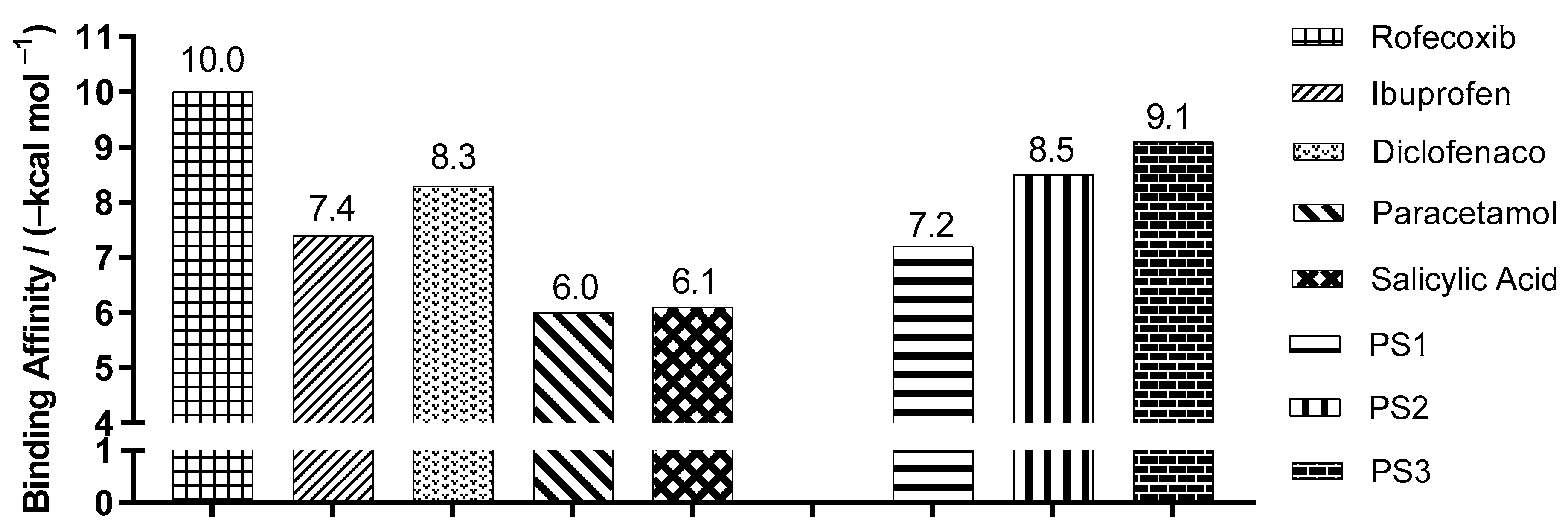
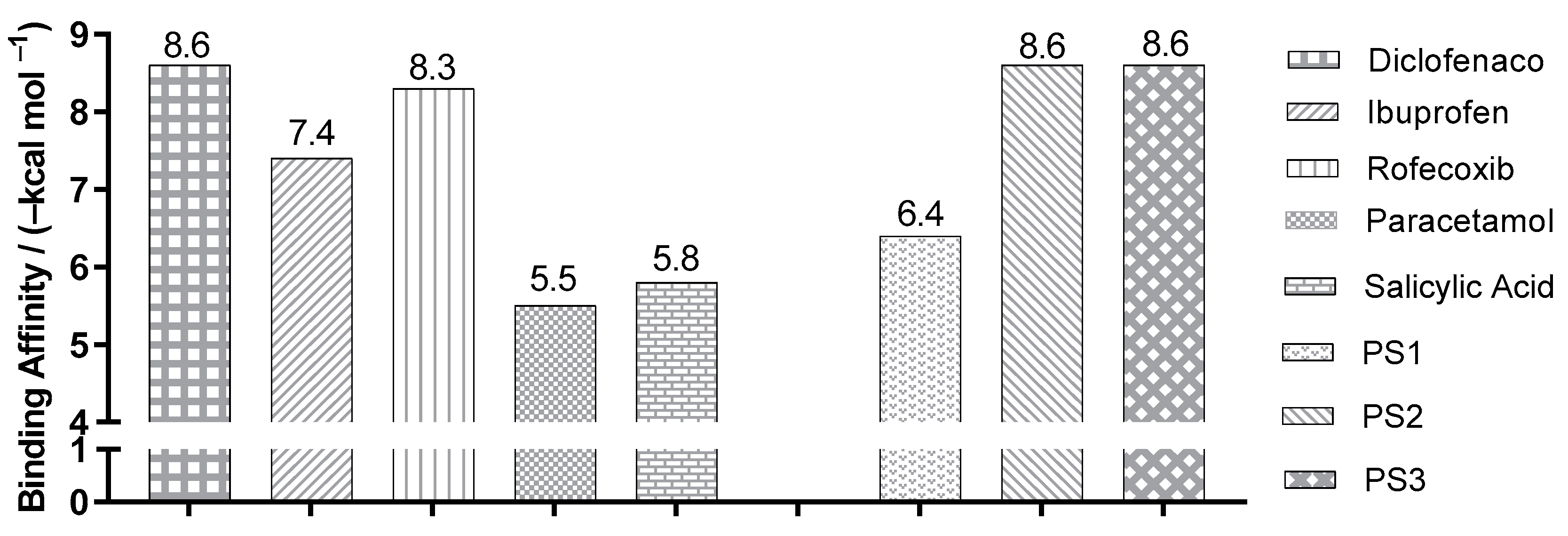
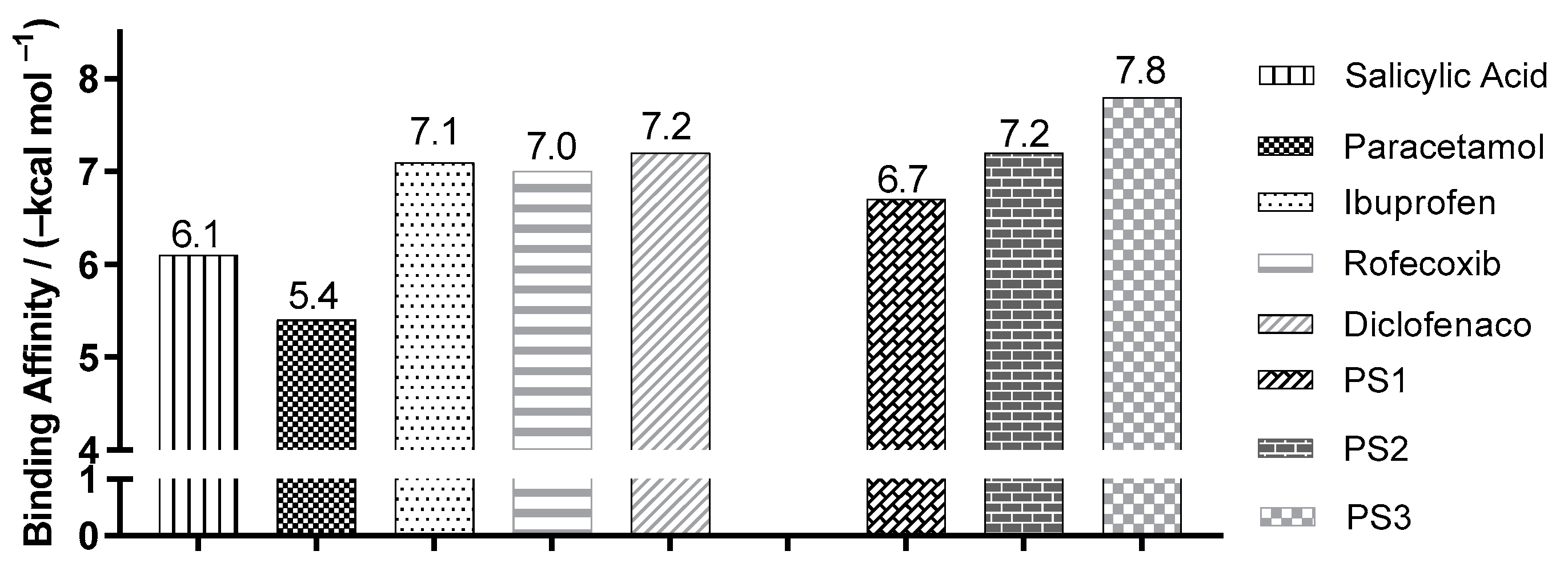
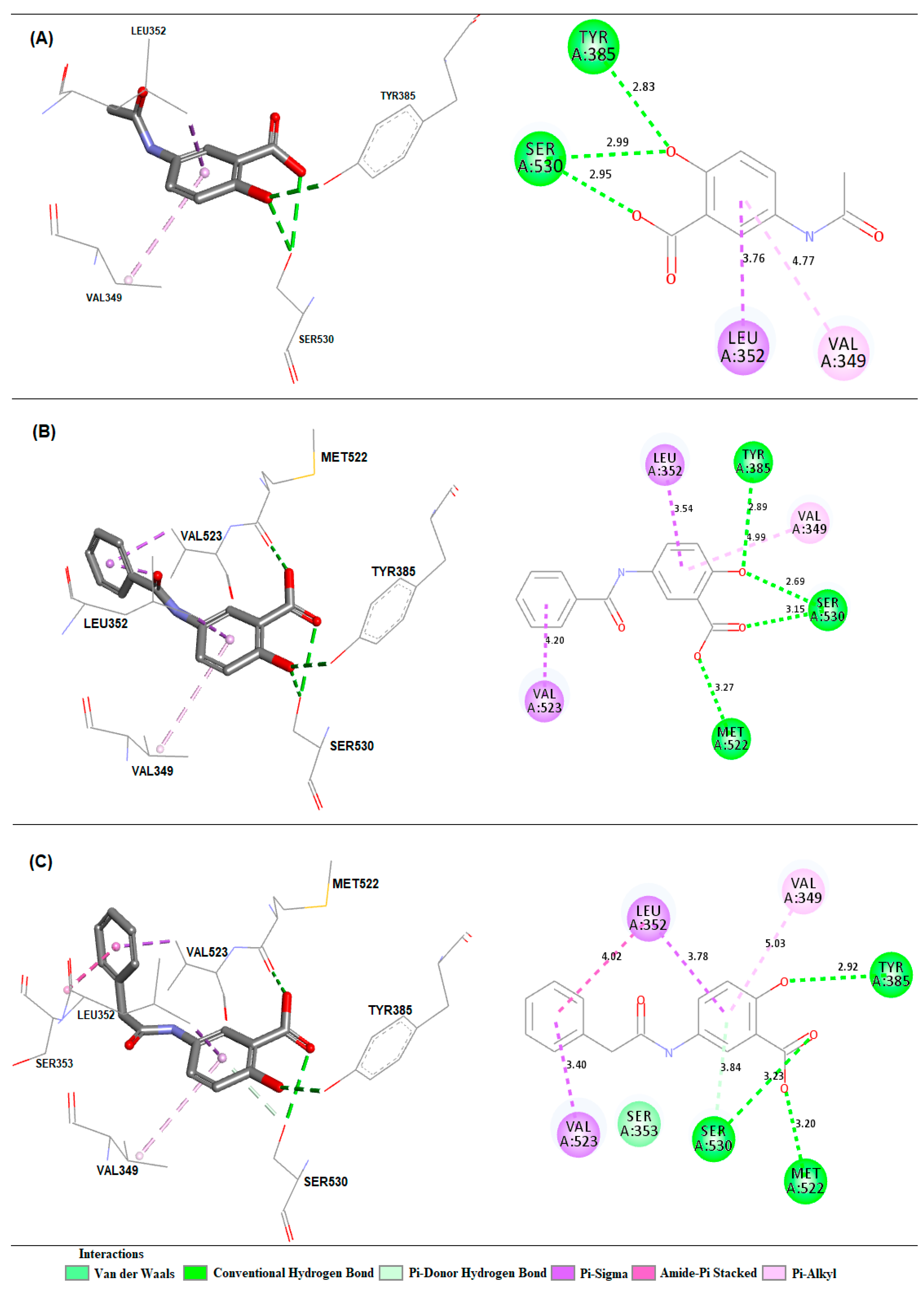
2.3. Molecular Docking Simulations
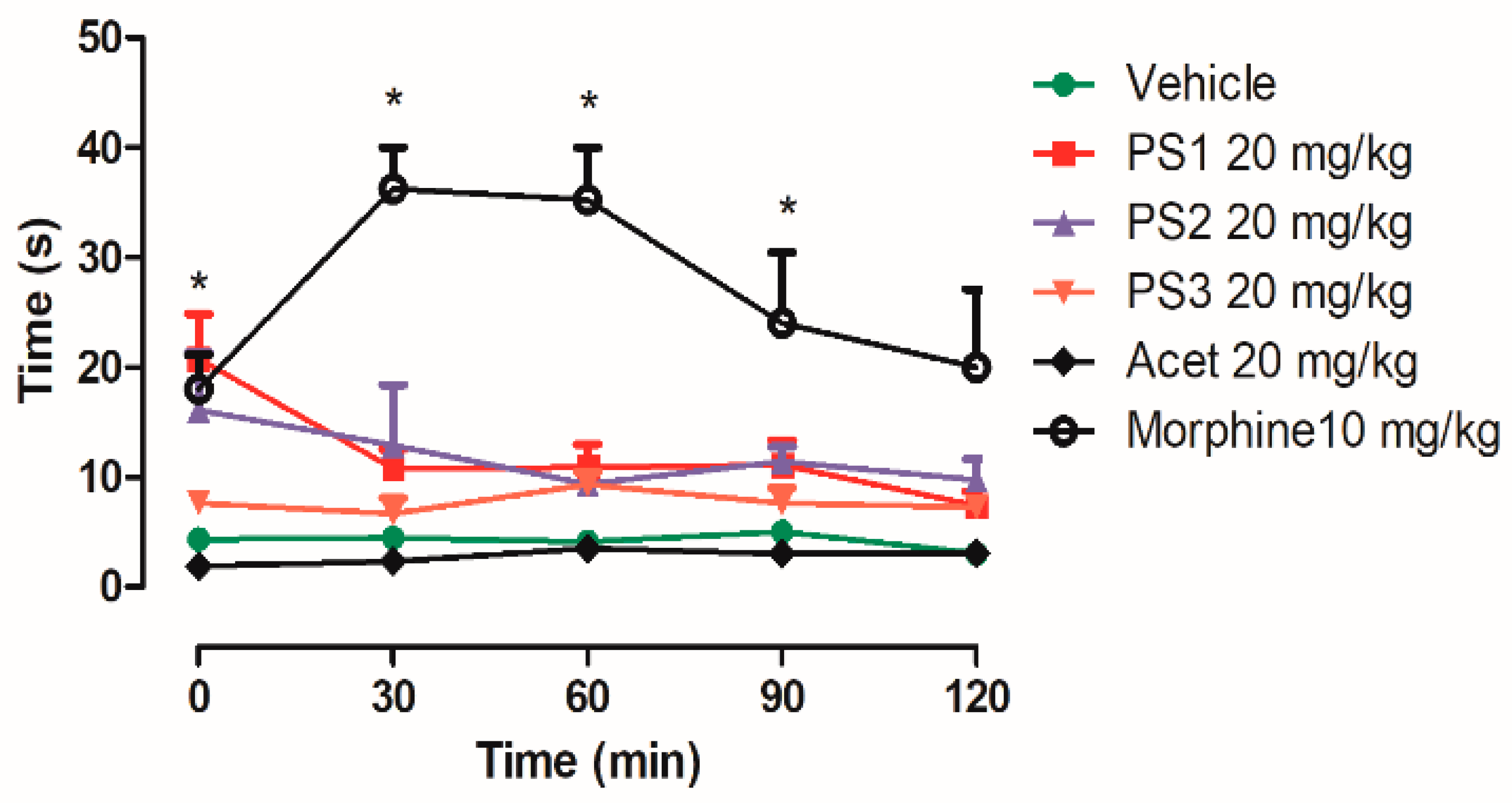
2.4. Anti-Nociceptive Activity
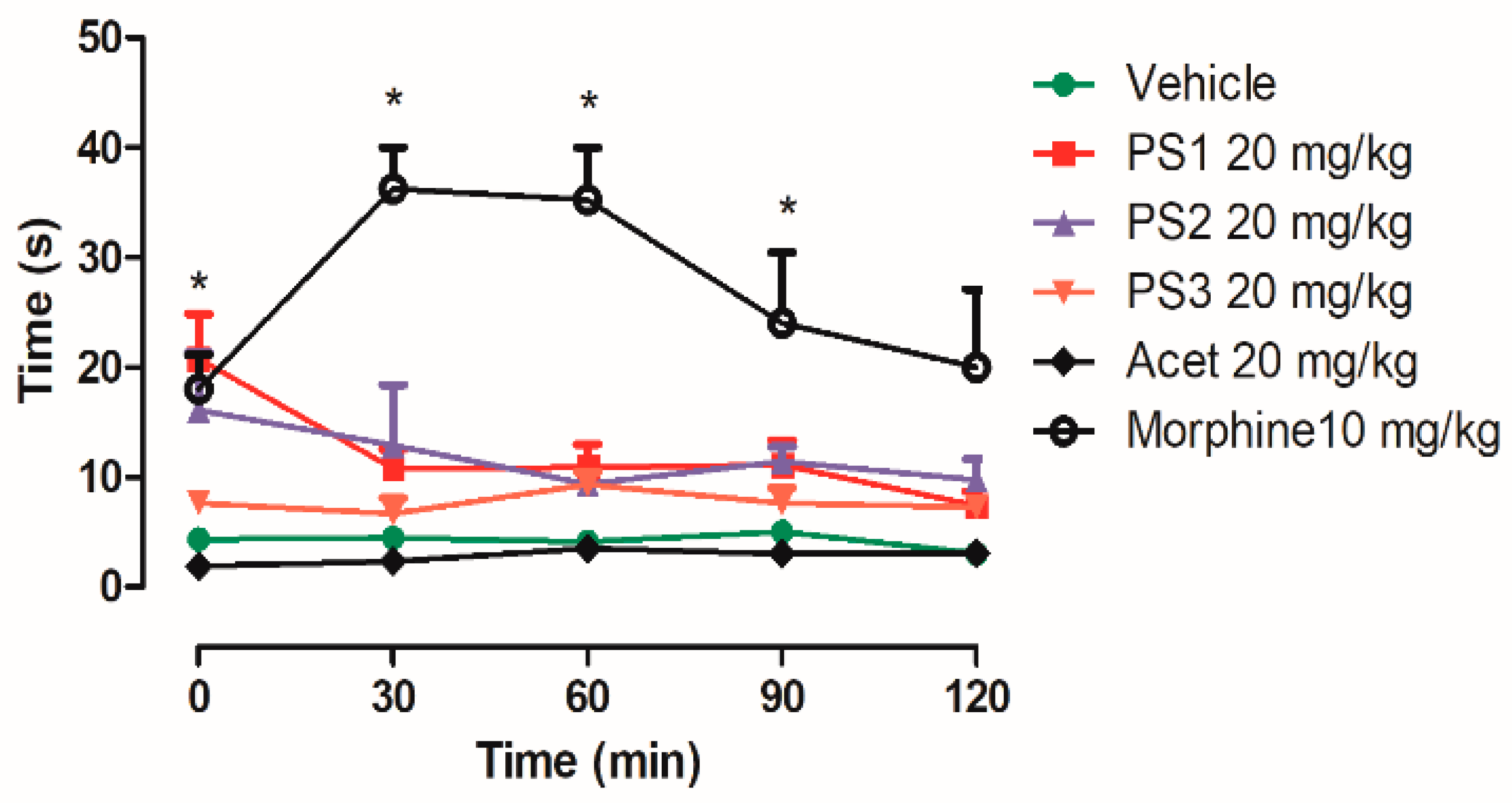
2.4.1. Central Analgesic Activity
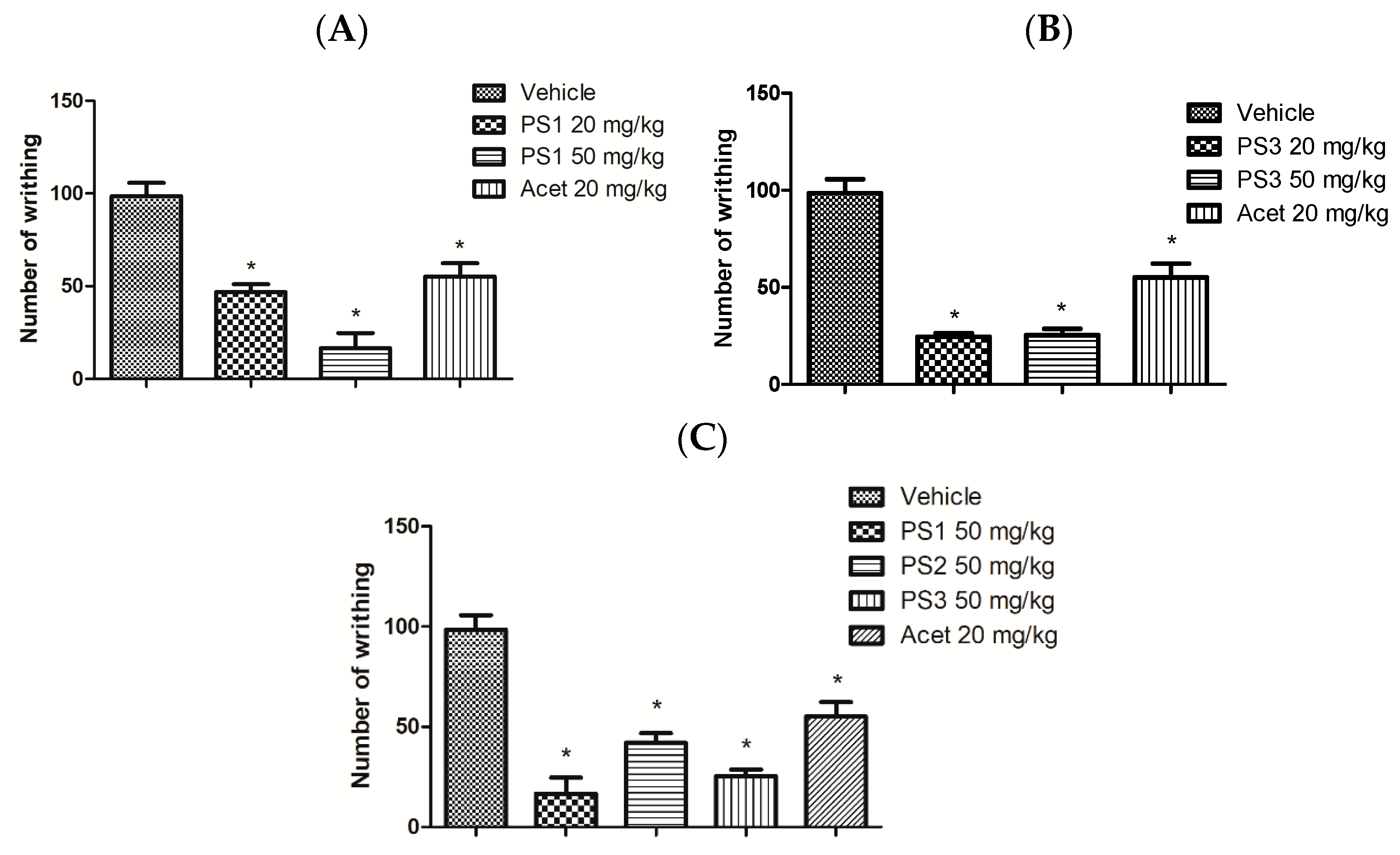
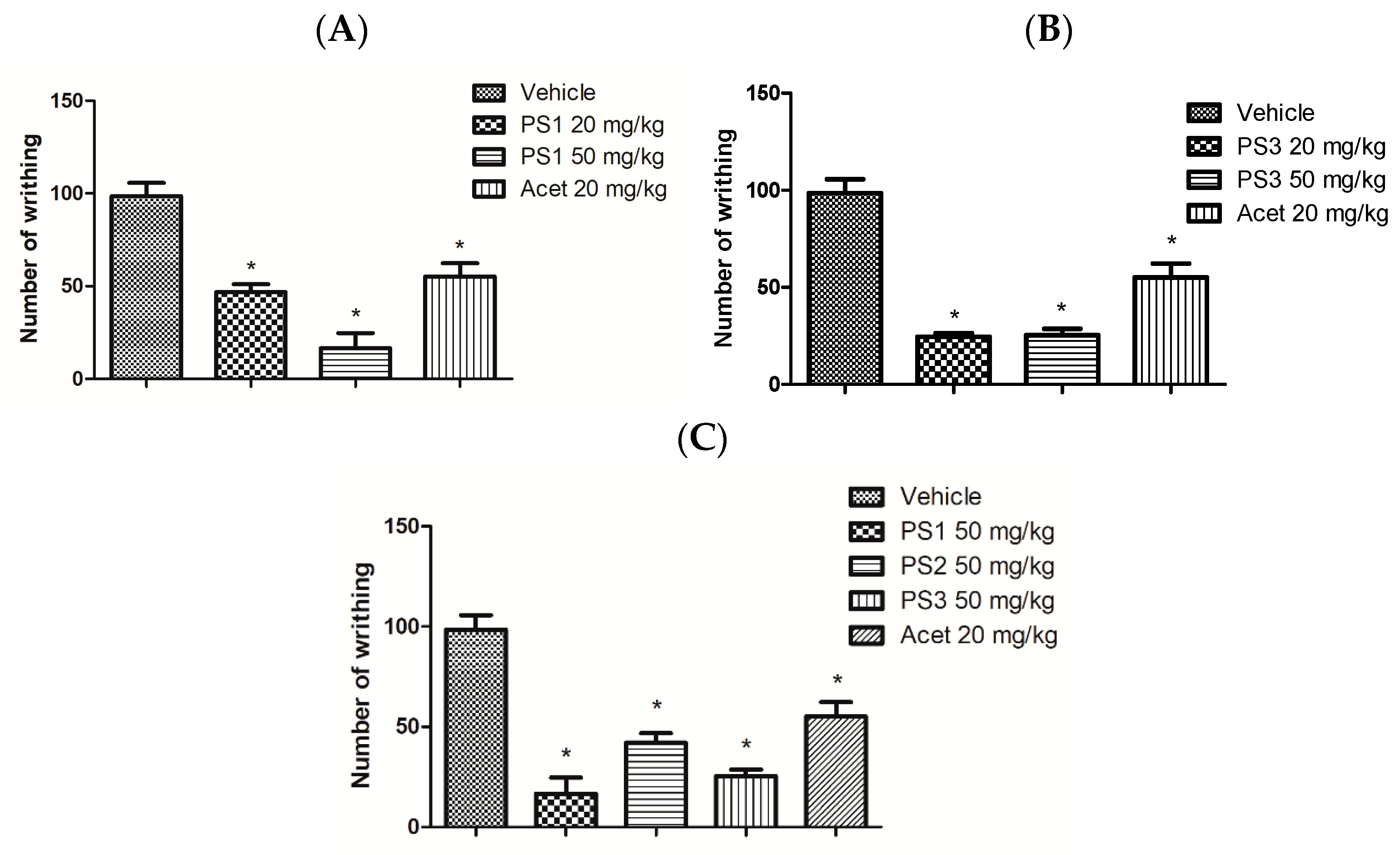
2.4.2. Peripheral Analgesic Activity
3. Discussion
3.1. Synthesis of 5-Acetamido-2-Hydroxy Benzoic Acid and Derivatives
3.2. In-Silico Study of Oral Bioavailability, Bioactivity, ADME and Toxicity Risk Assessment
3.3. Molecular Docking Simulations
3.4. Anti-Nociceptive Activity
4. Materials and Methods
4.1. Chemicals and Equipment
4.2. Synthetic Methodology
4.3. In-Silico Study of Oral Bioavailability, Bioactivity, ADME and Toxicity Risk Assessment
4.4. Molecular Docking Simulations
4.5. Anti-Nociceptive Activity
4.5.1. Animals
4.5.2. Writhing Test Induced by Acetic Acid
4.5.3. Hot Plate
4.5.4. Ethical Aspects
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mansouri, M.T.; Hemmati, A.A.; Naghizadeh, B.; Mard, S.A.; Rezaie, A.; Ghorbanzadeh, B. A study of the mechanisms underlying the anti-inflammatory effect of ellagic acid in carrageenan-induced paw edema in rats. Indian J. Pharmacol. 2015, 47, 292–298. [Google Scholar] [PubMed]
- Richy, F.; Bruyere, O.; Ethgen, O.; Rabenda, V.; Bouvenot, G.; Audran, M.; Herrero-Beaumont, G.; Moore, A.; Eliakim, R.; Haim, M.; et al. Time dependent risk of gastrointestinal complications induced by non-steroidal anti-inflammatory drug use: A consensus statement using a meta-analytic approach. Ann. Rheum. Dis. 2004, 63, 759–766. [Google Scholar] [CrossRef]
- Beck, P.L.; Xavier, R.; Lu, N.; Nanda, N.N.; Dinauer, M.; Podolsky, D.K.; Seed, B. Mechanisms of NSAID-induced gastrointestinal injury defined using mutant mice. Gastroenterology 2000, 119, 699–705. [Google Scholar] [CrossRef]
- Chan, F.K.L.; Graham, D.Y. Prevention of non-steroidal anti-inflammatory druggastrointestinal complications—Review and recommendations basedon risk assessment. Aliment. Pharmacol. Ther. 2004, 19, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Borges, R.S. Planejamento, Síntese e Avaliação Antioxidante de Inibidores Fenólicos da PGES Derivados da Associação p-Aminofenol e Salicilatos; Tese de Doutorado em Neurociências e Biologia Celular, UFPA: Belém, Brazil, 2007. [Google Scholar]
- Borges, R.S.; Alves, C.N.; Nascimento, J.L.M. Aplicação de Derivados da Associação Molecular como Antiagregantes Plaquetários e Inibidores de Radicais Livres. PI1001434-9, 20 January 2010. [Google Scholar]
- Borges, R.S.; Pereira, G.A.N.; Vale, J.K.L.; França, L.C.S.; Monteiro, M.C.; Alves, C.N.; Silva, A.B.F.d. Design and evaluation of 4-aminophenol and salicylate derivatives as free-radical scavenger. Chem. Biol. Drug Des. 2013, 81, 414–419. [Google Scholar] [CrossRef]
- Borges, R.S.; Castle, S.L. The antioxidant properties of salicylate derivatives: A possible new mechanism of anti-inflammatory activity. Bioorganic Med. Chem. Lett. 2015, 25, 4808–4811. [Google Scholar] [CrossRef] [PubMed]
- Guedes, K.M.M.; Borges, R.S.; Fontes-Júnior, E.A.; Silva, A.S.B.; Fernandes, L.M.P.; Cartágenes, S.C.; Pinto, A.C.G.; Silva, M.L.; Queiroz, L.M.D.; Vieira, J.L.F.; et al. Salicytamide: A New Anti-inflammatory Designed Drug Candidate. Inflammation 2018, 41, 1349–1360. [Google Scholar] [CrossRef] [PubMed]
- Hawkey, C.J. COX-1 and COX-2 inhibitors. Best Pract. Res. Clin. Gastroenterol. 2001, 15, 801–820. [Google Scholar] [CrossRef]
- Tanhehco, E.J. Potassium channel modulators as anti-inflammatory agents. Expert. Opin. Ther. Pat. 2001, 11, 1137–1145. [Google Scholar] [CrossRef]
- Van De Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef]
- Roy, S.; Samant, L.; Chowdhary, A. In silico pharmacokinetics analysis and ADMET of phytochemicals of Datura metel Linn. and Cynodon dactylon Linn. J. Chem. Pharm. Res. 2015, 7, 385–388. [Google Scholar]
- Cunha, E.L.; Santos, C.F.; Braga, F.S.; Costa, J.S.; Silva, R.C.; Favacho, H.A.; Hage-Melim, L.I.; Carvalho, J.C.; Silva, C.H.; Santos, C.B. Computational Investigation of Antifungal Compounds Using Molecular Modeling and Prediction of ADME/Tox Properties. J. Comput. Theor. Nanosci. 2015, 12, 3682–3691. [Google Scholar] [CrossRef]
- Yee, S. In vitro permeability across caco-2 cells (colonic) can predict in vivo (small intestinal) absorption in man-fact or myth. Pharm. Res. 1997, 14, 763–766. [Google Scholar] [CrossRef]
- Zhao, Y.H.; Le, J.; Abraham, M.H.; Hersey, A.; Eddershaw, P.J.; Luscombe, C.N.; Platts, J.A. Evaluation of human intestinal absorption data and subsequent derivation of a quantitative structure activity relationship (QSAR) with the Abraham descriptors. J. Pharm. Sci. 2001, 90, 749–784. [Google Scholar] [CrossRef] [PubMed]
- Yazdanian, M.; Glynn, S.L.; Wright, J.L.; Hawi, A. Correlating Partitioning and Caco-2 Cell Permeability of Structurally Diverse Small Molecular Weight Compounds. Pharm. Res. 1998, 15, 1490–1494. [Google Scholar] [CrossRef]
- Irvine, J.D.; Takahashi, L.; Lockhart, K.; Cheong, J.; Tolan, J.W.; Selick, H.E.; Grove, J.R. MDCK (Madin–Darby canine kidney) cells: A tool for membrane permeability screening. J. Pharm. Sci. 1999, 88, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Costa, J.S.; Costa, K.S.L.; Cruz, J.V.; Ramos, R.S.; Silva, L.B.; Brasil, D.S.B.; Silva, C.H.T.P.; Santos, C.B.R.; Macêdo, W.J.C. Virtual screening and statistical analysis in the design of new caffeine analogues molecules with potential epithelial anticancer activity. Curr. Pharm. Des. 2018, 24, 576–594. [Google Scholar] [CrossRef]
- Cruz, J.V.; Neto, M.F.A.; Silva, L.B.; Ramos, R.; Costa, J.; Brasil, D.S.B.; Lobato, C.C.; Costa, G.V.; Bittencourt, J.A.H.M.; Silva, C.H.T.P.; et al. Identification of novel protein kinase receptor type 2 inhibitors using pharmacophore and structure-based virtual screening. Molecules 2018, 23, 453. [Google Scholar] [CrossRef]
- Alberga, D.; Trisciuzzi, D.; Mansouri, K.; Mangiatordi, G.F.; Nicolotti, O. Prediction of Acute Oral Systemic Toxicity Using a Multifingerprint Similarity Approach. Toxicol. Sci. 2018, 167, 484–495. [Google Scholar] [CrossRef]
- Orlando, B.J.; Lucido, M.J.; Malkowski, M.G. The structure of ibuprofen bound to cyclooxygenase-2. J. Struct. Biol. 2015, 189, 62–66. [Google Scholar] [CrossRef]
- Orlando, B.J.; Malkowski, M.G. Crystal structure of rofecoxib bound to human cyclooxygenase-2. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2016, 72, 772–776. [Google Scholar] [CrossRef] [PubMed]
- Rowlinson, S.W.; Kiefer, J.R.; Prusakiewicz, J.J.; Pawlitz, J.L.; Kozak, K.R.; Kalgutkar, A.S.; Stallings, W.C.; Kurumbail, R.G.; Marnett, L.J. A novel mechanism of cyclooxygenase-2 inhibition involving interactions with Ser-530 and Tyr-385. J. Biol. Chem. 2003, 278, 45763–45769. [Google Scholar] [CrossRef] [PubMed]
- Lucido, M.J.; Orlando, B.J.; Vecchio, A.J.; Malkowski, M.G. Crystal structure of aspirin-acetylated human cyclooxygenase-2: Insight into the formation of products with reversed stereochemistry. Biochemistry 2016, 55, 1226–1238. [Google Scholar] [CrossRef] [PubMed]
- Cera, E.D. Thermodynamic Theory of Site-Specific Binding Processes in Biological Macromolecules; Cambridge University Press: Cambridge, UK; Washington University: St Louis, MO, USA, 1995. [Google Scholar]
- Gohlke, H.; Klebe, G. Approaches to the description and prediction of the binding affinity of small-molecule ligands to macromolecular receptors. Angew. Chem. Int. Ed. 2002, 41, 2644–2676. [Google Scholar] [CrossRef]
- Sportoletti, G.; Testi, V. Amino-Salicylic Acid Derivatives and Pharmaceutical Compositions. WO 86/03199 A1, 26 November 1985. [Google Scholar]
- Serhan, C.N. Resolution phase of inflammation: Novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu. Rev Immunol. 2007, 25, 101–137. [Google Scholar] [CrossRef]
- Cherezov, V.; Rosenbaum, D.M.; Hanson, M.A.; Rasmussen, S.G.; Thian, F.S.; Kobilka, T.S.; Choi, H.J.; Kuhn, P.; Weis, W.I.; Kobilka, B.K.; et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 2007, 318, 1258–1265. [Google Scholar] [CrossRef]
- Gurevich, V.V.; Gurevich, E.V. Molecular Mechanisms of GPCR Signaling:A Structural Perspective. Int. J. Mol. Sci. 2017, 18, 2519. [Google Scholar] [CrossRef]
- Filmore, D. It’s a GPCR world. Mod. Drug Discov. 2004, 28, 24–26. [Google Scholar]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Tang, T.; Gong, T.; Jiang, W.; Zhou, R. GPCRs in NLRP3 Inflammasome Activation, Regulation, and Therapeutics. Trends Pharmacol. Sci. 2018, 39, 798–811. [Google Scholar] [CrossRef]
- Bermudez, M.; Nguyen, T.N.; Omieczynski, C.; Wolber, G. Strategies for the discovery of biased GPCR ligands. Drug Discov. Today 2019, 24, 1031–1037. [Google Scholar] [CrossRef]
- Muratspahić, E.; Freissmuth, M.; Gruber, C.W. Nature-Derived Peptides: A Growing Niche for GPCR Ligand Discovery. Trends Pharmacol. Sci. 2019, 40, 309–316. [Google Scholar] [CrossRef]
- Ashok, S.R.; Shivananda, M.K.; Manikandan, A.; Chandrasekaran, R. Discovery and synthesis of 2-amino-1-methyl-1H-imidazol-4(5H)-ones as GPCR ligands; an approach to develop breast cancer drugs via GPCR associated PAR1 and PI3Kinase inhibition mechanism. Bioorg. Chem. 2019, 86, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Bantscheff, M.; Scholten, A.; Heck, A.J.R. Revealing promiscuous drug-target interactions by chemical proteomics. Drug Discov. Today 2009, 14, 1021–1029. [Google Scholar] [CrossRef]
- Hu, Y.; Gupta-Ostermann, D.; Bajoratha, J. Exploring Compound Promiscuity Patterns and Multi-Target Activity Spaces. Comput. Struct. Biotechnol. J. 2014, 9, e201401003. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Ye, R.D. Role of G protein-coupled receptors in inflammation. Acta Pharmacol. Sin. 2012, 33, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Vane, J.R. Inhibition of Prostaglandin Synthesis as a Mechanism of Action for Aspirin-like Drugs. Nat. New Biol. 1971, 231, 232–235. [Google Scholar] [CrossRef]
- Vane, J.R.; Botting, R.M. Mechanism of action of aspirin-like drugs. Semin. Arthritis Rheum. 1997, 26, 2–10. [Google Scholar] [CrossRef]
- Ortiz, M.; Granados, S.V.; Castañeda, H.G. Possible Activation of Inward Rectifier-and G Protein-Coupled K^+ Channels in the Antinociception Induced by Non-steroidal Anti-inflammatory Drugs. Proc. West. Pharmacol. 2006, 49, 141–144. [Google Scholar]
- Wulff, H.; Palle, C. Recent developments in ion channel pharmacology. Channels 2015, 9, 335. [Google Scholar] [CrossRef] [PubMed]
- Gfeller, D.; Michielin, O.; Zoete, V. Shaping the interaction landscape of bioactive molecules. Bioinformatics 2013, 29, 3073–3079. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ma, C.; Wipf, P.; Liu, H.; Su, W.; Xie, X.-Q. TargetHunter: An in silico target identification tool for predicting therapeutic potential of small organic molecules based on chemogenomic database. AAPS J. 2013, 15, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, G.; Coletto, L.; Sciorati, C.; Bozzolo, E.; Manunta, P.; Rovere-Querini, P.; Manfredi, A. Ion Channels and Transporters in Inflammation: Special Focus on TRP Channels and TRPC6. Cells 2018, 7, 70. [Google Scholar] [CrossRef]
- Roberts, M.S. Dermal Absorption and Toxicity Assessment, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Pratt, W.B. The entry, distribution, and elimination of drugs. In Principles of Drug Action: The Basis of Pharmacology, 3rd ed.; Pratt, W.B., Taylor, P., Eds.; Churchill Livingstone: New York, NY, USA, 1990. [Google Scholar]
- Ajay, A.; Bemis, G.W.; Murcko, M.A. Designing libraries with CNS activity. J. Med. Chem. 1999, 42, 4942–4951. [Google Scholar] [CrossRef]
- Ma, X.L.; Chen, C.; Yang, J. Predictive model of blood-brain barrier penetration of organic compounds. Acta Pharmacol. Sin. 2005, 26, 500–512. [Google Scholar] [CrossRef]
- Geneve, J.E.A.N.; Hayat-Bonan, B.; Labbe, G.; Degott, C.; Letteron, P.; Freneaux, E.; Pessayre, D. Inhibition of mitochondrial beta-oxidation of fatty acids by pirprofen. Role in microvesicular steatosis due to this nonsteroidal anti-inflammatory drug. J. Pharmacol. Exp. Ther. 1987, 242, 1133–1137. [Google Scholar]
- Dahl, S.L.; Ward, J.R. Pharmacology, clinical efficacy, and adverse effects of the nonsteroidal anti-inflammatory agent benoxaprofen. Pharmacother. J. Hum. Pharmacol. Drug Ther. 1982, 2, 354–365. [Google Scholar] [CrossRef]
- Zimmerman, H.J. Hepatotoxicity: The Adverse Effects of Drugs and Other Chemicals on the Liver, 2nd ed.; Lippincott Williams & Wilkins: Dallas, TX, USA, 1999. [Google Scholar]
- Li, C.; Grillo, M.P.; Benet, L.Z. In vivo mechanistic studies on the metabolic activation of 2-phenylpropionic acid in rat. J. Pharmacol. Exp. Ther. 2003, 305, 250–256. [Google Scholar] [CrossRef]
- Boelsterli, U.A. Diclofenac-induced liver injury: A paradigm of idiosyncratic drug toxicity. Toxicol. Appl. Pharmacol. 2003, 192, 307–322. [Google Scholar] [CrossRef]
- Dong, J.Q.; Liu, J.; Smith, P.C. Role of benoxaprofen and flunox-aprofen acyl glucuronides in covalent binding to rat plasmaand liver proteins in vivo. Biochem. Pharmacol. 2005, 70, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, T.; Hayashi, N.; Maizumi, H.; Huff, J.; Barrett, J.C. Benzene-,catechol-, hydroquinone- and phenol-induced cell transformation, gene mutations, chromosome aberrations, aneuploidy, sister chro-matid exchanges and unscheduled DNA synthesis in Syrian hamster embryo cells. Mutat. Res. Fundam. Mol. Mech. Mutagen. 1997, 373, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Bolton, J.L.; Trush, M.A.; Penning, T.M.; Dryhurst, G.; Monks, T.J. Role of quinones in toxicology. Chem. Res. Toxicol. 2000, 13, 135–160. [Google Scholar] [CrossRef] [PubMed]
- Rannug, U.; Holme, J.A.; Hongslo, J.K.; Šrám, R.J. An evalution of the genetic toxicityof paracetamol. Mutat. Res. Fundam. Mol. Mech. Mutagen. 1995, 327, 179–200. [Google Scholar] [CrossRef] [PubMed]
- Gujral, J.S.; Knight, T.R.; Farhood, A.; Bajt, M.L.; Jaeschke, H. Mode of cell death after acetaminophen overdose in mice: Apoptosis or oncotic necrosis? Toxicol. Sci. 2002, 67, 322–328. [Google Scholar] [CrossRef]
- Calder, I.C.; Hart, S.J.; Smail, M.C.; Tange, J.D. Hepatotoxicity of phenacetin and paracetamol in the Gunn rat. Pathology 1981, 13, 757–762. [Google Scholar] [CrossRef]
- Kalgutkar, A.S.; Gardner, I.; Obach, R.S.; Shaffer, C.L.; Callegari, E.; Henne, K.R.; O’Donnell, J.P. A comprehensive listing of bioactivation pathways of organic functional groups. Curr. Drug Metab. 2005, 6, 161–225. [Google Scholar] [CrossRef]
- Nelson, S.D.; Forte, A.J.; McMurtry, R.J. Decreased toxicity of the N-methyl analogs of acetaminophen and phenacetin. Res. Commun. Chem. Pathol. Pharmacol. 1978, 22, 61–71. [Google Scholar]
- Bursulaya, B.D.; Totrov, M.; Abagyan, R.; Brooks, C.L. Comparative study of several algorithms for flexible ligand docking. J. Comput. Aided. Mol. Des. 2003, 17, 755–763. [Google Scholar] [CrossRef]
- Cole, J.C.; Murray, C.W.; Nissink, J.W.M.; Taylor, R.D.; Taylor, R. Comparing protein–ligand docking programs is difficult. Proteins Struct. Funct. Bioinf. 2005, 60, 325–332. [Google Scholar] [CrossRef]
- Hevener, K.E.; Zhao, W.; Ball, D.M.; Babaoglu, K.; Qi, J.; White, S.W.; Lee, R.E. Validation of molecular docking programs for virtual screening against dihydropteroate synthase. J. Chem. Inf. Model. 2009, 49, 444–460. [Google Scholar] [CrossRef] [PubMed]
- Kontoyianni, M.; McClellan, L.M.; Sokol, G.S. Evaluation of Docking Performance: Comparative Data on Docking Algorithms. J. Med. Chem. 2004, 47, 558–565. [Google Scholar] [CrossRef]
- Nissink, J.W.M.; Murray, C.; Hartshorn, M.; Verdonk, M.L.; Cole, J.C.; Taylor, R. A new test set for validating predictions of protein-ligand interaction. Proteins Struct. Funct. Bioinf. 2002, 49, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Barcellos, M.P.; Santos, C.B.R.; Federico, L.B.; Almeida, P.F.D.; Silva, C.H.D.P.; Taft, C.A. Pharmacophore and structure-based drug design, molecular dynamics and admet/tox studies to design novel potential pad4 inhibitors. J. Biomol. Struct. Dyn. 2019, 37, 966–981. [Google Scholar] [CrossRef] [PubMed]
- Borges, R.S.; Palheta, I.C.; Ota, S.S.B.; Morais, R.B.; Barros, V.A.; Ramos, R.S.; Silva, R.C.; Costa, J.S.; Silva, C.H.T.P.; Campos, J.M.; et al. Toward of Safer Phenylbutazone Derivatives by Exploration of Toxicity Mechanism. Molecules 2019, 24, 143. [Google Scholar] [CrossRef]
- Costa, J.S.; Ramos, R.S.; Costa, K.S.L.; Brasil, D.S.B.; Silva, C.H.T.P.; Ferreira, E.F.B.; Borges, R.S.; Campos, J.M.; Macêdo, W.J.C.; Santos, C.B.R. An in silico study of the antioxidant ability for two caffeine analogs using molecular docking and quantum chemical methods. Molecules 2018, 23, 2801. [Google Scholar] [CrossRef] [PubMed]
- Ramos, R.D.S.; Costa, J.D.S.; Silva, R.C.; Costa, G.V.; Rodrigues, A.B.L.; Rabelo, É.D.M.; Santos, C.B.R.D. Identification of Potential Inhibitors from Pyriproxyfen with Insecticidal Activity by Virtual Screening. Pharmaceuticals 2019, 12, 20. [Google Scholar] [CrossRef]
- Esser, R.; Berry, C.; Du, Z.; Dawson, J.; Fox, A.; Fujimoto, R.A.; Haston, W.; Kimble, E.F.; Koehler, J.; Peppard, J.; et al. Preclinical Pharmacology of Lumiracoxib: A Novel Selective Inhibitor of Cyclooxygenase-2. Br. J. Pharmacol. 2005, 144, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Kurumbail, R.G.; Stevens, A.M.; Gierse, J.K.; McDonald, J.J.; Stegeman, R.A.; Pak, J.Y.; Gildehaus, D.; Miyashiro, J.M.; Penning, T.D.; Seibert, K.; et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 1996, 384, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Marnett, L.J. Cyclooxygenase mechanisms. Curr. Opin. Chem. Biol. 2000, 4, 545–552. [Google Scholar] [CrossRef]
- House, H.O. Modern Organic Reactions; The Benjamin Publishing Co.: Amsterdam, The Netherlands, 1972. [Google Scholar]
- Patani, G.A.; LaVoie, E.J. Bioisosterism: A Rational Approach in Drug Design. Chem. Rev. 1996, 96, 3147–3176. [Google Scholar] [CrossRef]
- Biagi, G.; Giorgi, I.; Livi, O.; Nardi, A.; Calderone, V.; Martelli, A.; Martinotti, E.; LeRoy, S.O. Synthesis and biological activity of novel substituted benzanilides as potassium channel activators. V. Eur. J. Med. Chem. 2004, 39, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, R.M.; Bassler, G.C.; Morril, T.C. Indentificação Espectrometria de Compostos Orgânicos; Guanabara Dois: Rio de Janeiro, Brazil, 1994. [Google Scholar]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimatesolubility and permeability in drug discovery and developmentqsettings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Drwal, M.N.; Banerjee, P.; Dunkel, M.; Wettig, M.R.; Preissner, R. ProTox: A web server for the in silico prediction of rodent oral toxicity. Nucleic Acids Res. 2014, 42, W53–W58. [Google Scholar] [CrossRef]
- Santos, C.B.R.; Ramos, R.S.; Ortiza, B.L.S.; Silva, G.M.; Giuliatti, S.; Navarrete, J.L.A.; Carvalho, J.C.T. Oil from the fruits of Pterodon emarginatus Vog.: A traditional anti-inflammatory. Study combining in vivo and in silico. J. Ethnopharmacol. 2018, 222, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Derek for Windows, version 10.0.2; User Guide. Lhasa Limited; Department of Chemistry, University of Leeds: Leeds, UK, 2007.
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar]
- BIOVIA Dassault Systèmes. BIOVIA Discovery Studio Visualizer, version 17.2; Dassault Systèmes: San Diego, CA, USA, 2017.
- Turner, G.W.; Tedesco, E.; Harris, K.D.; Johnston, R.L.; Kariuki, B.M. Implementation of lamarckian concepts in a genetic algorithm for structure solution from powder diffraction data. Chem. Phys. Lett. 2000, 321, 183–190. [Google Scholar] [CrossRef]
- Bittencourt, J.A.H.M.; Neto, M.F.A.; Lacerda, P.S.; Bittencourt, R.C.V.S.; Silva, R.C.; Lobato, C.C.; Silva, L.B.; Leite, F.A.; Zuliani, J.P.; Rosa, J.M.C.; et al. In silico evaluation of ibuprofen and two benzoylpropionic acid derivatives with potential anti-inflammatory activity. Molecules 2019, 24, 1476. [Google Scholar] [CrossRef]
- dos Santos, K.L.B.; Cruz, J.N.; Silva, L.B.; Ramos, R.S.; Neto, M.F.A.; Lobato, C.C.; Ota, S.S.B.; Leite, F.H.A.; Borges, R.S.; da Silva, C.H.T.P.; et al. Identification of Novel Chemical Entities for Adenosine Receptor Type 2A Using Molecular Modeling Approaches. Molecules 2020, 25, 1245. [Google Scholar] [CrossRef]
- Santos, C.B.R.; Santos, K.L.B.; Cruz, J.N.; Leite, F.H.A.; Borges, R.S.; Taft, C.A.; Campos, J.M.; Silva, C.H.T.P. Molecular modeling approaches of selective adenosine receptor type 2A agonists as potential anti-inflammatory drugs. J. Biomol. Struct. Dyn. 2020, 38, 3115–3127. [Google Scholar] [CrossRef]
- Leão, R.P.; Cruz, J.V.; da Costa, G.V.; Cruz, J.N.; Ferreira, E.F.B.; Silva, R.C.; de Lima, L.R.; Borges, R.S.; dos Santos, G.B.; Santos, C.B.R. Identification of New Rofecoxib-Based Cyclooxygenase-2 Inhibitors: A Bioinformatics Approach. Pharmaceuticals 2020, 13, 209. [Google Scholar] [CrossRef]
- Araújo, P.H.F.; Ramos, R.S.; da Cruz, J.N.; Silva, S.G.; Ferreira, E.F.B.; de Lima, L.R.; Macêdo, W.J.C.; Espejo-Román, J.M.; Campos, J.M.; Santos, C.B.R. Identification of Potential COX-2 Inhibitors for the Treatment of Inflammatory Diseases Using Molecular Modeling Approaches. Molecules 2020, 25, 4183. [Google Scholar] [CrossRef]
- Cruz, J.V.; Giuliatti, S.; Alves, L.B.; Silva, R.C.; Ferreira, E.F.B.; Kimani, N.M.; Silva, C.H.T.P.; de Souza, J.S.N.; Espejo-Román, J.M.; Santos, C.B.R. Identification of novel potential cyclooxygenase-2 inhibitors using ligand- and structure-based virtual screening approaches. J. Biomol. Struct. Dyn. 2021, 39, 5386–5408. [Google Scholar] [CrossRef]
- Koster, R.; Anderson, M.; Ej, D.B. Acetic acid for analgesic screening. Fed. Proc. 1959, 18, 412. [Google Scholar]
- Woolfe, G.; MacDonald, A.D. The evaluation of the analgesic action of pethidine hydrochloride (demerol). J. Pharmacol. Exp. Ther. 1944, 80, 300–307. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | MW 1 (<500 Da) | HBA 2 (≤10) | HBD 3 (≤5) | LogP (≤5) 4 | MPSA (Å2) 5 | MV (Å3) 6 | NRB 7 |
|---|---|---|---|---|---|---|---|
| Ibuprofen | 206.28 | 2 | 1 | 3.46 | 37.30 | 211.19 | 4 |
| Diclofenac | 296.15 | 3 | 2 | 4.57 | 49.33 | 238.73 | 4 |
| Acetaminophen | 151.16 | 3 | 2 | 0.68 | 49.33 | 140.01 | 1 |
| Salicylic acid | 137.11 | 3 | 1 | −1.81 | 60.36 | 116.32 | 1 |
| Rofecoxib | 314.36 | 4 | 0 | 0.71 | 60.45 | 264.79 | 3 |
| PS1 | 208.21 | 5 | 3 | 1.06 | 86.62 | 167.01 | 2 |
| PS2 | 210.19 | 5 | 3 | 2.74 | 86.62 | 221.86 | 3 |
| PS3 | 271.27 | 5 | 3 | 2.83 | 86.62 | 238.66 | 4 |
| Compounds | GPCR | Ion Channel Modulator | Kinase Inhibitor | Nuclear Receptor Ligand | Protease Inhibitor | Enzyme Inhibitor |
|---|---|---|---|---|---|---|
| Ibuprofen | −0.17 | −0.01 | −0.72 | 0.05 | −0.21 | 0.12 |
| Diclofenac | 0.14 | 0.20 | 0.17 | 0.09 | −0.10 | 0.25 |
| Acetaminophen | −1.05 | −0.54 | −1.04 | −1.21 | −1.20 | −0.68 |
| Rofecoxib | 0.20 | −0.18 | −0.18 | 0.12 | 0.26 | 0.61 |
| Salicylic Acid | −1.00 | −0.41 | −1.26 | −1.26 | −1.13 | −0.48 |
| PS1 | −0.67 | −0.38 | −0.70 | −0.64 | −0.76 | −0.33 |
| PS2 | −0.20 | −0.21 | −0.13 | −0.15 | −0.25 | −0.04 |
| PS3 | −0.10 | −0.19 | −0.15 | −0.04 | −0.14 | −0.02 |
| Compounds | Absorption | Distribuition | |||
|---|---|---|---|---|---|
| HIA 1 | Pcaco-2 2 | PMDCK 3 | PPB (%) 4 | CBrain/CBlood 5 | |
| Ibuprofen | 98.38 | 21.20 | 136.48 | 88.24 | 1.26 |
| Diclofenac | 95.95 | 24.53 | 51.46 | 91.95 | 1.39 |
| Acetaminophen | 88.23 | 18.78 | 15.43 | 0.00 | 0.61 |
| Salicylic Acid | 86.59 | 20.43 | 25.36 | 7.31 | 0.43 |
| Rofecoxib | 98.22 | 2.72 | 11.27 | 0.00 | 0.61 |
| PS1 | 75.97 | 19.93 | 5.17 | 14.34 | 0.35 |
| PS2 | 91.29 | 18.29 | 51.99 | 69.05 | 0.50 |
| PS3 | 91.95 | 20.70 | 31.04 | 66.41 | 0.15 |
| Compounds | Toxicity Prediction Alert (Lhasa Prediction) | Toxicophoric Group | Toxicity Alert | LD50 Toxic 1 | Toxicity Class 2 |
|---|---|---|---|---|---|
| Ibuprofen | Hepatotoxicity in human, mouse and rat | 2-arylacetic or 3-arylpropionic acid | PLAUSIBLE | 299 | III |
| Alpha-substituted propionic acid or ester | |||||
| Diclofenac | Hepatotoxicity in human | 2-arylacetic or 3-arylpropionic acid | CERTAIN | 53 | III |
| Nephrotoxicity in human, mouse and rat | Aryl or fulvenyl acetic or 2-propionic acid derivative | PLAUSIBLE | |||
| Acetaminophen | Chromosome damage in vitro in human | Phenol | CERTAIN | 338 | III |
| Phenol | PROBABLE | ||||
| Hepatotoxicity in human, mouse and rat | Para-aminophenol or derivative | CERTAIN | |||
| Rofecoxib | - | - | NO ALERTS | 4500 | V |
| Salicylic Acid | - | - | NO ALERTS | 480 | IV |
| PS1 | Hepatotoxicity in human, mouse and rat | Salicylic acid or analog | PLAUSIBLE | 2800 | V |
| Para-Aminophenol or derivative | |||||
| PS2 | Carcinogenicity in mouse and rat | Alkylaryl or bisaryl carboxylic acid or precursor | PLAUSIBLE | 2400 | V |
| Hepatotoxicity in human, mouse and rat | Salicylic acid or analog | ||||
| Peroxisome proliferation in mouse and rat | Para-aminophenol or derivative | ||||
| PS3 | Hepatotoxicity in human, mouse and rat | Salicylic acid or analog | PLAUSIBLE | 2175 | V |
| Para-aminophenol or derivative |
| Enzyme COX2 | Ligand | Experimental Binding Affinity * (kcal/mol) | Ki (nM) | Docking Predicted Binding Affinity (kcal/mol) | Resolution (Å) |
|---|---|---|---|---|---|
| (PDB ID 4PH9) | Ibuprofen | −7.3 | 7.2.103 [22] | −7.6 | 1.81 |
| (PDB ID 5KIR) | Rofecoxib | −9.2 | 310 [23] | −10.0 | 2.69 |
| (PDB ID 1PXX) | Diclofenac | −11.3 | 1.104 [24] | −8.6 | 2.90 |
| (PDB ID 5F1A) | salicylic acid | −6.7 | 1.104 [25] | −6.1 | 2.38 |
| (PDB ID 4PH9) | Ibuprofen | −7.3 | 7.2.103 [22] | −7.6 | 1.81 |
| Enzyme COX2 | Inhibitor * | Coordinates of the Grid Center (Angstrom) | Grid Dimensions (Angstrom) |
|---|---|---|---|
| (PDB ID 4PH9) | Ibuprofen | X = 12.58 Y = 24.20 Z = 25.33 | X = 17 Y = 17 Z = 16 |
| (PDB ID 5KIR) | Rofecoxib | X = 23.63 Y = 1.30 Z = 34.07 | X = 18 Y = 19 Z = 18 |
| (PDB ID 1PXX) | Diclofenac | X = 27.16 Y = 24.45 Z = 15.30 | X = 18 Y = 19 Z = 18 |
| (PDB ID 5F1A) | Salicylic acid | X = 41.72 Y = 24.24 Z = 240.03 | X = 18 Y = 17 Z = 17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, C.B.R.; Lobato, C.C.; Ota, S.S.B.; Silva, R.C.; Bittencourt, R.C.V.S.; Freitas, J.J.S.; Ferreira, E.F.B.; Ferreira, M.B.; Silva, R.C.; De Lima, A.B.; et al. Analgesic Activity of 5-Acetamido-2-Hydroxy Benzoic Acid Derivatives and an In-Vivo and In-Silico Analysis of Their Target Interactions. Pharmaceuticals 2023, 16, 1584. https://doi.org/10.3390/ph16111584
Santos CBR, Lobato CC, Ota SSB, Silva RC, Bittencourt RCVS, Freitas JJS, Ferreira EFB, Ferreira MB, Silva RC, De Lima AB, et al. Analgesic Activity of 5-Acetamido-2-Hydroxy Benzoic Acid Derivatives and an In-Vivo and In-Silico Analysis of Their Target Interactions. Pharmaceuticals. 2023; 16(11):1584. https://doi.org/10.3390/ph16111584
Chicago/Turabian StyleSantos, Cleydson B. R., Cleison C. Lobato, Sirlene S. B. Ota, Rai C. Silva, Renata C. V. S. Bittencourt, Jofre J. S. Freitas, Elenilze F. B. Ferreira, Marília B. Ferreira, Renata C. Silva, Anderson B. De Lima, and et al. 2023. "Analgesic Activity of 5-Acetamido-2-Hydroxy Benzoic Acid Derivatives and an In-Vivo and In-Silico Analysis of Their Target Interactions" Pharmaceuticals 16, no. 11: 1584. https://doi.org/10.3390/ph16111584
APA StyleSantos, C. B. R., Lobato, C. C., Ota, S. S. B., Silva, R. C., Bittencourt, R. C. V. S., Freitas, J. J. S., Ferreira, E. F. B., Ferreira, M. B., Silva, R. C., De Lima, A. B., Campos, J. M., Borges, R. S., & Bittencourt, J. A. H. M. (2023). Analgesic Activity of 5-Acetamido-2-Hydroxy Benzoic Acid Derivatives and an In-Vivo and In-Silico Analysis of Their Target Interactions. Pharmaceuticals, 16(11), 1584. https://doi.org/10.3390/ph16111584