

Tannic Acid and Ethyl Gallate Potentialize Paclitaxel Effect on Microtubule Dynamics in Hep3B Cells
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
2.1. TA and EG Treatment Enhance the Inhibitory Effect of PTX on the Viability of Hep3B HCC Cancer Cells
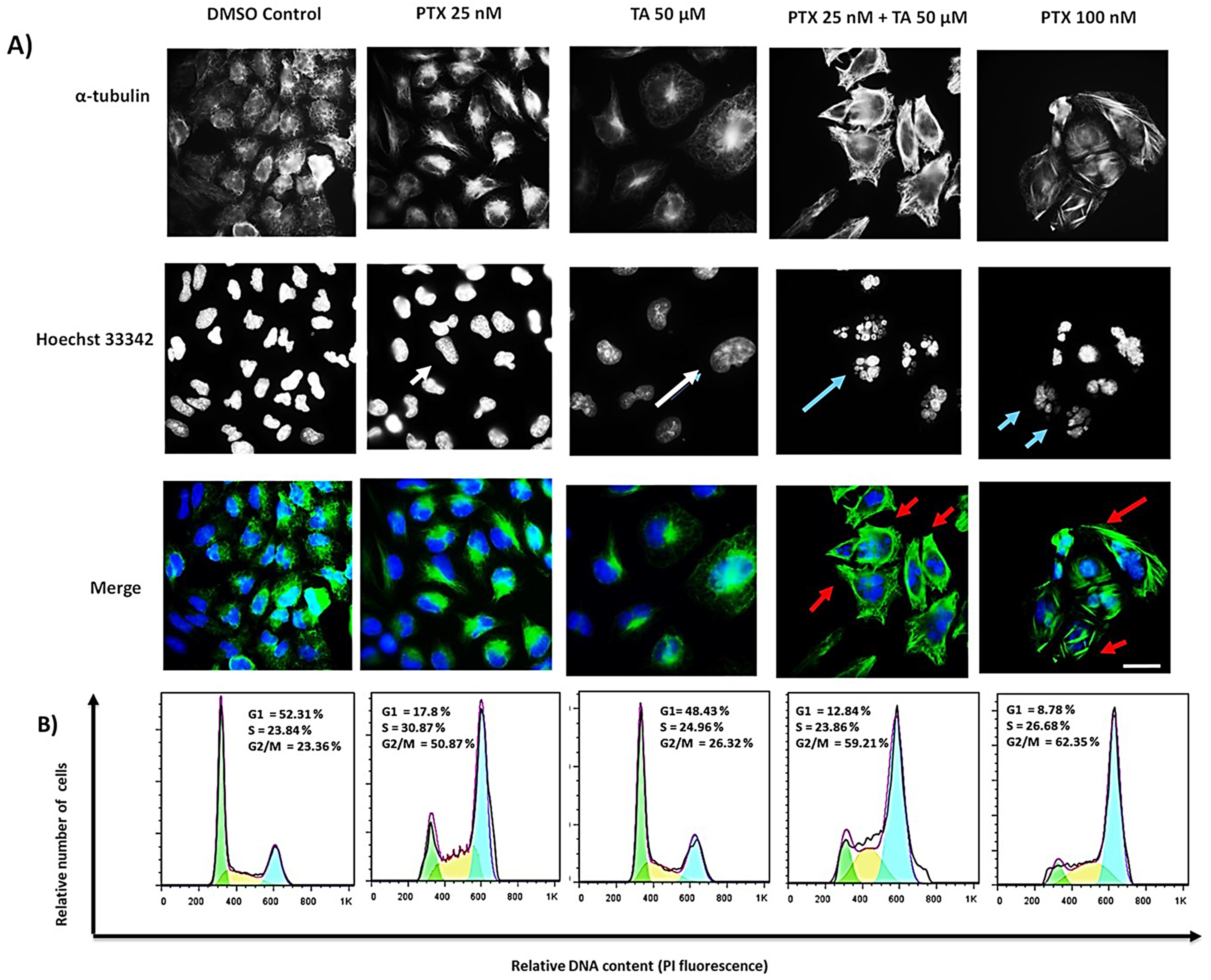
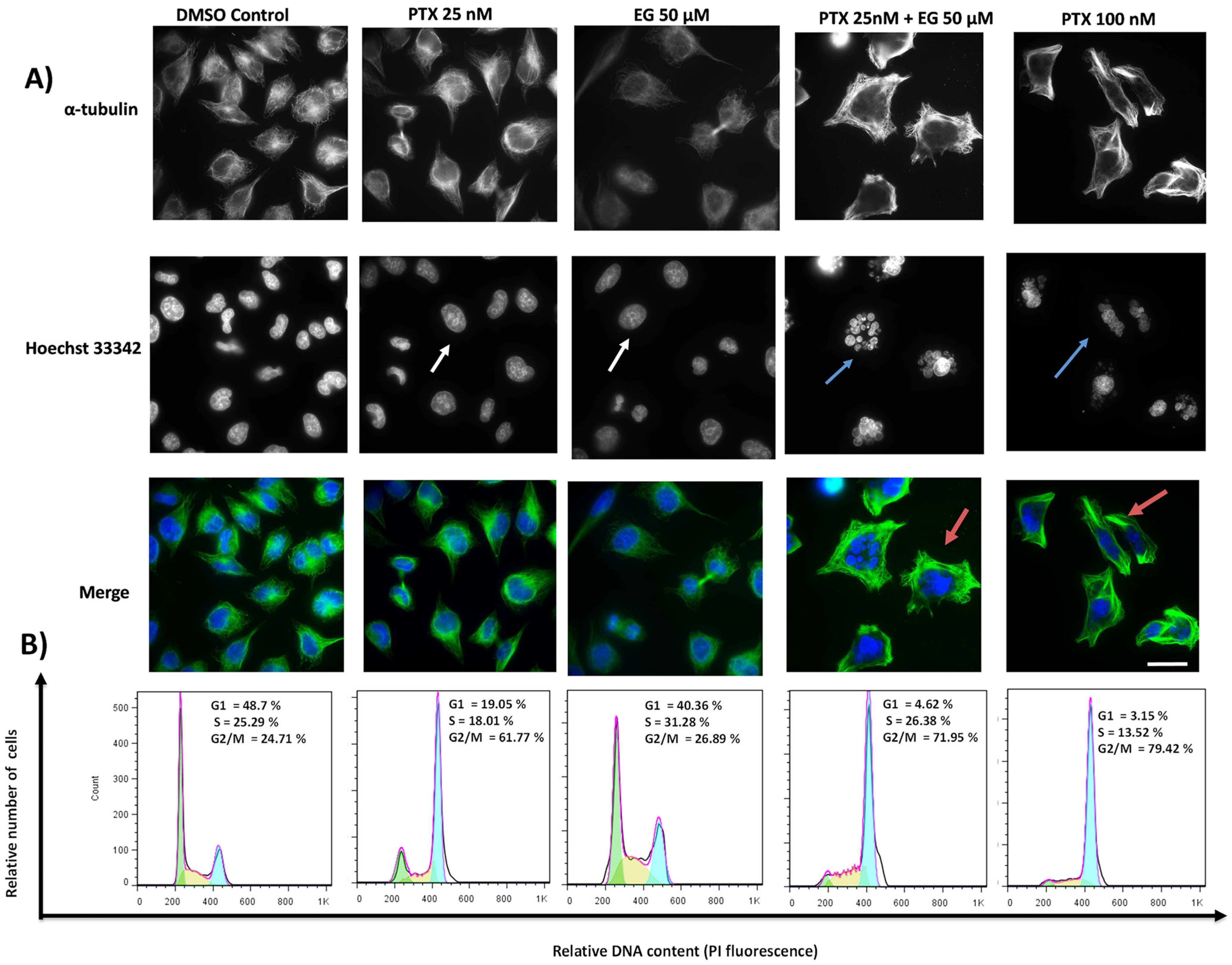
2.2. TA and EG Treatment Potentiates the Effect of PTX on Microtubule Stabilization and the G2/M Phase of the Cell Cycle in HEP3B Cells
2.3. Modulation of Tubulin Assembly by Ligand
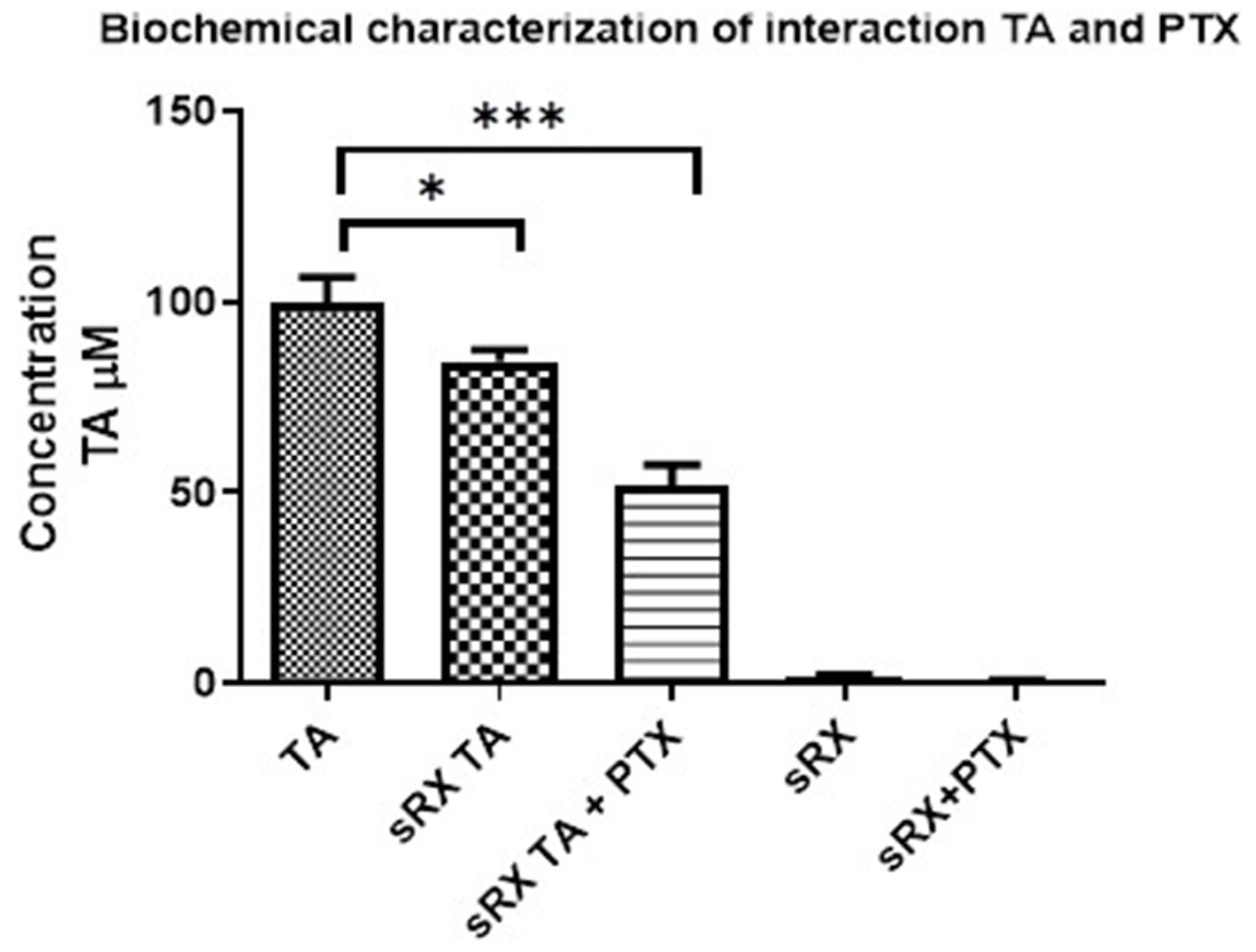
2.4. Biochemical Characterization of TA and Tubulin Interaction
2.5. Taxane Displacement Assay
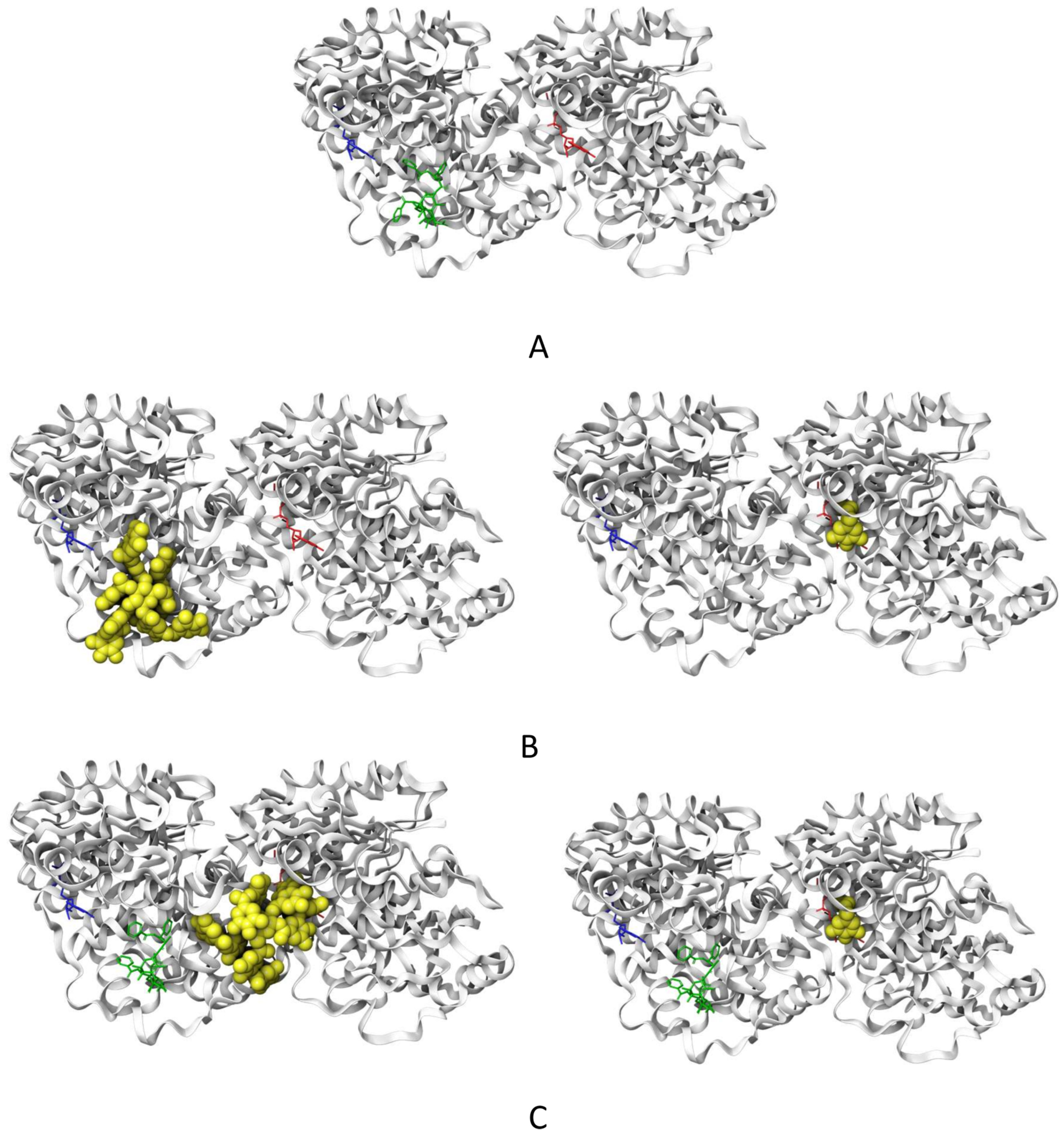
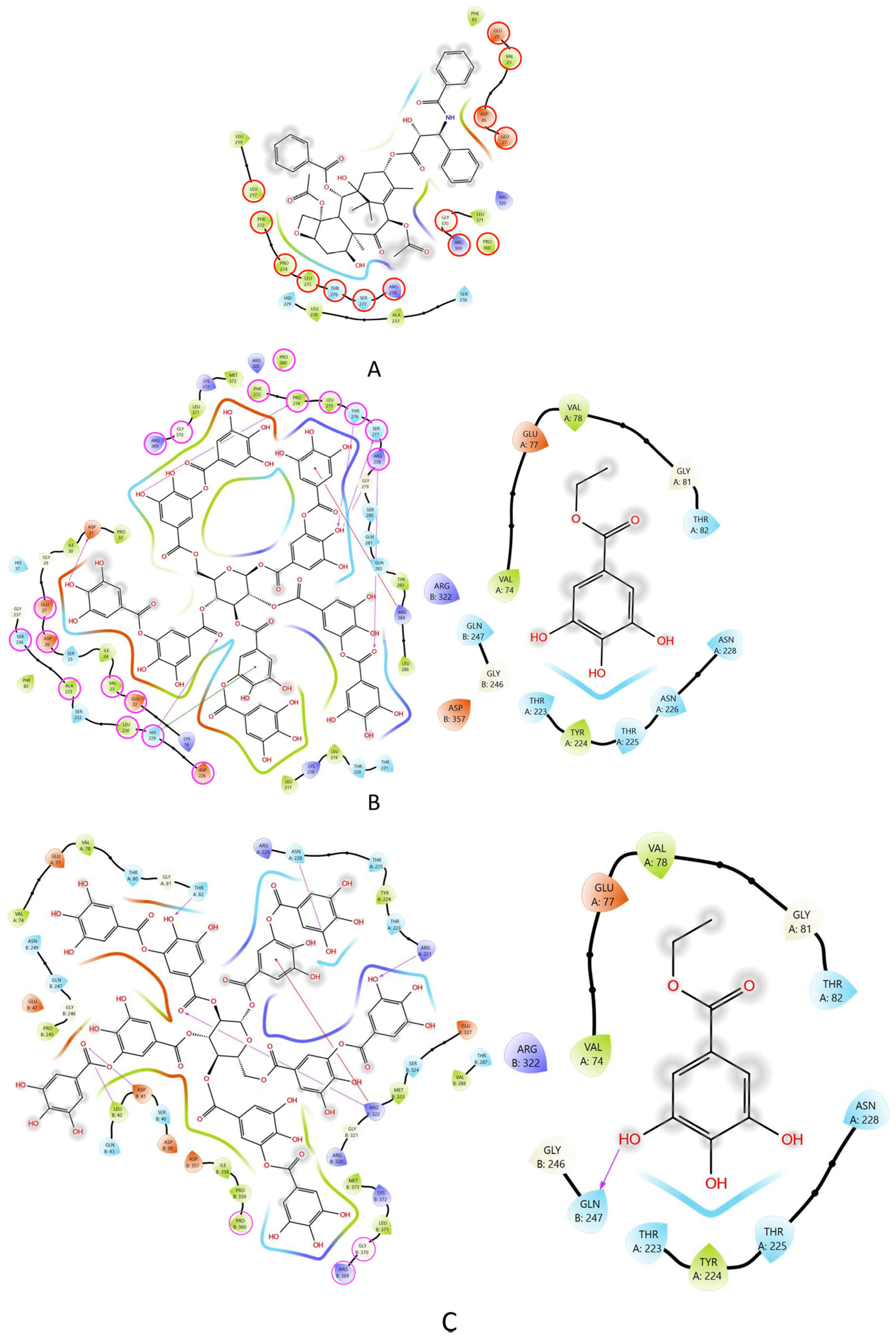
2.6. Molecular Docking of TA and EG to Tubulin
2.7. Effect of TA, EG, and PTX on Protein Kinase Activity
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Cytotoxicity Assay
4.3. Indirect Immunofluorescence
4.4. Cell Cycle Analysis
4.5. Ligand-Induced Tubulin Assembly
4.6. Biochemical Characterization of TA and Tubulin Interaction
4.7. Taxane Displacement Assay
4.8. Molecular Docking
4.9. Western Blotting
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chidambaranathan-Reghupaty, S.; Fisher, P.B.; Sarkar, D. Hepatocellular Carcinoma (HCC): Epidemiology, Etiology, and Molecular Classification. Adv. Cancer Res. 2021, 149, 1–61. [Google Scholar] [PubMed]
- Rowinsky, E.K.; Cazenave, L.A.; Donehower, R.C. Taxol: A Novel Investigational Antimicrotubule Agent. J. Natl. Cancer Inst. 1990, 82, 1247–1259. [Google Scholar] [CrossRef] [PubMed]
- Weaver, B.A. How Taxol/Paclitaxel Kills Cancer Cells. Mol. Biol. Cell 2014, 25, 2677–2681. [Google Scholar] [CrossRef] [PubMed]
- Samaan, T.M.A.; Samec, M.; Liskova, A.; Kubatka, P.; Büsselberg, D. Paclitaxel’s Mechanistic and Clinical Effects on Breast Cancer. Biomolecules 2019, 9, 789. [Google Scholar] [CrossRef]
- Parker, A.L.; Teo, W.S.; McCarroll, J.A.; Kavallaris, M. An Emerging Role for Tubulin Isotypes in Modulating Cancer Biology and Chemotherapy Resistance. Int. J. Mol. Sci. 2017, 18, 1434. [Google Scholar] [CrossRef]
- Nawara, H.M.; Afify, S.M.; Hassan, G.; Zahra, M.H.; Seno, A.; Seno, M. Paclitaxel-Based Chemotherapy Targeting Cancer Stem Cells from Mono- to Combination Therapy. Biomedicines 2021, 9, 500. [Google Scholar] [CrossRef]
- Jin, C.; Li, H.; He, Y.; He, M.; Bai, L.; Cao, Y.; Song, W.; Dou, K. Combination Chemotherapy of Doxorubicin and Paclitaxel for Hepatocellular Carcinoma In Vitro and In Vivo. J. Cancer Res. Clin. Oncol. 2010, 136, 267–274. [Google Scholar] [CrossRef]
- Markman, M.; Mekhail, T.M. Paclitaxel in Cancer Therapy. Expert. Opin. Pharmacother. 2002, 3, 755–766. [Google Scholar] [CrossRef]
- Yang, X.; Tian, X.; Zhao, P.; Wang, Z.; Sun, X. Paclitaxel Inhibits Hepatocellular Carcinoma Tumorigenesis by Regulating the circ_0005785/miR-640/GSK3β Pathway. Cell Biol. Int. 2023, 47, 1170–1182. [Google Scholar] [CrossRef]
- Huzil, J.T.; Chen, K.; Kurgan, L.; Tuszynski, J.A. The Roles of β-Tubulin Mutations and Isotype Expression in Acquired Drug Resistance. Cancer Inform. 2007, 3, 159–181. [Google Scholar] [CrossRef]
- Bae, T.; Weon, K.Y.; Lee, J.W.; Eum, K.H.; Kim, S.; Choi, J.W. Restoration of Paclitaxel Resistance by CDK1 Intervention in Drug-Resistant Ovarian Cancer. Carcinogenesis 2015, 36, 1561–1571. [Google Scholar] [CrossRef] [PubMed]
- Duran, G.E.; Wang, Y.C.; Moisan, F.; Francisco, E.B.; Sikic, B.I. Decreased Levels of Baseline and Drug-Induced Tubulin Polymerisation Are Hallmarks of Resistance to Taxanes in Ovarian Cancer Cells and Are Associated with Epithelial-to-Mesenchymal Transition. Br. J. Cancer 2017, 116, 1318–1328. [Google Scholar] [CrossRef]
- Kavallaris, M. Microtubules and Resistance to Tubulin-Binding Agents. Nat. Rev. Cancer 2010, 10, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhu, G.; Getzenberg, R.H.; Veltri, R.W. The Upregulation of PI3K/Akt and MAP Kinase Pathways Is Associated with Resistance of Microtubule-Targeting Drugs in Prostate Cancer. J. Cell. Biochem. 2015, 116, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Verret, B.; Cortes, J.; Bachelot, T.; Andre, F.; Arnedos, M. Efficacy of PI3K Inhibitors in Advanced Breast Cancer. Ann. Oncol. 2019, 30 (Suppl. S10), x12–x20. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Wu, J.; Chen, Y.; Nie, J.; Chen, C. Activation of PI3K/AKT/mTOR Pathway Causes Drug Resistance in Breast Cancer. Front. Pharmacol. 2021, 12, 628690. [Google Scholar] [CrossRef]
- Islam, B.U.; Suhail, M.; Khan, M.K.; Zughaibi, T.A.; Alserihi, R.F.; Zaidi, S.K.; Tabrez, S. Polyphenols as Anticancer Agents: Toxicological Concern to Healthy Cells. Phytother. Res. 2021, 35, 6063–6079. [Google Scholar] [CrossRef]
- Bouyahya, A.; Omari, N.E.; Bakrim, S.; Hachlafi, N.E.; Balahbib, A.; Wilairatana, P.; Mubarak, M.S. Advances in Dietary Phenolic Compounds to Improve Chemosensitivity of Anti-cancer Drugs. Cancers 2022, 14, 4573. [Google Scholar] [CrossRef]
- Bona, N.P.; Pedra, N.S.; Azambuja, J.H.; Soares, M.S.; Spohr, L.; Gelsleichter, N.E.; de Meine, B.M.; Sekine, F.G.; Mendonça, L.T.; de Oliveira, F.H.; et al. Tannic Acid Elicits Selective Antitumoral Activity In Vitro and Inhibits Cancer Cell Growth in a Preclinical Model of Glioblastoma Multiforme. Metab. Brain Dis. 2020, 35, 283–293. [Google Scholar] [CrossRef]
- Youness, R.; Kamel, R.; Elkasabgy, N.A.; Shao, P.; Farag, M.A. Recent Advances in Tannic Acid (Gallotannin) Anticancer Activities and Drug Delivery Systems for Efficacy Improvement; A Comprehensive Review. Molecules 2021, 26, 1486. [Google Scholar] [CrossRef]
- Mohan, S.; Thiagarajan, K.; Chandrasekaran, R. In Vitro Evaluation of Antiproliferative Effect of Ethyl Gallate against Human Oral Squamous Carcinoma Cell Line KB. Nat. Prod. Res. 2015, 29, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Carranza, J.N.; Alvarez, L.; Marquina-Bahena, S.; Salas-Vidal, E.; Cuevas, V.; Jiménez, E.W.; Veloz, G.R.A.; Carraz, M.; González-Maya, L. Phenolic Compounds Isolated from Caesalpinia coriaria Induce S and G2/M Phase Cell Cycle Arrest Differentially and Trigger Cell Death by Interfering with Microtubule Dynamics in Cancer Cell Lines. Molecules 2017, 22, 666. [Google Scholar] [CrossRef] [PubMed]
- Oosawa, F.; Asakura, S. Thermodynamics of the Polymerization of Protein; Academic Press: London, UK, 1975. [Google Scholar]
- Buey, R.M.; Barasoain, I.; Jackson, E.; Meyer, A.; Giannakakou, P.; Paterson, I.; Mooberry, S.; Andreu, J.M.; Díaz, J.F. Microtubule Interactions with Chemically Diverse Stabilizing Agents: Thermodynamics of Binding to the Paclitaxel Site Predicts Cytotoxicity. Chem. Biol. 2005, 12, 1269–1279. [Google Scholar] [CrossRef] [PubMed]
- Löwe, J.; Li, H.; Downing, K.H.; Nogales, E. Refined Structure of Alpha Beta-Tubulin at 3.5 A Resolution. J. Mol. Biol. 2001, 313, 1045–1057. [Google Scholar] [CrossRef]
- Prota, A.E.; Lucena-Agell, D.; Ma, Y.; Estevez-Gallego, J.; Li, S.; Bargsten, K.; Josa-Prado, F.; Altmann, K.H.; Gaillard, N.; Kamimura, S.; et al. Structural insight into the stabilization of microtubules by taxanes. Elife 2023, 12, 84791. [Google Scholar] [CrossRef]
- Field, J.J.; Díaz, J.F.; Miller, J.H. The binding sites of microtubule-stabilizing agents. Chem. Biol. 2013, 20, 301–315. [Google Scholar] [CrossRef]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef]
- Platanias, L.C. Map kinase signaling pathways and hematologic malignancies. Blood 2003, 101, 4667–4679. [Google Scholar] [CrossRef]
- Gigant, B.; Wang, C.; Ravelli, R.B.G.; Roussi, F.; Steinmetz, M.O.; Curmi, P.A.; Sobel, A.; Knossow, M. Structural basis for the regulation of tubulin by vinblastine. Nature 2005, 435, 519–522. [Google Scholar] [CrossRef]
- Prota, A.E.; Bargsten, K.; Northcote, P.T.; Marsh, M.; Altmann, K.H.; Miller, J.H.; Díaz, J.F.; Steinmetz, M.O. Structural basis of microtubule stabilization by laulimalide and peloruside A. Angew Chem. Int. Ed. Engl. 2014, 53, 1621–1625. [Google Scholar] [CrossRef]
- Jordan, M.A. Mechanism of action of antitumor drugs that interact with microtubules and tubulin. Curr. Med. Chem. Anticancer Agents 2002, 2, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.L.; Horbinski, C.; Garzotto, M.; Qian, D.Z.; Beer, T.M.; Kyprianou, N. Tubulin-Targeting Chemotherapy Impairs Androgen Receptor Activity in Prostate Cancer. Cancer Res. 2010, 70, 7992–8002. [Google Scholar] [CrossRef] [PubMed]
- Duran, G.E.; Wang, Y.C.; Francisco, E.B.; Rose, J.C.; Martinez, F.J.; Coller, J.; Brassard, D.; Vrignaud, P.; Sikic, B.I. Mechanisms of resistance to cabazitaxel. Mol. Cancer Ther. 2015, 14, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Basu, D.; Majumdar, S.; Mandal, N.; Dastidar, S.G. Mechanisms of influence of the microtubule over-stabilizing ligands on the structure and intrinsic dynamics of α,β-Tubulin. Comput. Biol. Chem. 2022, 96, 107617. [Google Scholar] [CrossRef] [PubMed]
- MacKeigan, J.P.; Taxman, D.J.; Hunter, D.; Earp, H.S., III; Graves, L.M.; Ting, J.P. Inactivation of the antiapoptotic phosphatidylinositol 3-kinase-Akt pathway by the combined treatment of taxol and mitogen-activated protein kinase kinase inhibition. Clin. Cancer Res. 2002, 8, 2091–2099. [Google Scholar]
- Okano, J.; Rustgi, A.K. Paclitaxel induces prolonged activation of the Ras/MEK/ERK pathway independently of activating the programmed cell death machinery. J. Biol. Chem. 2001, 276, 19555–19564. [Google Scholar] [CrossRef]
- McDaid, H.M.; Horwitz, S.B. Selective potentiation of paclitaxel (taxol)-induced cell death by mitogen-activated protein kinase kinase inhibition in human cancer cell lines. Mol. Pharmacol. 2001, 60, 290–301. [Google Scholar] [CrossRef]
- Domina, A.M.; Vrana, J.A.; Gregory, M.A.; Hann, S.R.; Craig, R.W. MCL1 is phosphorylated in the PEST region and stabilized upon ERK activation in viable cells, and at additional sites with cytotoxic okadaic acid or taxol. Oncogene 2004, 23, 5301–5315. [Google Scholar] [CrossRef]
- De Inés, C.; Leynadier, D.; Barasoain, I.; Peyrot, V.; Briand, C.; Rener, G.A.; Temple, C., Jr. Inhibition of microtubules and cell cycle arrest by a new 1-deaza-7,8-dihydropteridine antitumor drug, CI 980, and by its chiral isomer, NSC 613863. Cancer Res. 1994, 1, 75–84. [Google Scholar]
- Buey, R.M.; Diaz, J.F.; Andreu, J.M.; O’Brate, A.; Giannakakou, P.; Nicolaou, K.C.; Sasmal, P.K.; Ritzen, A.; Namoto, K. Interaction of epothilone analogs with the paclitaxel binding site: Relationship between binding affinity, microtubule stabilization, and cytotoxicity. Chem. Biol. 2004, 11, 225–236. [Google Scholar]
- Díaz, J.F.; Andreu, J.M. Assembly of purified GDP-tubulin into microtubules induced by taxol and taxotere: Reversibility, ligand stoichiometry, and competition. Biochemistry 1993, 32, 2747–2755. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- MarvinSketch (Version 22.22, Calculation Module Developed by ChemAxon). 2022. Available online: http://www.chemaxon.com/products/marvin/marvinsketch/ (accessed on 1 December 2022).
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Maestro. Schrödinger Release; Maestro, Schrödinger, LLC.: New York, NY, USA, 2023. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Affinitty Energy (Kcal/mol) |
|---|---|
| PTX (62%) a | −9.0 +/− 0.2 |
| TA (cluster 1) (30%) a | −8.6 +/− 0.5 |
| TA (cluster 2) (20%) a | −8.4 +/− 0.4 |
| EG (77%) a | −6.5 +/− 0.1 |
| Ligand | Affinitty Energy (Kcal/mol) |
|---|---|
| Tannic Acid (23%) a | −8.7 +/− 0.6 |
| Ethyl Gallate (68%) a | −6.5 +/− 0.1 |
| Residue | Tubulin | 1JFF | REDOCKING | TA | TA + PTX | EG | EG + PTX | Ligands [27] |
|---|---|---|---|---|---|---|---|---|
| VAL74 | α | * | * | * | ||||
| GLU77 | α | * | * | * | ||||
| VAL78 | α | * | * | * | ||||
| THR80 | α | * | ||||||
| GLY81 | α | * | * | * | ||||
| THR82 | α | * | * | * | ||||
| ARG221 | α | * | ||||||
| THR223 | α | * | * | * | ||||
| TYR224 | α | * | * | * | ||||
| THR225 | α | * | * | * | ||||
| ASN226 | α | * | ||||||
| ASN228 | α | * | * | * | ||||
| ARG229 | α | * | ||||||
| LYS19 | β | * | L2 | |||||
| GLU22 | β | * | * | * | L2 | |||
| VAL23 | β | * | * | * | L2, L3, PTX | |||
| ILE24 | β | * | ||||||
| SER25 | β | * | ||||||
| ASP26 | β | * | * | * | PTX | |||
| GLU27 | β | * | * | * | L2, L3 | |||
| GLY29 | β | * | ||||||
| ILE30 | β | * | ||||||
| ASP31 | β | * | ||||||
| PRO32 | β | * | ||||||
| HIS37 | β | * | ||||||
| ASP39 | β | * | ||||||
| SER40 | β | * | ||||||
| ASP41 | β | * | ||||||
| LEU42 | β | * | ||||||
| GLN43 | β | * | ||||||
| GLU47 | β | * | ||||||
| GLU55 | PTX | |||||||
| PHE83 | β | * | * | |||||
| CYS213 | β | * | L1, L2, L3 | |||||
| LEU217 | β | * | * | * | L1, L2, L3, PTX | |||
| LYS218 | β | * | ||||||
| LEU219 | β | * | * | L1, L2, L3, PTX | ||||
| THR220 | β | * | ||||||
| THR221 | β | * | ||||||
| ASP226 | β | * | * | L1, L2, L3, EPOA | ||||
| ASN228 | β | ZMP | ||||||
| HIS229 | β | * | * | * | L1, L2, L3, PTX, ZMP | |||
| LEU230 | β | * | * | * | L1, L2, L3, PTX | |||
| SER232 | β | * | L2 | |||||
| ALA233 | β | * | * | * | L1, L2, L3, PTX | |||
| SER236 | β | * | * | * | L2, L3, PTX | |||
| GLY237 | β | * | L2 | |||||
| PRO245 | β | * | ||||||
| GLY246 | β | * | * | * | ||||
| GLN247 | β | * | * | * | ||||
| ASN249 | β | * | ||||||
| PHE272 | β | * | * | * | L1, L2, L3, PTX | |||
| PRO274 | β | * | * | * | L1, L2, L3, PTX | |||
| LEU275 | β | * | * | * | L1, L2, L3, PTX | |||
| THR276 | β | * | * | * | L1, L2, L3, PTX, EPOA, ZMP | |||
| SER277 | β | * | * | * | L1, L2, L3, PTX | |||
| ARG278 | β | * | * | * | L1, L2, L3, PTX | |||
| GLY279 | β | * | ||||||
| SER280 | β | * | ||||||
| GLN281 | β | * | PTX, EPOA | |||||
| GLN282 | β | * | L3 | |||||
| TYR283 | β | * | ||||||
| ARG284 | β | * | PTX | |||||
| LEU286 | β | * | PTX | |||||
| THR287 | β | * | ||||||
| VAL288 | β | * | ||||||
| LEU291 | β | PTX | ||||||
| GLN294 | β | PEL | ||||||
| PHE296 | β | LAU, PEL | ||||||
| ASP297 | β | PEL | ||||||
| ALA298 | β | PEL | ||||||
| PRO307 | β | PEL | ||||||
| ARG308 | β | LAU, PEL | ||||||
| TYR312 | β | LAU, PEL | ||||||
| ARG320 | β | * | * | * | L2, L3 | |||
| GLY321 | β | * | ||||||
| ARG322 | β | * | * | * | ||||
| MET323 | β | * | ||||||
| SER324 | β | * | ||||||
| GLU327 | β | * | ||||||
| VAL335 | β | LAU, PEL | ||||||
| ASN339 | β | LAU, PEL | ||||||
| SER341 | β | LAU | ||||||
| TYR342 | β | LAU, PEL | ||||||
| PHE343 | β | LAU | ||||||
| ILE347 | β | LAU | ||||||
| ASP357 | β | * | * | |||||
| ILE358 | β | * | ||||||
| PRO359 | β | * | ||||||
| PRO360 | β | * | * | * | * | L2, L3, PTX | ||
| ARG369 | β | * | * | * | * | L1, L2, L3, PTX | ||
| GLY370 | β | * | * | * | * | L1, L2, L3, PTX | ||
| LEU371 | β | * | * | * | L1, L2, L3, PTX | |||
| LYS372 | β | * | * | |||||
| MET373 | β | * | * | |||||
| SER374 | β | PTX |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Carranza, J.N.; Redondo-Horcajo, M.; Barasoain, I.; Escobar-Aguilar, E.A.; Millán-Pacheco, C.; Alvarez, L.; Salas Vidal, E.; Diaz, J.F.; Gonzalez-Maya, L. Tannic Acid and Ethyl Gallate Potentialize Paclitaxel Effect on Microtubule Dynamics in Hep3B Cells. Pharmaceuticals 2023, 16, 1579. https://doi.org/10.3390/ph16111579
Sánchez-Carranza JN, Redondo-Horcajo M, Barasoain I, Escobar-Aguilar EA, Millán-Pacheco C, Alvarez L, Salas Vidal E, Diaz JF, Gonzalez-Maya L. Tannic Acid and Ethyl Gallate Potentialize Paclitaxel Effect on Microtubule Dynamics in Hep3B Cells. Pharmaceuticals. 2023; 16(11):1579. https://doi.org/10.3390/ph16111579
Chicago/Turabian StyleSánchez-Carranza, Jessica Nayelli, Mariano Redondo-Horcajo, Isabel Barasoain, Ever Angel Escobar-Aguilar, César Millán-Pacheco, Laura Alvarez, Enrique Salas Vidal, J. Fernando Diaz, and Leticia Gonzalez-Maya. 2023. "Tannic Acid and Ethyl Gallate Potentialize Paclitaxel Effect on Microtubule Dynamics in Hep3B Cells" Pharmaceuticals 16, no. 11: 1579. https://doi.org/10.3390/ph16111579
APA StyleSánchez-Carranza, J. N., Redondo-Horcajo, M., Barasoain, I., Escobar-Aguilar, E. A., Millán-Pacheco, C., Alvarez, L., Salas Vidal, E., Diaz, J. F., & Gonzalez-Maya, L. (2023). Tannic Acid and Ethyl Gallate Potentialize Paclitaxel Effect on Microtubule Dynamics in Hep3B Cells. Pharmaceuticals, 16(11), 1579. https://doi.org/10.3390/ph16111579