
Recent Progress in Synthesis, POM Analyses and SAR of Coumarin-Hybrids as Potential Anti-HIV Agents—A Mini Review

 ,
,  ,
,  , , ,
, , ,
Abstract
:
1. Introduction
2. Bibliometric Analysis
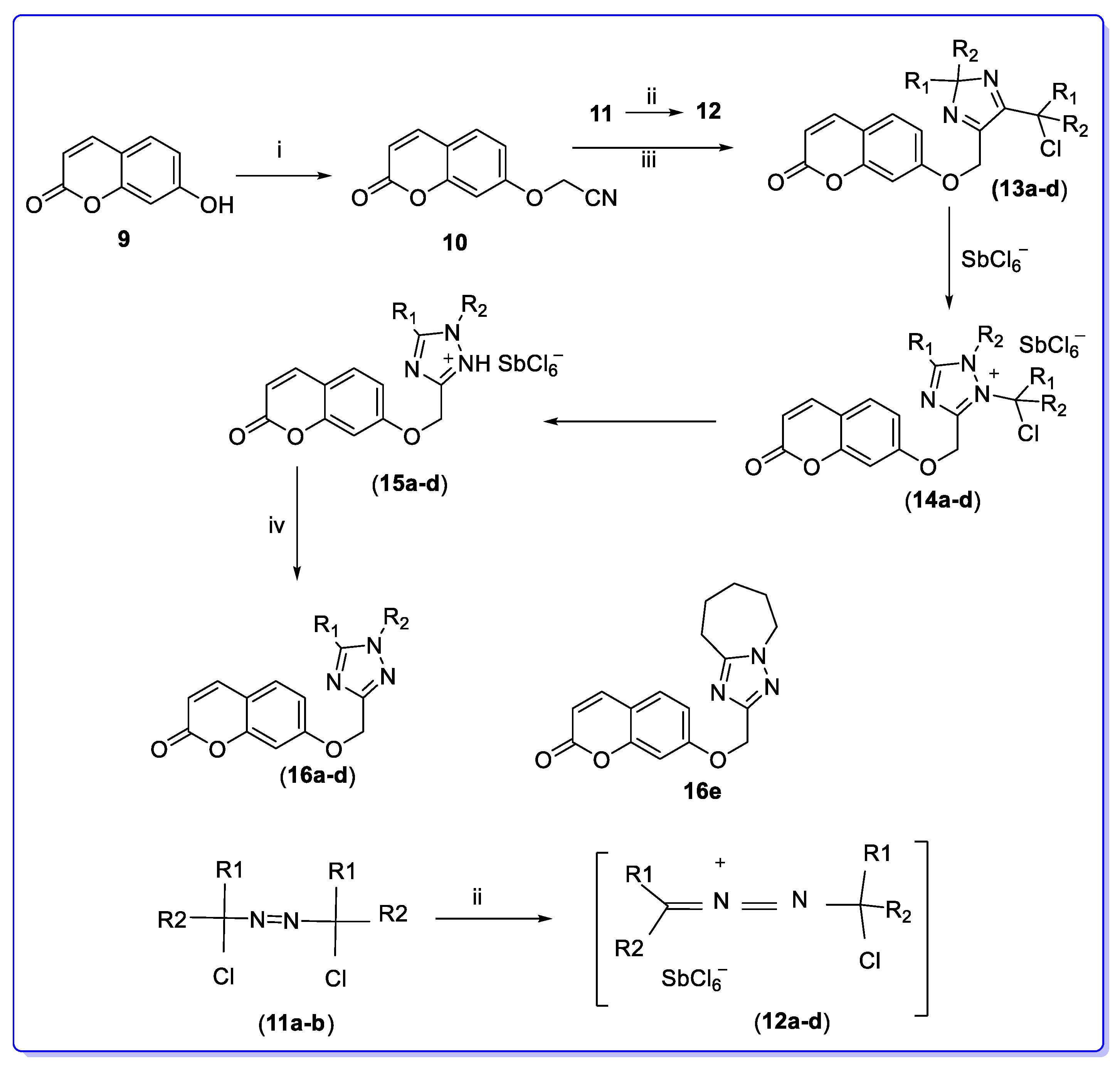
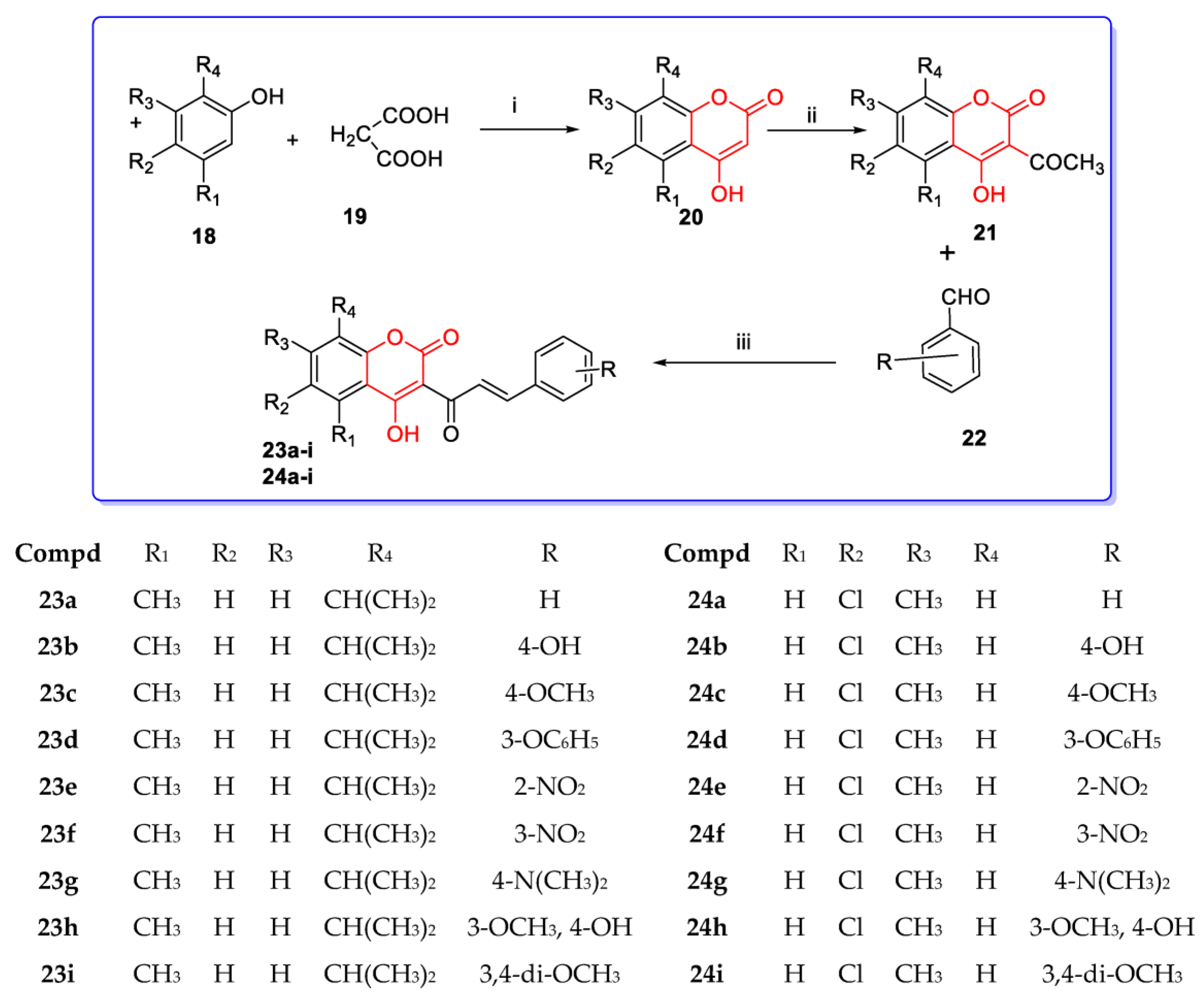
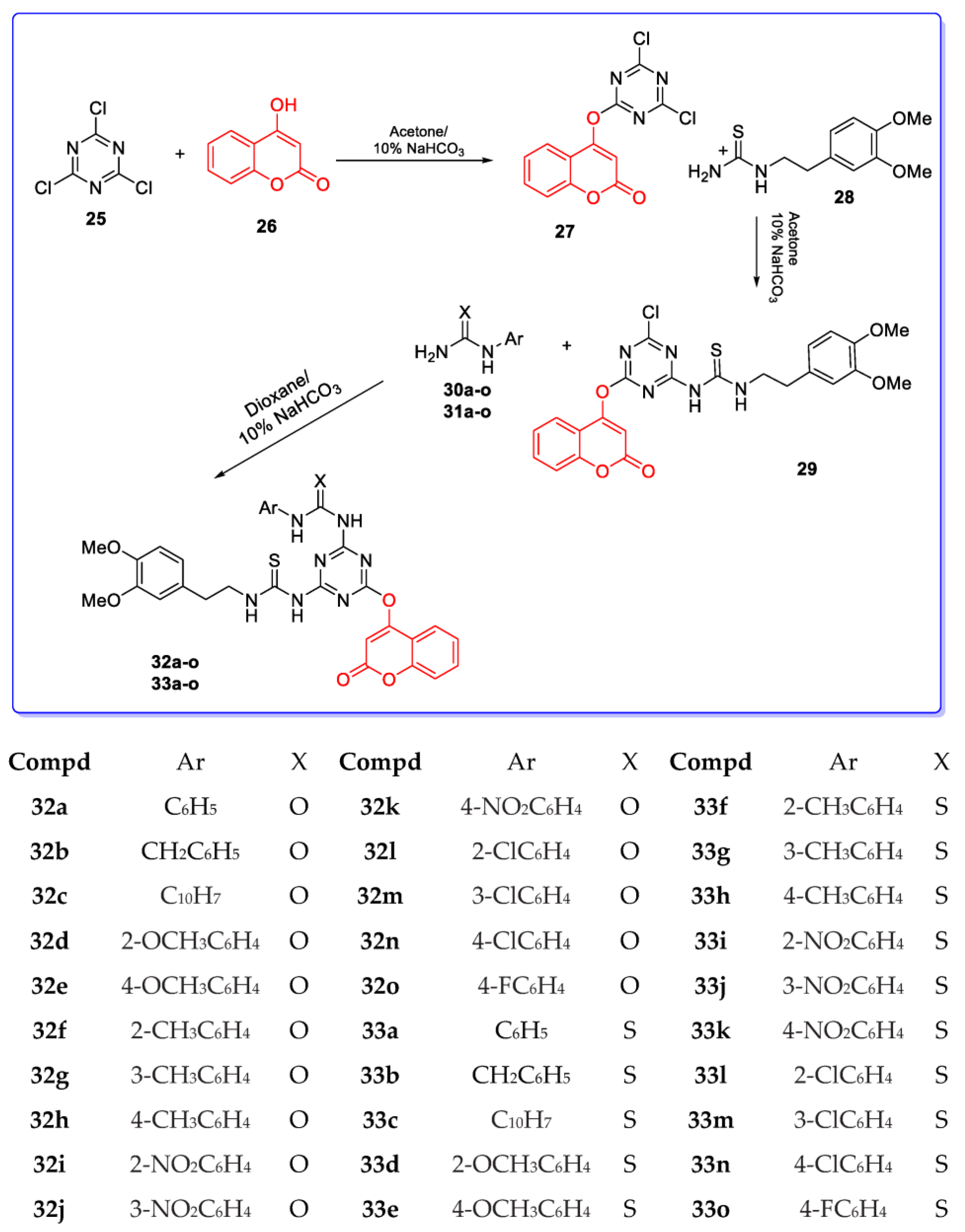
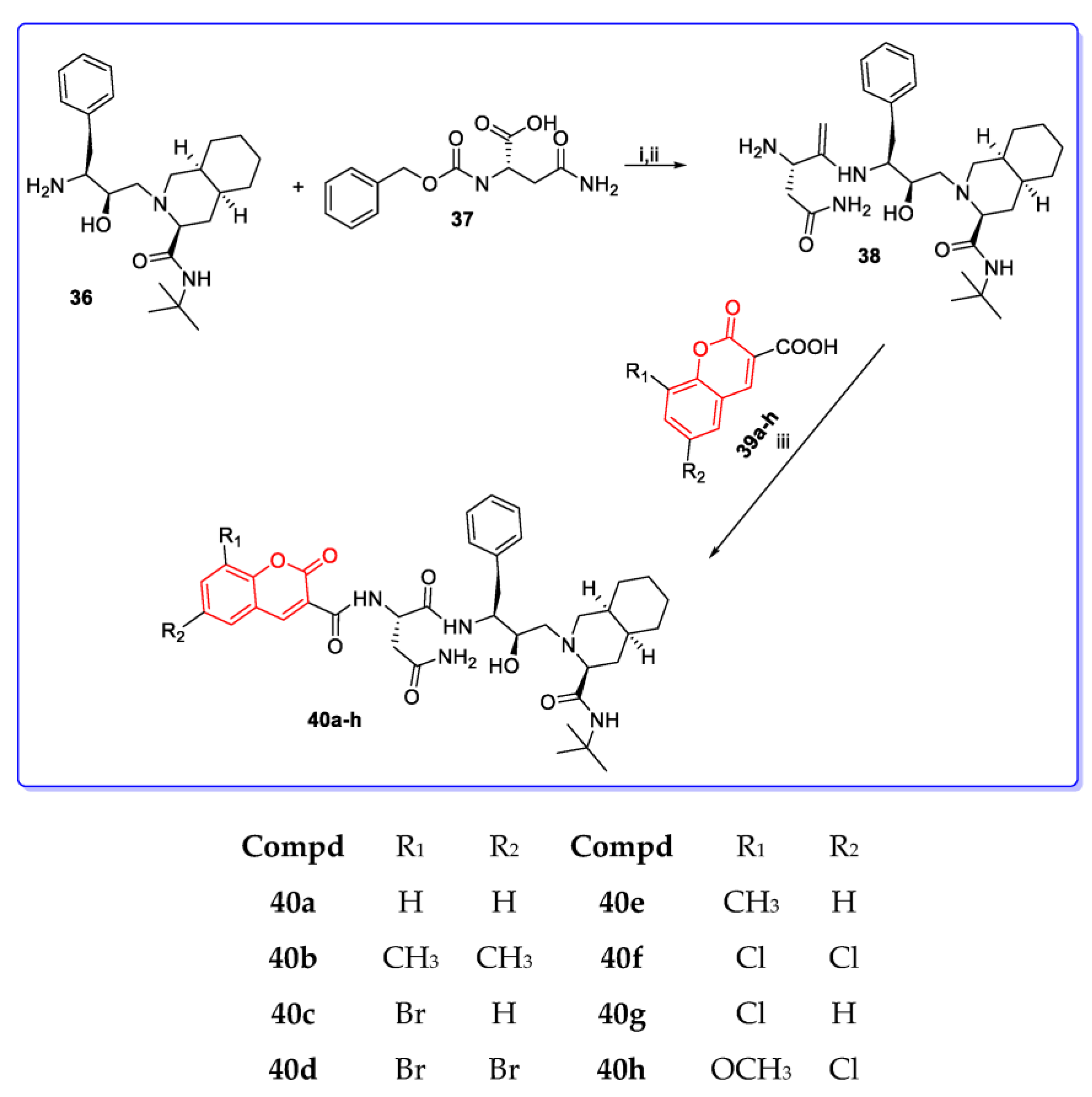
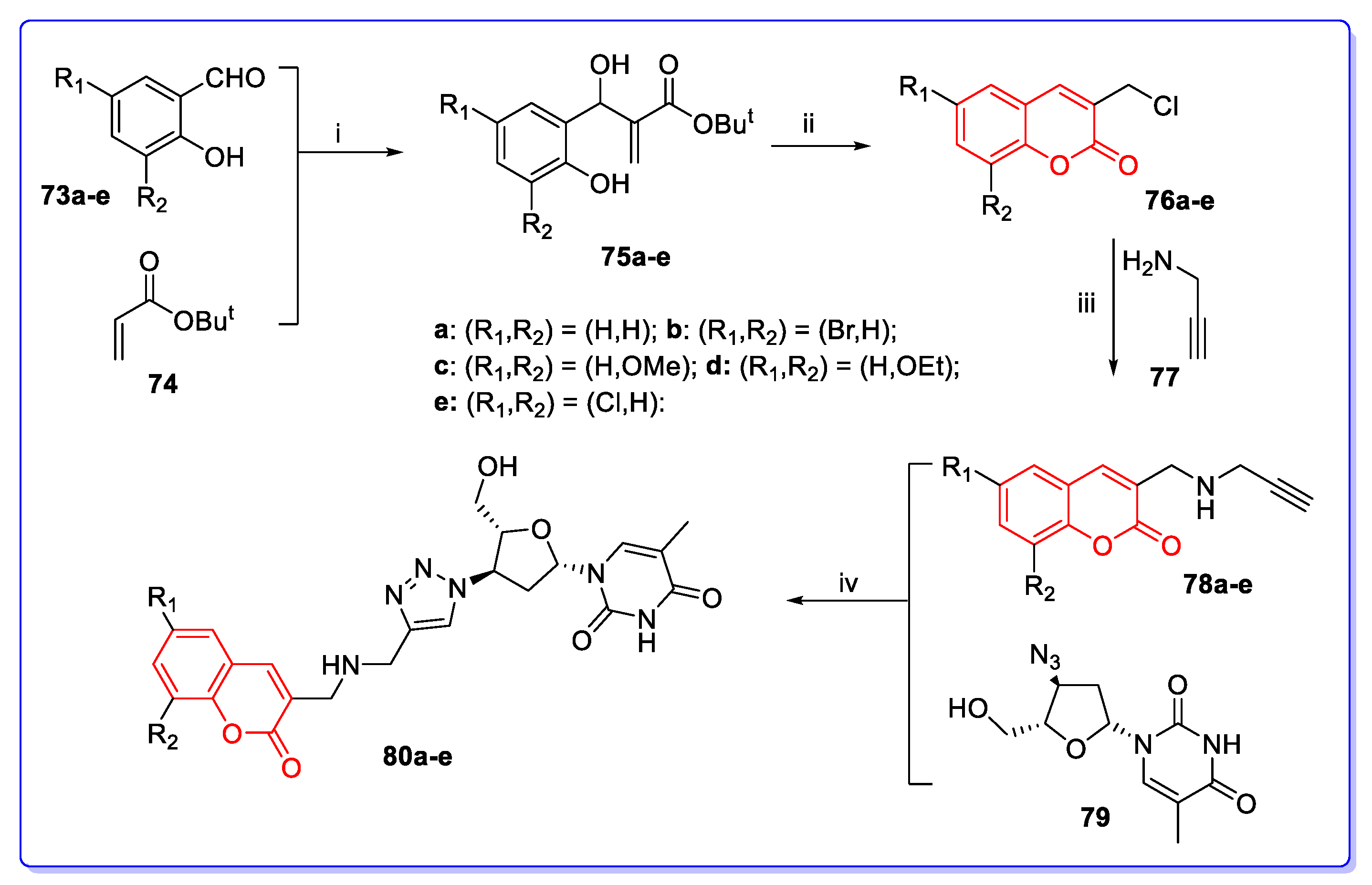
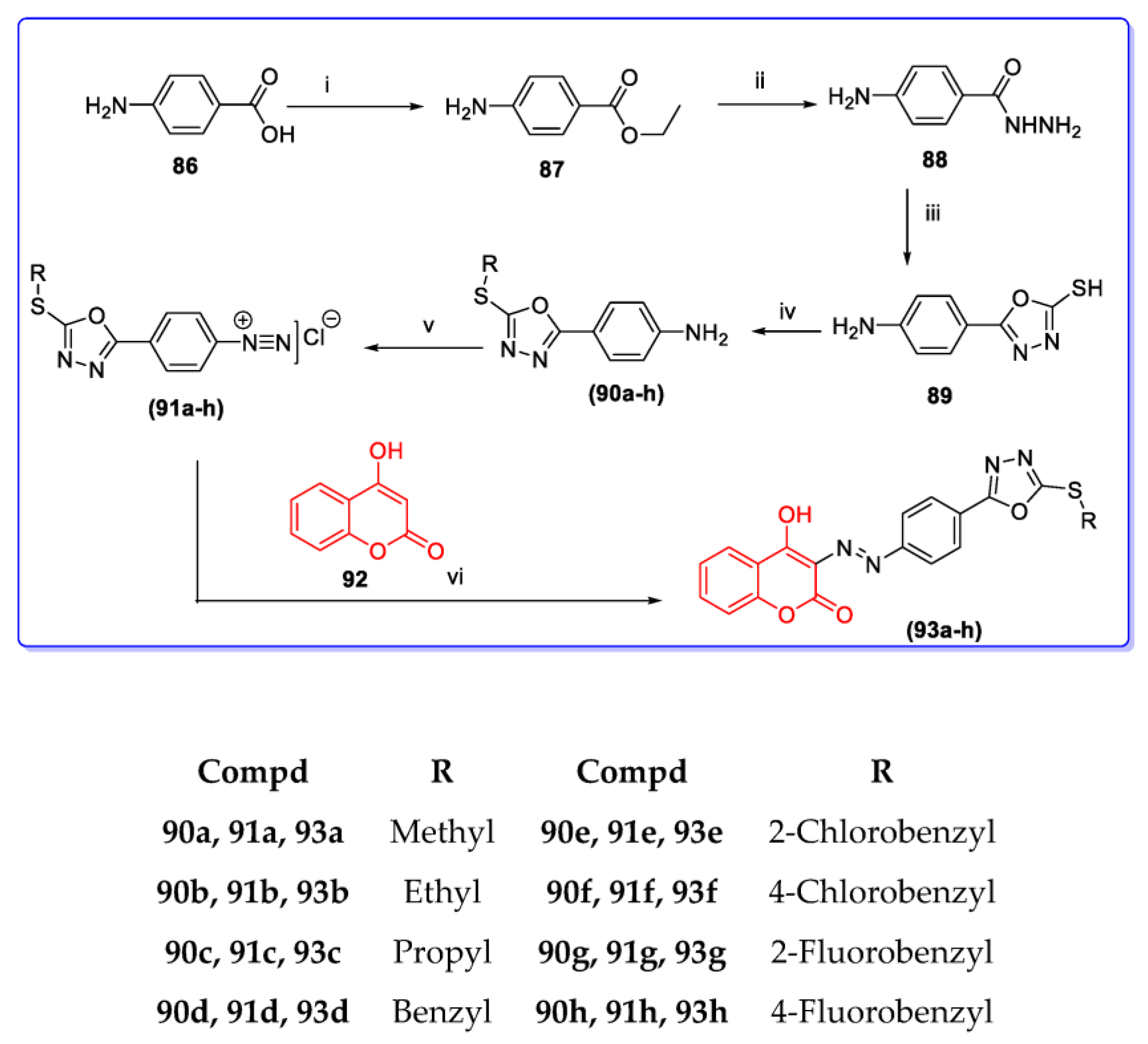
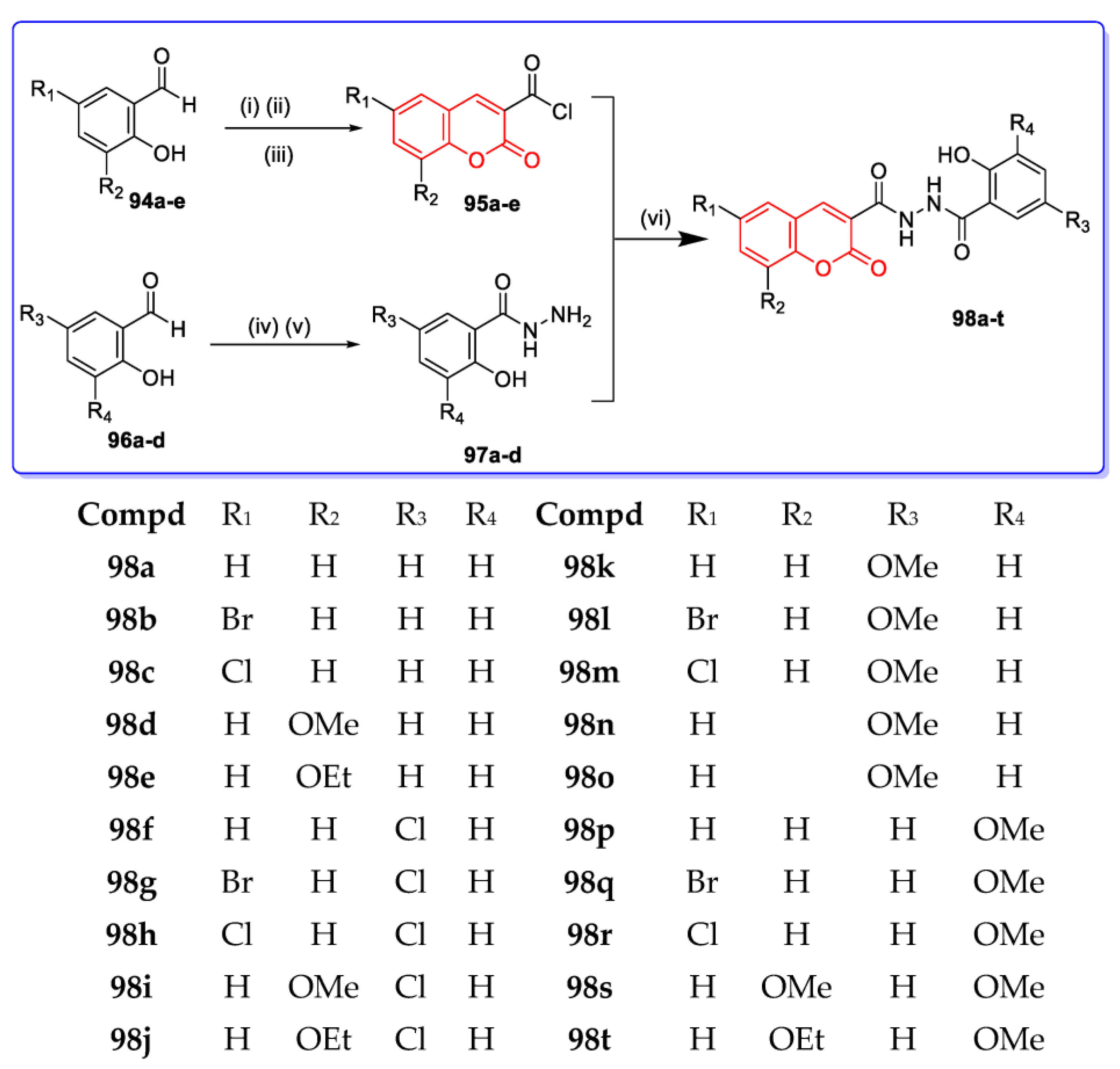
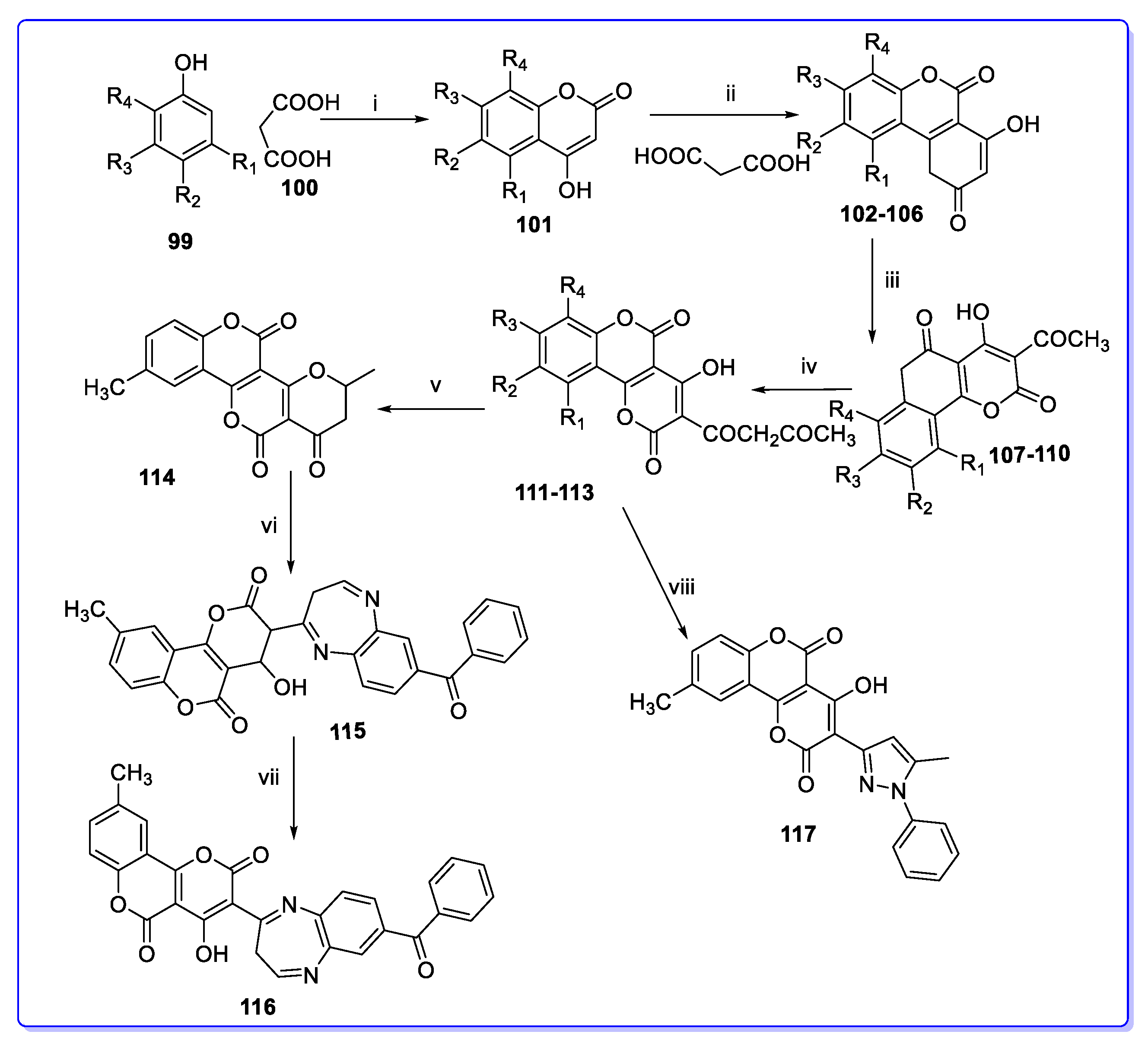
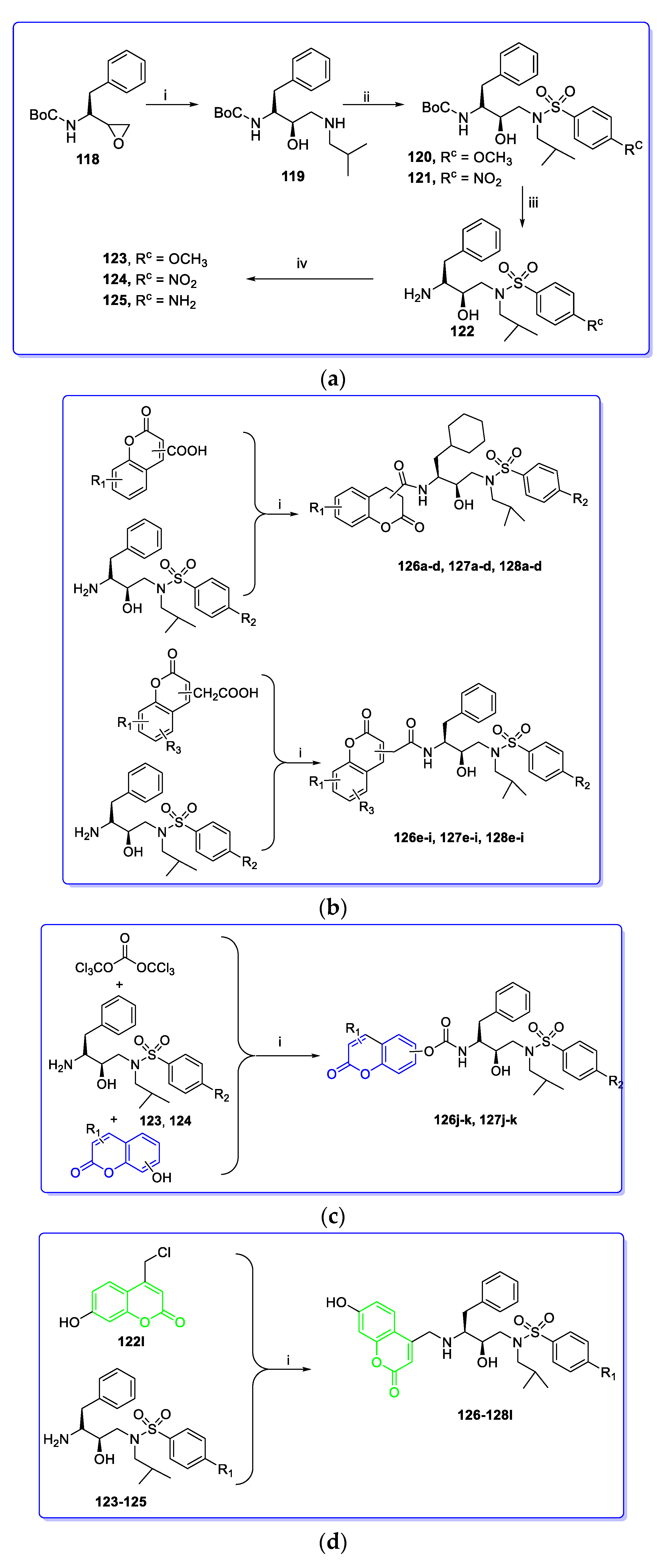
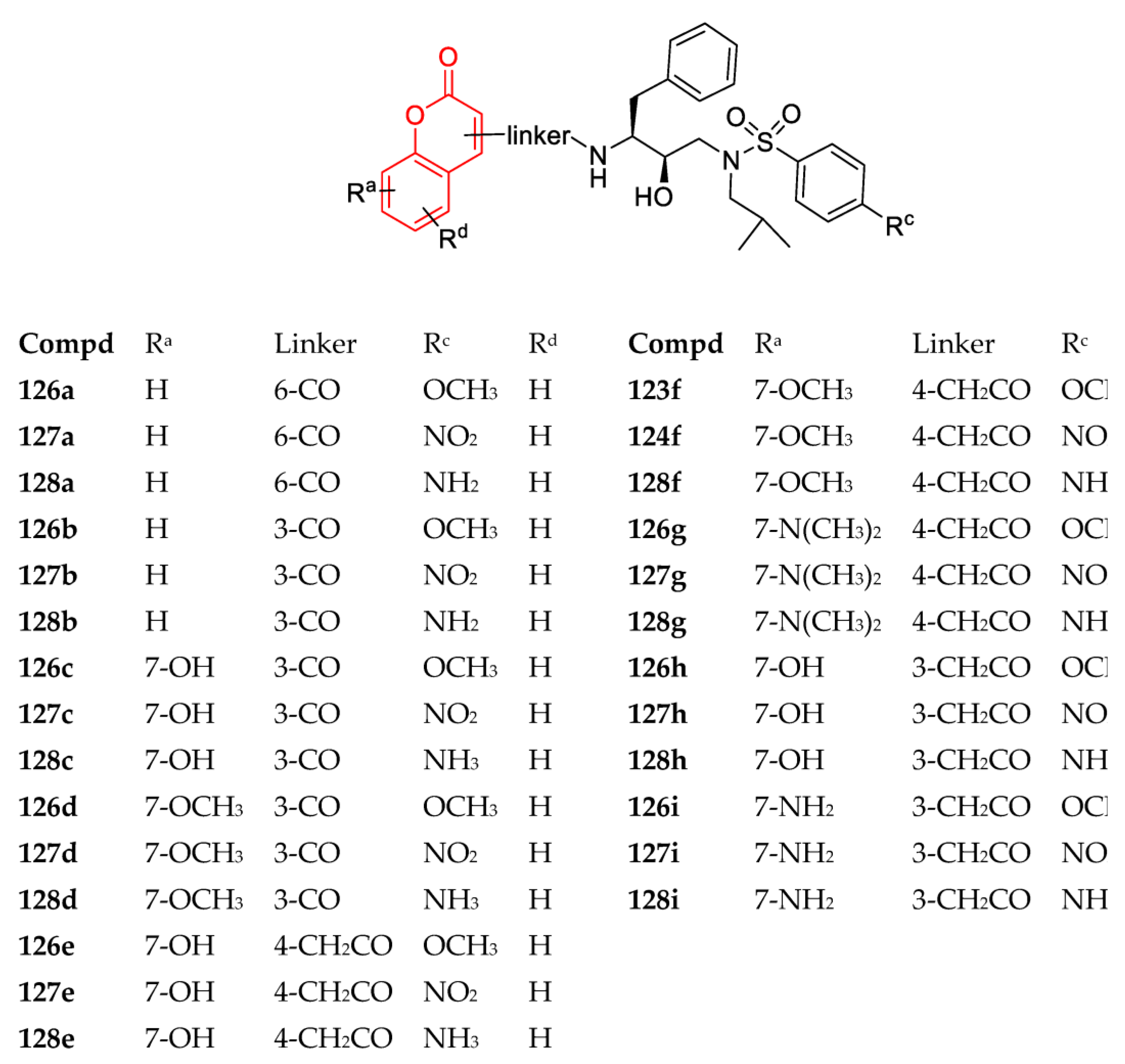
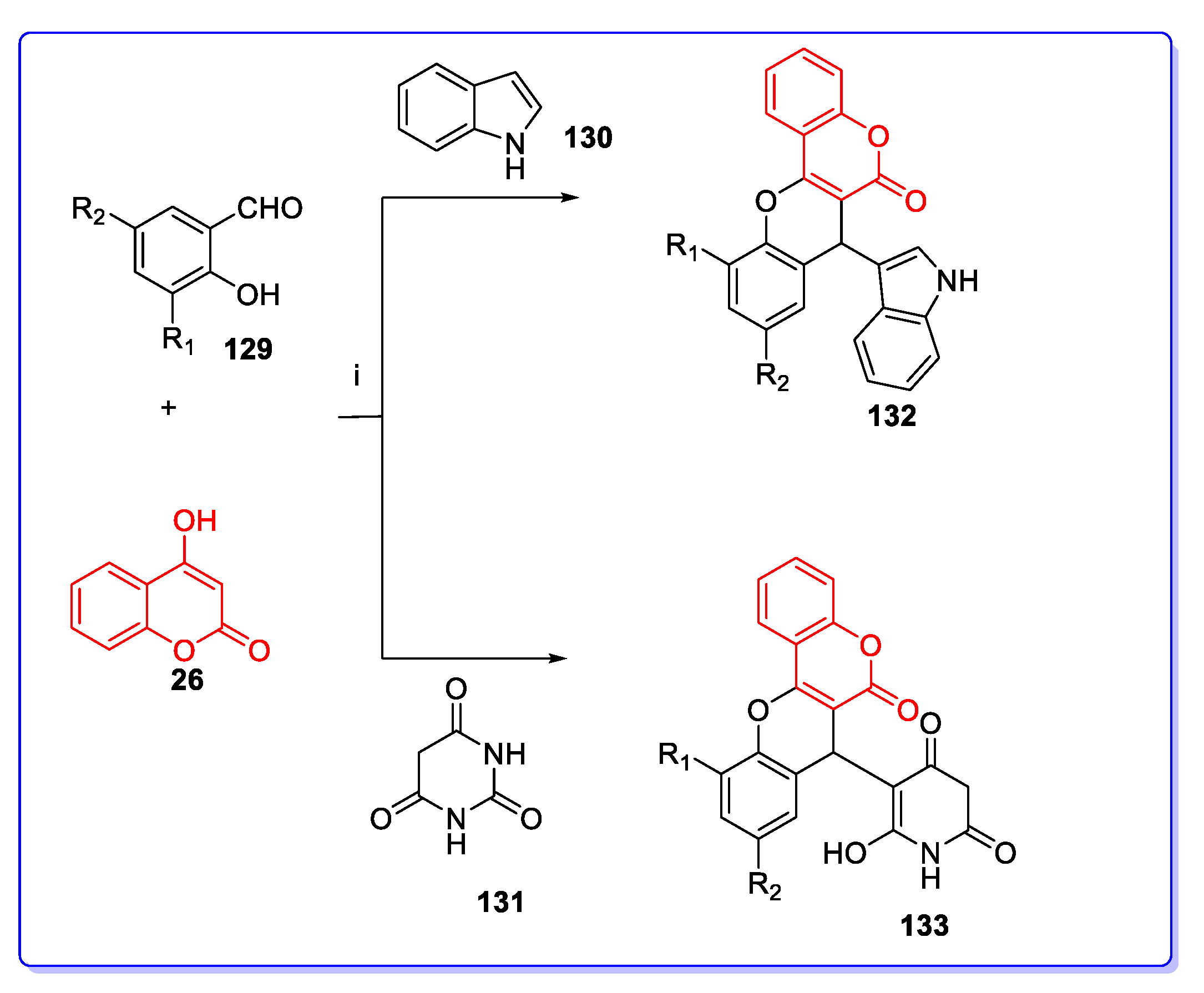
3. Coumarin Hybrids
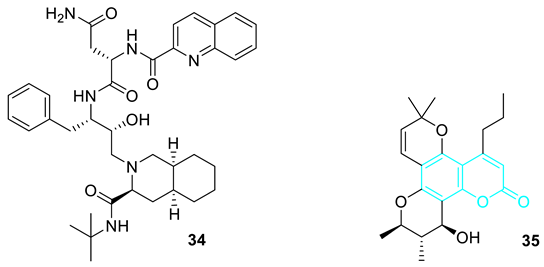
4. Coumarin Hybrids with Weak Activity against HIV Infections
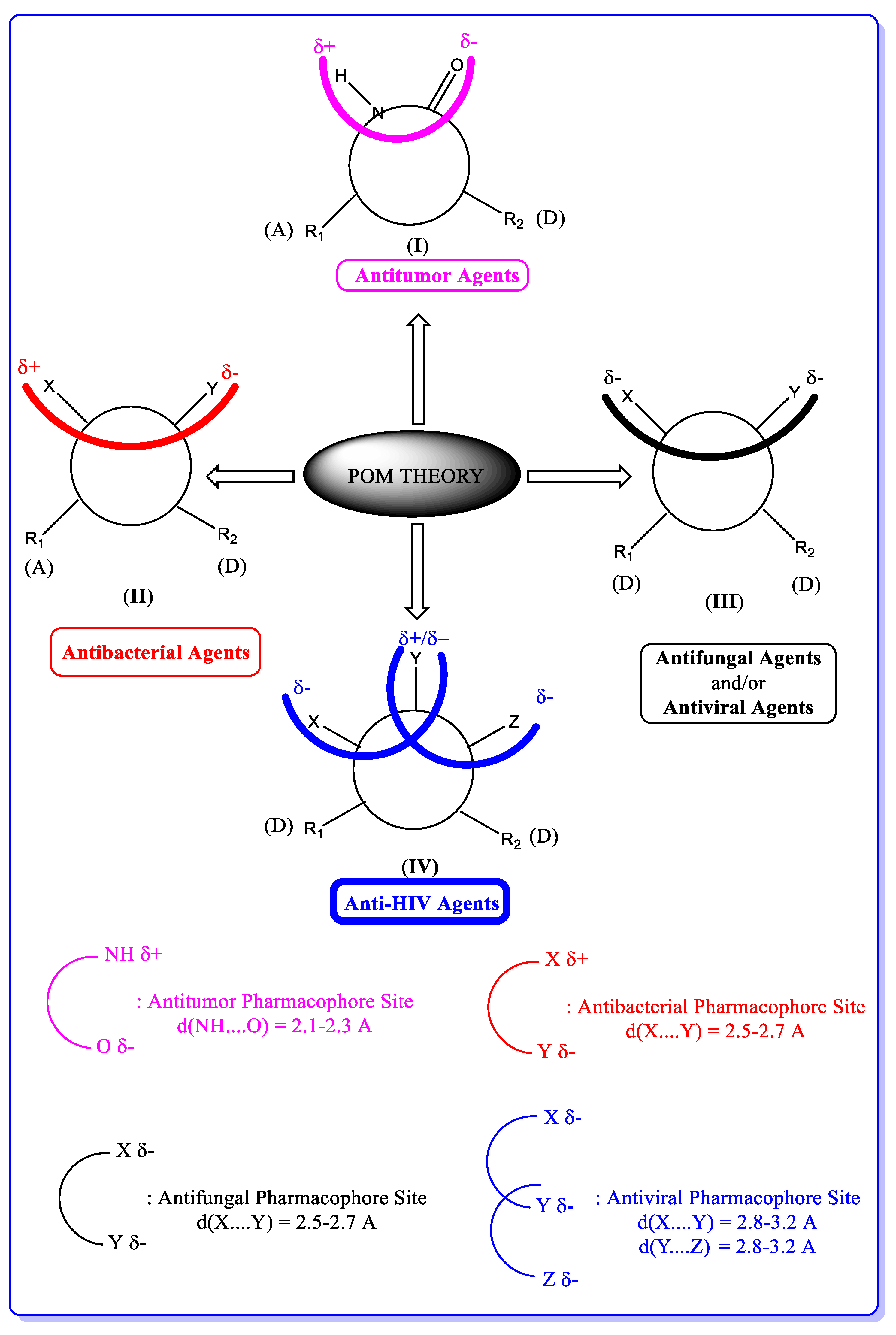
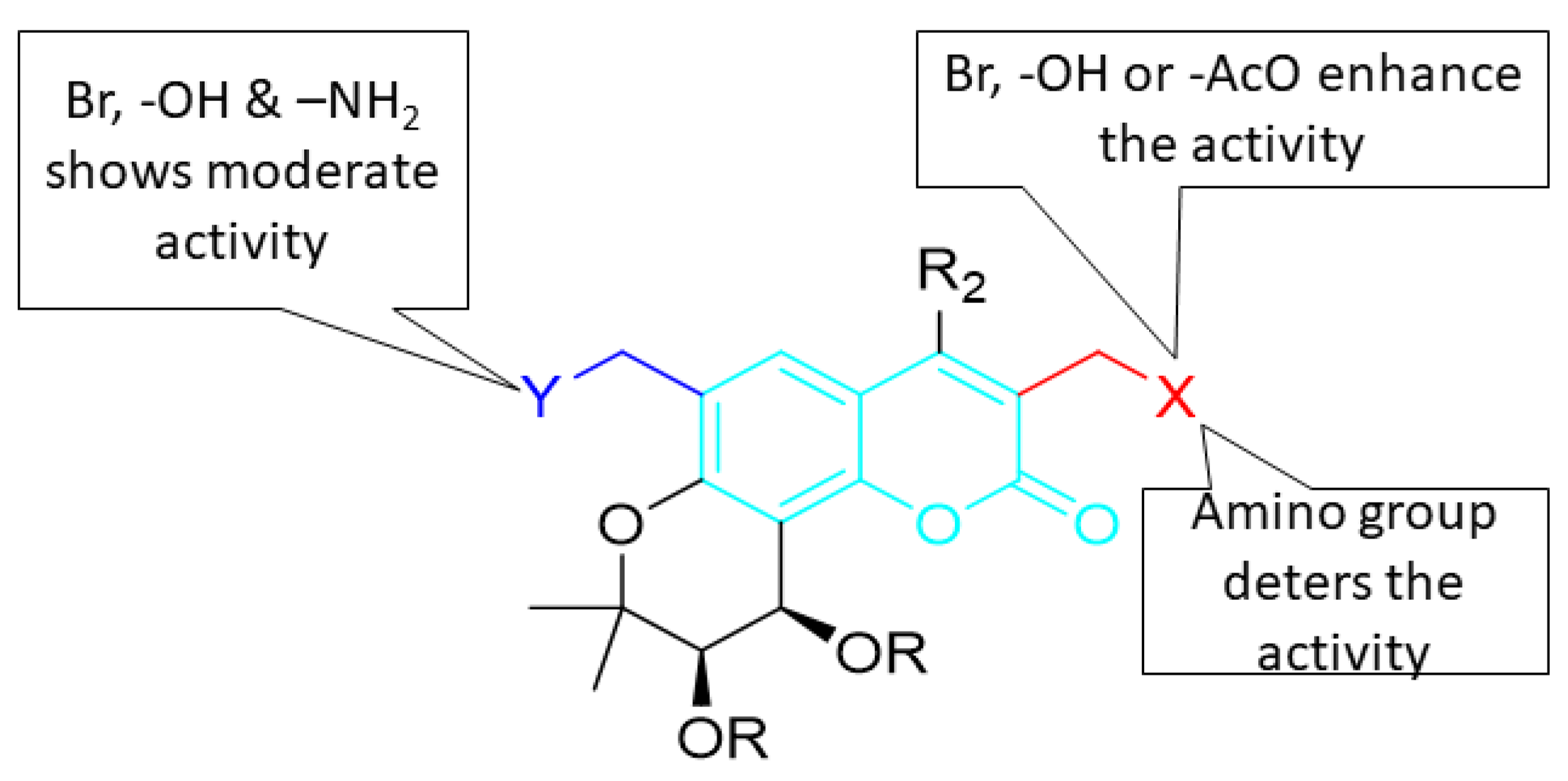
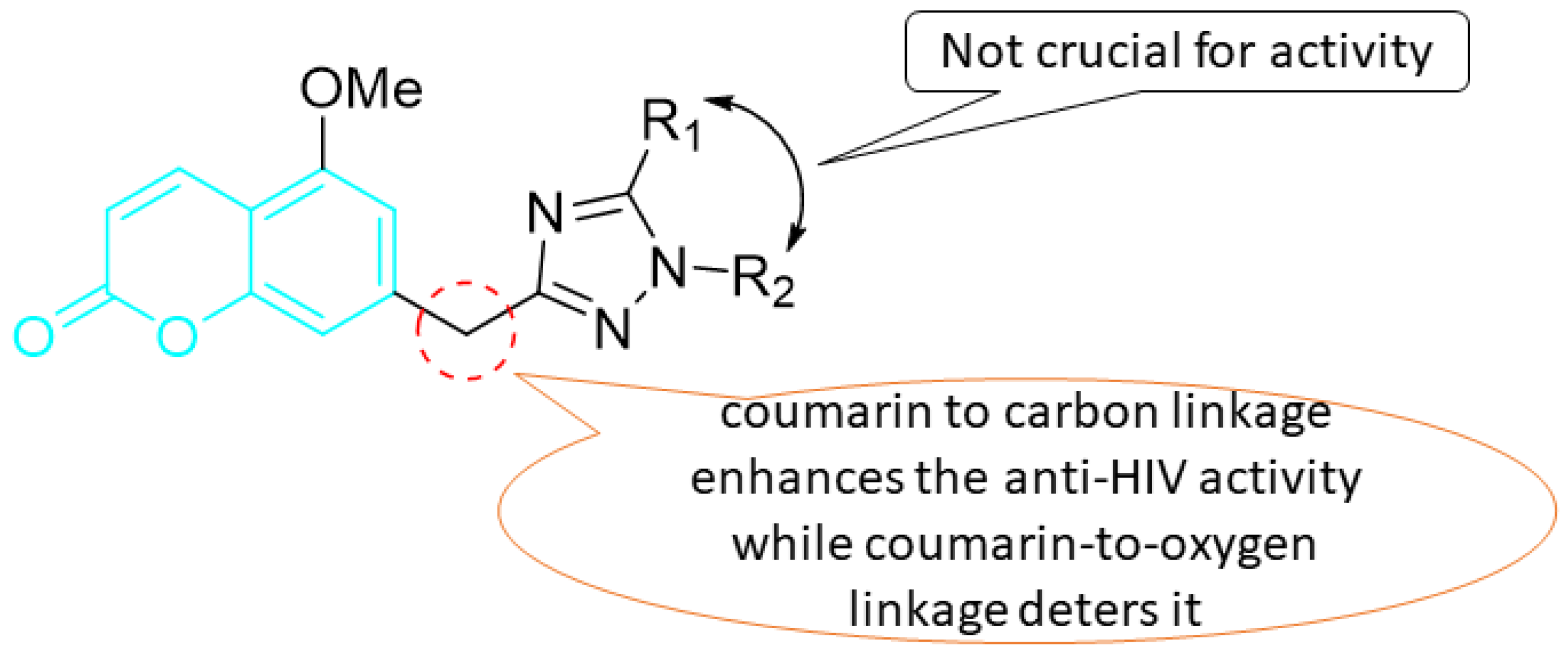
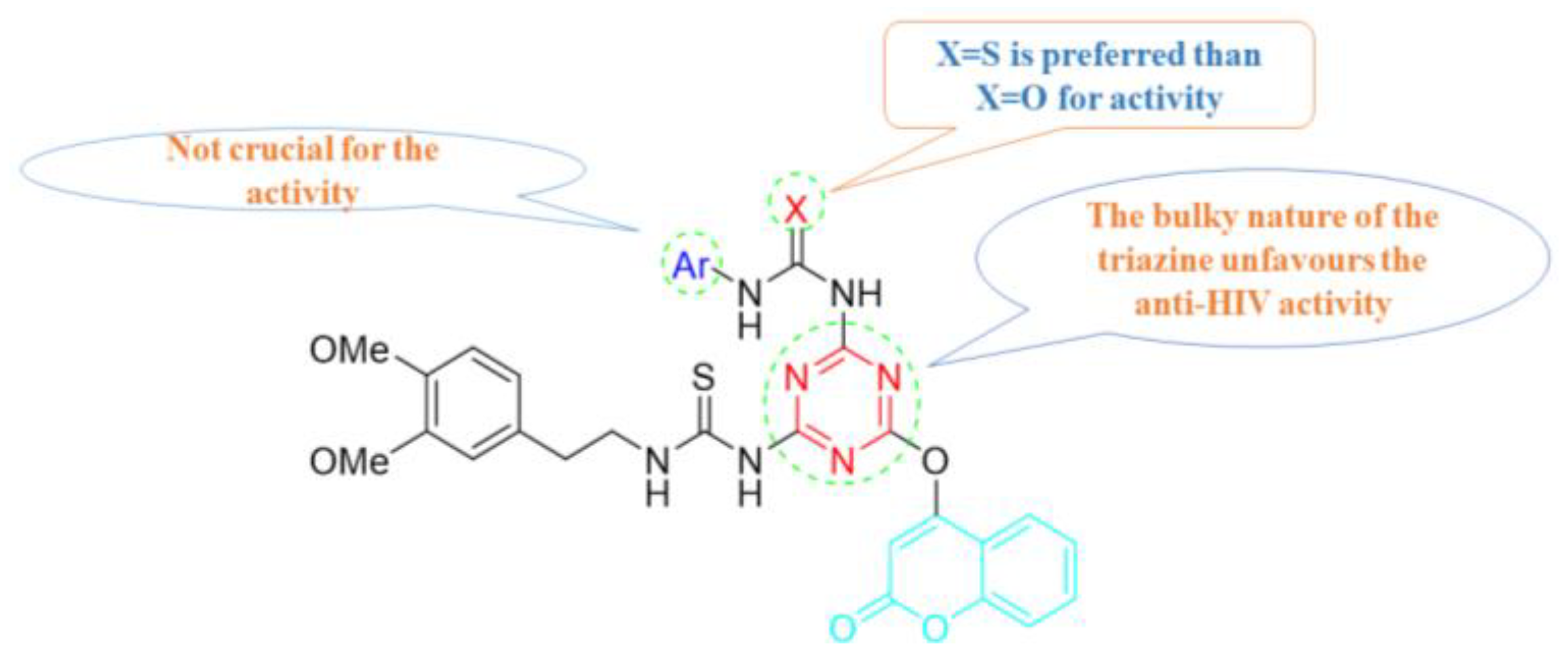
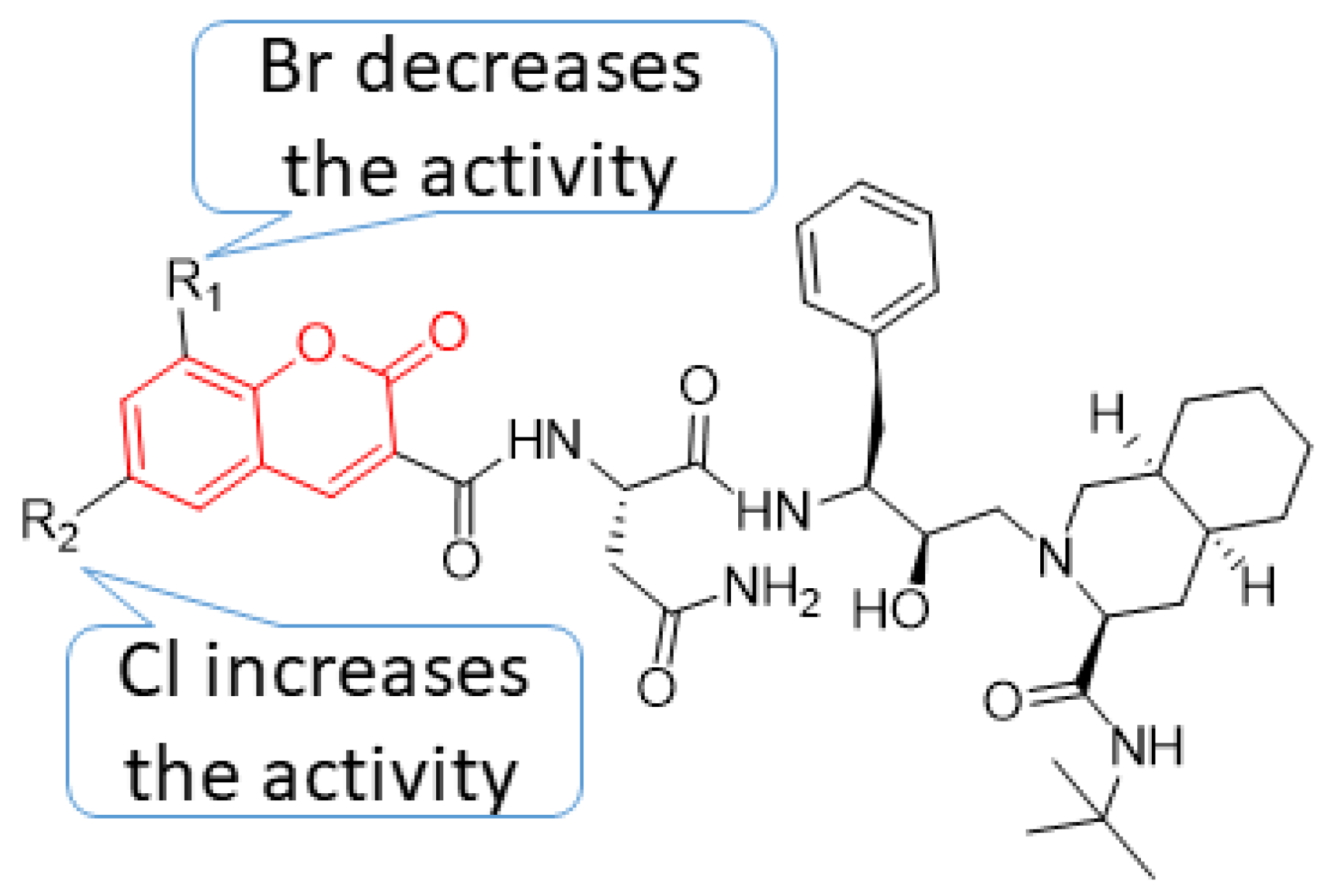
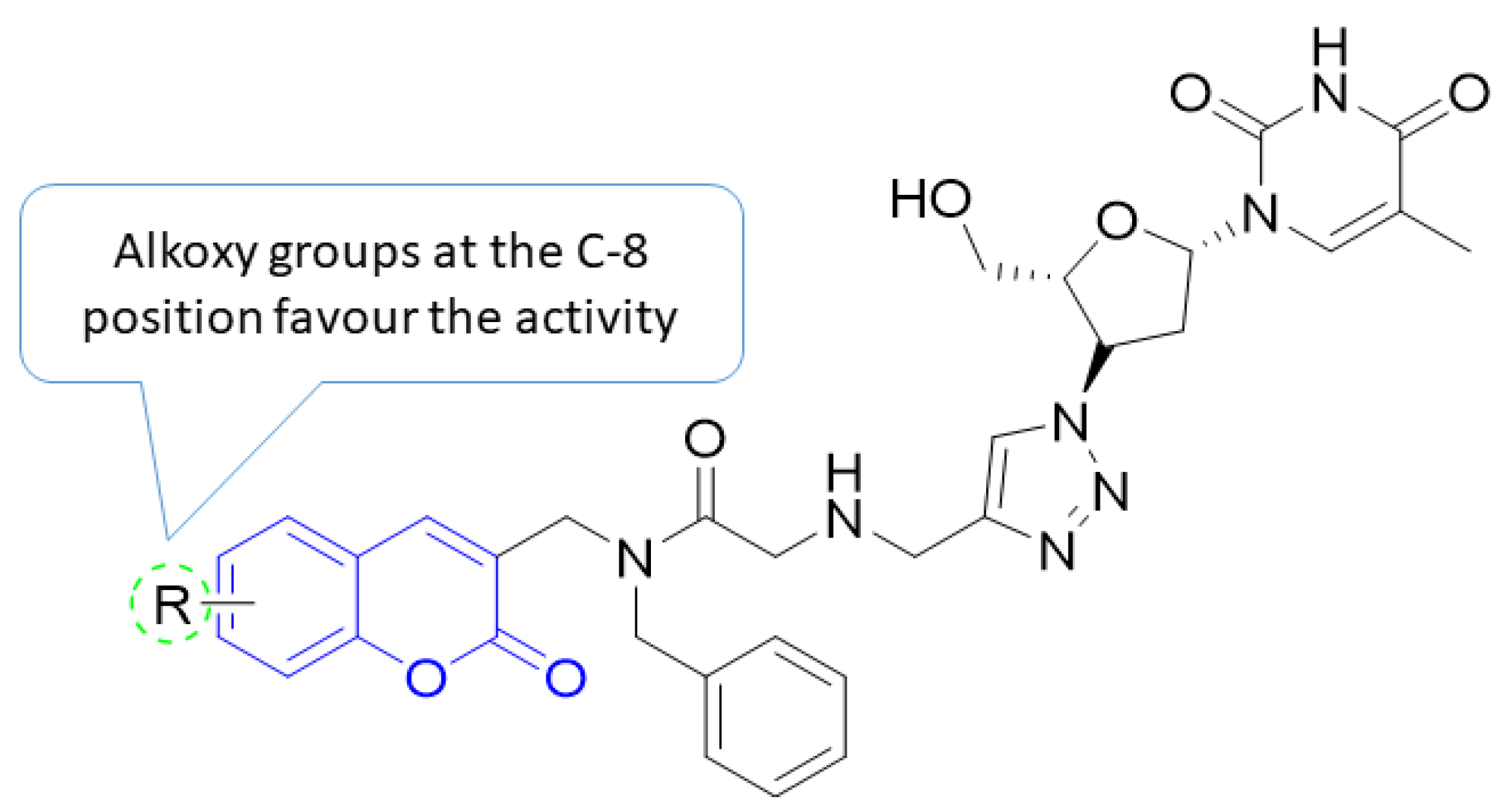
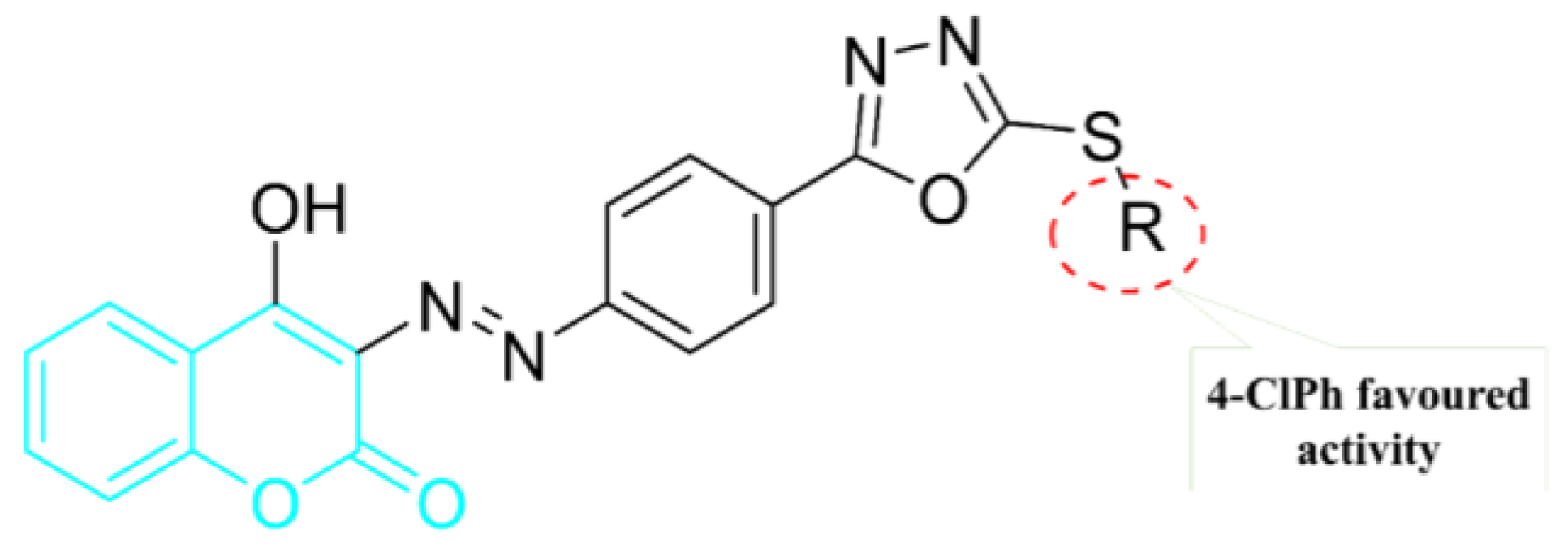
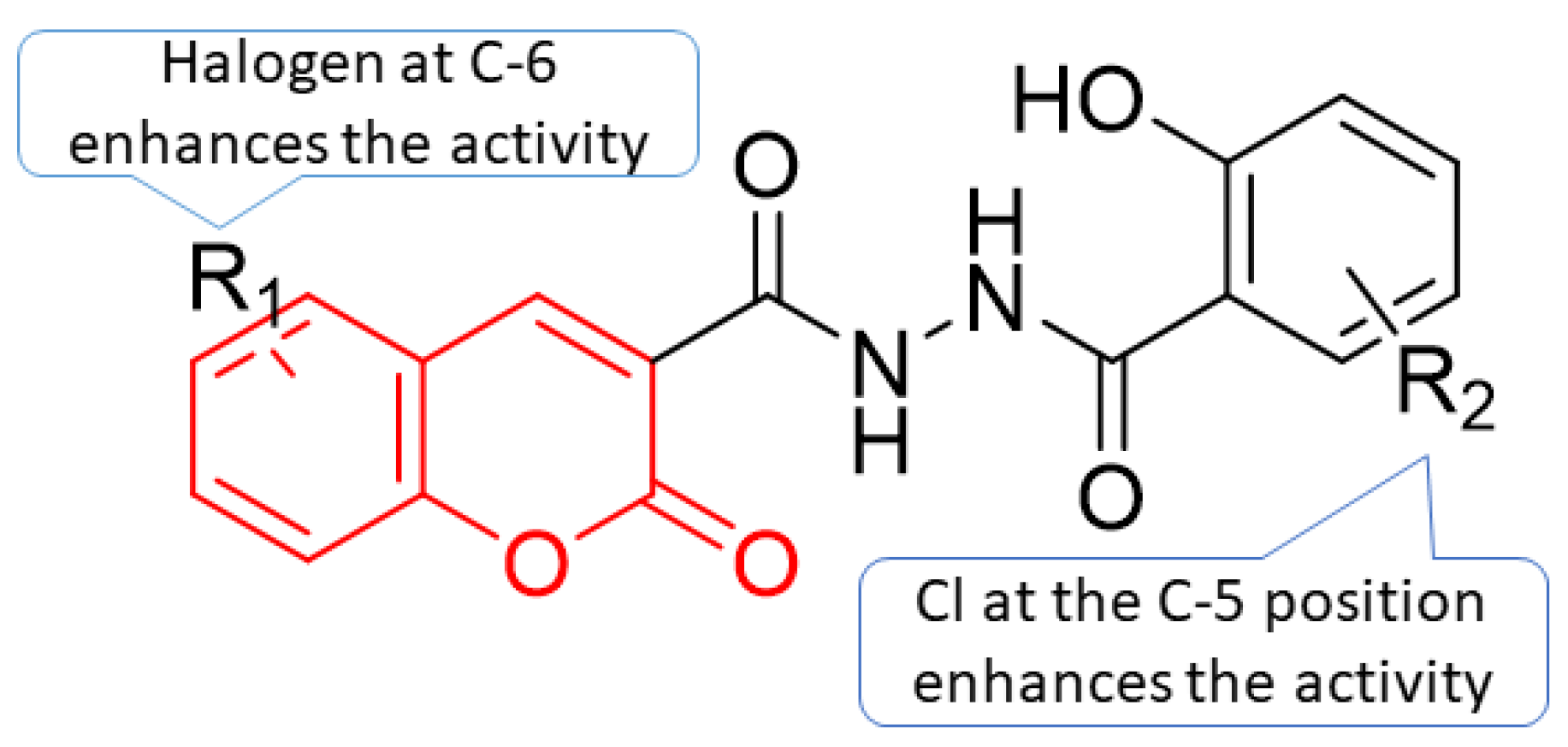
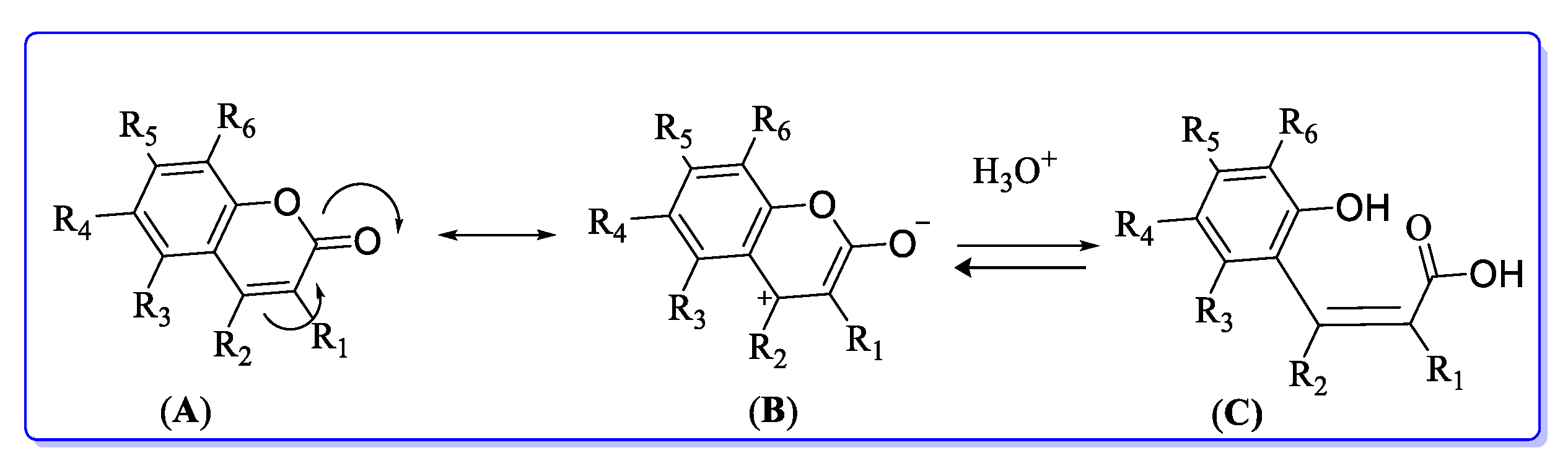
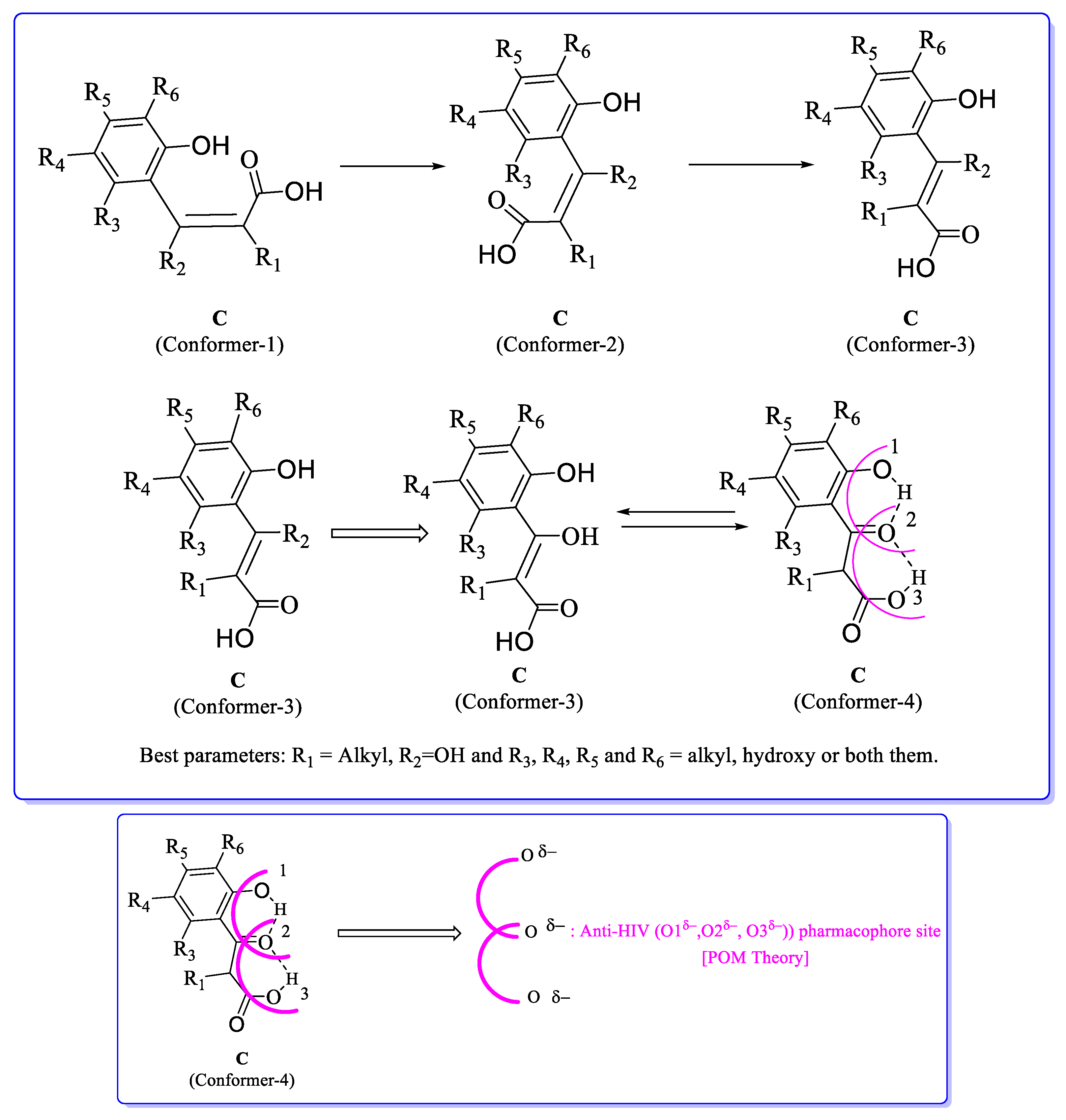
5. POM Analyses: Identification of Anti-HIV Pharmacophore Sites
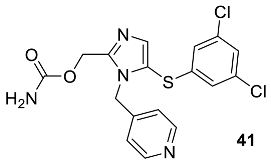
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
6. Conclusions and Future Viewpoint
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| POM | Petra/Osiris/Molinspiration |
| SAR | Structure–Activity Relationship |
| HIV | Human Immunodeficiency Syndrome |
| AIDS | Acquired Immunodeficiency Syndrome |
| HAART | Highly Active Antiretroviral Therapy |
| RT | Reverse Transcriptase |
| WHO | World Health Organisation |
| WOS | Web of Science |
| ART | Antiretroviral therapy |
| cART | Combined Antiretroviral Therapy |
| IN | Integrase |
| DNA | Deoxyribonucleic acid |
| VPR | Viral Protein Regulator |
| DCK | Hydroxymethyl (3′R,4′R)-3′,4′-di-O-(S)-camphanoyl-(+)-cis-khellactone |
| AZT | Azidothymidine |
| PETT | Phenyl Ethyl Thiazolyl Thiourea |
| MTT | 4,5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide |
| TEA | Triethylamine |
| DMF | Dimethyl formamide |
| EFV | Efavirenz |
| DABCO | 1,4-diazabicyclo[2.2.2]octane/triethylenediamine |
| PR | Protease |
| DMLs | Designed Multifunctional Ligands |
| EDCI | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| DMAP | 4-Dimethylaminopyridine |
| BTC | Bis(trichloromethyl)carbonate |
| DIEA | N,N-Diisopropylethylamine |
| DCM | Dimethylamonopyridine |
| DCC | Dicyclocarbodiimide |
| WT | Wild-Type |
| DFT | Density Functional Theory |
| NCI | American National Cancer Institute |
| TAAF | Tunisian–American Association for Cancer Research and Training Foundation |
References
- Mohamed, H.; Gurrola, T.; Berman, R.; Collins, M.; Sariyer, I.K.; Nonnemacher, M.R.; Wigdahl, B. Targeting CCR5 as a Component of an HIV-1 Therapeutic Strategy. Front. Immunol. 2022, 12, 816515. [Google Scholar] [CrossRef]
- Yu, D.; Suzuki, M.; Xie, L.; Morris-natschke, S.L.; Lee, K. Recent Progress in the Development of Coumarin Derivatives as Potent. Med. Res. Rev. 2003, 23, 322–345. [Google Scholar] [CrossRef]
- Kumar, V.; Joshi, H.C.; Pandey, I.P. Reverse Transcriptase Inhibition: A Way to Defeat HIV. HIV AIDS Rev. 2022, 21, 3–9. [Google Scholar] [CrossRef]
- Xu, Z.; Chen, Q.; Zhang, Y.; Liang, C. Coumarin-Based Derivatives with Potential Anti-HIV Activity. Fitoterapia 2021, 150, 104863. [Google Scholar] [CrossRef] [PubMed]
- Tariq, M.; Sirajuddin, M.; Ali, S.; Khalid, N.; Tahir, M.N.; Khan, H.; Ansari, T.M. Pharmacological Investigations and Petra/Osiris/Molinspiration (POM) Analyses of Newly Synthesized Potentially Bioactive Organotin(IV) Carboxylates. J. Photochem. Photobiol. B 2016, 158, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Suleiman, M.; Hasan, A.H.; Murugesan, S.; Amran, S.I.; Jamalis, J. Advances in the Synthesis of Diarylpyrimidine as Potent Non-Nucleoside Reverse Transcriptase Inhibitors: Biological Activities, Molecular Docking Studies and Structure-Activity Relationship: A Critical Review. Curr. Org. Chem. 2023, 27, 661–691. [Google Scholar] [CrossRef]
- Falagas, M.E.; Pitsouni, E.I.; Malietzis, G.A.; Pappas, G. Comparison of PubMed, Scopus, Web of Science, and Google Scholar: Strengths and Weaknesses. FASEB J. 2008, 22, 338–342. [Google Scholar] [CrossRef]
- Cunningham, W.E.; Tisnado, D.M.; Hu Lui, H.; Nakazono, T.T.; Carlisle, D.M. The Effect of Hospital Experience on Mortality among Patients Hospitalized with Acquired Immunodeficiency Syndrome in California. Am. J. Med. 1999, 107, 137–143. [Google Scholar] [CrossRef]
- Kitahata, M.M.; Koepsell, T.D.; Deyo, R.A.; Maxwell, C.L.; Dodge, W.T.; Wagner, E.H. Physicians’ Experience with the Acquired Immunodeficiency Syndrome as a Factor in Patients’ Survival. N. Engl. J. Med. 1996, 334, 701–707. [Google Scholar] [CrossRef]
- Olomola, T.O.; Klein, R.; Mautsa, N.; Sayed, Y.; Kaye, P.T. Synthesis and Evaluation of Coumarin Derivatives as Potential Dual-Action HIV-1 Protease and Reverse Transcriptase Inhibitors. Bioorg. Med. Chem. 2013, 21, 1964–1971. [Google Scholar] [CrossRef]
- Thompson, M.A.; Aberg, J.A.; Hoy, J.F.; Telenti, A.; Benson, C.; Cahn, P.; Eron, J.J.; Gü Nthard, H.F.; Hammer, S.M.; Reiss, P.; et al. Antiretroviral Treatment of Adult HIV Infection 2012 Recommendations of the International Antiviral Society-USA Panel. JAMA 2012, 308, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Post, F.A.; Moyle, G.J.; Stellbrink, H.J.; Domingo, P.; Podzamczer, D.; Fisher, M.; Norden, A.G.; Cavassini, M.; Rieger, A.; Khuong-Josses, M.-A.; et al. Randomized Comparison of Renal Effects, Efficacy, and Safety with Once-Daily Abacavir/Lamivudine Versus Tenofovir/Emtricitabine, Administered with Efavirenz, in Antiretroviral-Naive, HIV-1-Infected Adults: 48-Week Results from the ASSERT Study. J. Acquir. Immune Defic. Syndr. 2010, 55, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Günthard, H.F.; Saag, M.S.; Benson, C.A.; Del Rio, C.; Eron, J.J.; Gallant, J.E.; Hoy, J.F.; Mugavero, M.J.; Sax, P.E.; Thompson, M.A.; et al. Antiretroviral Drugs for Treatment and Prevention of HIV Infection in Adults: 2016 Recommendations of the International Antiviral Society-USA Panel. JAMA—J. Am. Med. Assoc. 2016, 316, 191–210. [Google Scholar] [CrossRef] [PubMed]
- Rodger, A.J.; Cambiano, V.; Bruun, T.; Vernazza, P.; Collins, S.; Van Lunzen, J.; Corbelli, G.M.; Estrada, V.; Geretti, A.M.; Beloukas, A.; et al. Sexual Activity without Condoms and Risk of HIV Transmission in Serodifferent Couples When the HIV-Positive Partner Is Using Suppressive Antiretroviral Therapy. JAMA—J. Am. Med. Assoc. 2016, 316, 171–181. [Google Scholar] [CrossRef]
- Read, P.J.; Mandalia, S.; Khan, P.; Harrisson, U.; Naftalin, C.; Gilleece, Y.; Anderson, J.; Hawkins, D.A.; Taylor, G.P.; De Ruiter, A. When Should HAART Be Initiated in Pregnancy to Achieve an Undetectable HIV Viral Load by Delivery? AIDS 2012, 26, 1095–1103. [Google Scholar] [CrossRef]
- Hoffman, R.M.; Black, V. Effects of Highly Active Antiretroviral Therapy Duration and Regimen on Risk for Mother-to-Child Transmission of HIV in Johannesburg, South Africa. J. Acquir. Immune Defic. Syndr. 2010, 54, 35–41. [Google Scholar] [CrossRef]
- Forbes, J.C.; Alimenti, A.M.; Singer, J.; Brophy, J.C.; Bitnun, A.; Samson, L.M.; Money, D.M.; Lee, T.C.K.; Lapointe, N.D.; Read, S.E. A National Review of Vertical HIV Transmission. AIDS 2012, 26, 757–763. [Google Scholar] [CrossRef]
- Esté, J.A.; Cihlar, T. Current Status and Challenges of Antiretroviral Research and Therapy. Antivir. Res. 2010, 85, 25–33. [Google Scholar] [CrossRef]
- Li, Z.; Kong, D.; Liu, Y.; Li, M. Pharmacological Perspectives and Molecular Mechanisms of Coumarin Derivatives against Virus Disease. Genes Dis. 2022, 9, 80–94. [Google Scholar] [CrossRef]
- World Health Assembly. 42 Forty-Second World Health Assembly, Geneva, 8–19 May 1989: Resolutions and Decisions, Annexes. 1989. Available online: https://apps.who.int/iris/handle/10665/171211 (accessed on 3 June 2022).
- World Health Organization; Onchocerciasis Control Programme in West Africa. Progress Report of the World Health Organization for 1989 (1 September 1988–31 August 1989); Onchocerciasis Control Programme in West Africa, 1989. Available online: https://apps.who.int/iris/handle/10665/345231 (accessed on 3 June 2022).
- Annunziata, F.; Pinna, C.; Dallavalle, S.; Tamborini, L.; Pinto, A. An Overview of Coumarin as a Versatile and Readily Accessible Scaffold with Broad-Ranging Biological Activities. Int. J. Mol. Sci. 2020, 21, 4618. [Google Scholar] [CrossRef]
- Borges, F.; Roleira, F.; Milhazes, N.; Santana, L.; Uriarte, E. Simple Coumarins and Analogues in Medicinal Chemistry: Occurrence, Synthesis and Biological Activity. Curr. Med. Chem. 2005, 12, 887–916. [Google Scholar] [CrossRef] [PubMed]
- Krogsgaard Thomsen, M.; Bokvist, K.; Høy, M.; Buschard, K.; Holst, J.J.; Lindström, P.; Gromada, J. Repaglinide at a Cellular Level. Diabetes Nutr. Metab. 2002, 15, 15–18. [Google Scholar] [PubMed]
- Hasan, A.H.; Murugesan, S.; Amran, S.I.; Chander, S.; Alanazi, M.M.; Ben Hadda, T.; Shakya, S.; Pratama, M.R.F.; Das, B.; Biswas, S.; et al. Novel Thiophene Chalcones-Coumarin as Acetylcholinesterase Inhibitors: Design, Synthesis, Biological Evaluation, Molecular Docking, ADMET Prediction and Molecular Dynamics Simulation. Bioorg. Chem. 2022, 119, 105572. [Google Scholar] [CrossRef]
- Ramli, N.; Mohd Sobani, S.S. Jurnal Teknologi. J. Teknol. 2013, 2, 19–25. [Google Scholar]
- Tang, T.; Liu, J.; Zuo, K.; Cheng, J.; Chen, L.; Lu, C.; Han, S.; Xu, J.; Jia, Z.; Ye, M.; et al. Genotype-Guided Dosing of Coumarin Anticoagulants: A Meta-Analysis of Randomized Controlled Trials. J. Cardiovasc. Pharmacol. Ther. 2015, 20, 387–394. [Google Scholar] [CrossRef]
- Jumal, J. Norhanis Sakinah Synthesis, Characterization, and Applications of Coumarin Derivatives: A Short Review. Malays. J. Sci. Health Technol. 2021, 7, 62–68. [Google Scholar] [CrossRef]
- Song, M.Q.; Min, W.; Wang, J.; Si, X.X.; Wang, X.J.; Liu, Y.W.; Shi, D.H. Design, Synthesis and Biological Evaluation of New Carbazole-Coumarin Hybrids as Dual Binding Site Inhibitors of Acetylcholinesterase. J. Mol. Struct. 2021, 1229, 129784. [Google Scholar] [CrossRef]
- Srivastav, V.K.; Tiwari, M. QSAR and Docking Studies of Coumarin Derivatives as Potent HIV-1 Integrase Inhibitors. Arab. J. Chem. 2017, 10, S1081–S1094. [Google Scholar] [CrossRef]
- Singh, V.K.; Srivastava, R.; Gupta, P.S.S.; Naaz, F.; Chaurasia, H.; Mishra, R.; Rana, M.K.; Singh, R.K. Anti-HIV Potential of Diarylpyrimidine Derivatives as Non-Nucleoside Reverse Transcriptase Inhibitors: Design, Synthesis, Docking, TOPKAT Analysis and Molecular Dynamics Simulations. J. Biomol. Struct. Dyn. 2021, 39, 2430–2446. [Google Scholar] [CrossRef]
- Reddy, D.S.; Kongot, M.; Kumar, A. Coumarin Hybrid Derivatives as Promising Leads to Treat Tuberculosis: Recent Developments and Critical Aspects of Structural Design to Exhibit Anti-Tubercular Activity. Tuberculosis 2021, 127, 102050. [Google Scholar] [CrossRef]
- Hu, Y.Q.; Xu, Z.; Zhang, S.; Wu, X.; Ding, J.W.; Lv, Z.S.; Feng, L.S. Recent Developments of Coumarin-Containing Derivatives and Their Anti-Tubercular Activity. Eur. J. Med. Chem. 2017, 136, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Song, S.; Wang, Z.; Sun, L.; Zhang, J.; Pannecouque, C.; de Clercq, E.; Zhan, P.; Liu, X. Design, Synthesis and Anti-HIV Evaluation of Novel 5-Substituted Diarylpyrimidine Derivatives as Potent HIV-1 NNRTIs. Bioorg. Med. Chem. 2021, 40, 116195. [Google Scholar] [CrossRef] [PubMed]
- Rojekar, S.; Fotooh Abadi, L.; Pai, R.; Mahajan, K.; Kulkarni, S.; Vavia, P.R. Multi-Organ Targeting of HIV-1 Viral Reservoirs with Etravirine Loaded Nanostructured Lipid Carrier: An in vivo Proof of Concept. Eur. J. Pharm. Sci. 2021, 164, 105916. [Google Scholar] [CrossRef] [PubMed]
- Jashari, A.; Hey-Hawkins, E.; Mikhova, B.; Draeger, G.; Popovski, E. An Improved Synthesis of 4-Chlorocoumarin-3-Sulfonyl Chloride and Its Reactions with Different Bidentate Nucleophiles to Give Pyrido[1′,2′:2,3]- and Thiazino[3′,2′:2,3]-1,2,4-Thiadiazino[6,5-c]Benzopyran-6-One 7,7-Dioxides. Molecules 2007, 12, 2017–2028. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, P.; Jain, P.; Rudrawar, S.; Jadhav, H.R.A. Quinoline, Coumarin and Other Heterocyclic Analogs Based HIV-1 Integrase Inhibitors. Curr. Drug Discov. Technol. 2017, 15, 2–19. [Google Scholar] [CrossRef]
- Mishra, S.; Pandey, A.; Manvati, S. Coumarin: An Emerging Antiviral Agent. Heliyon 2020, 6, e03217. [Google Scholar] [CrossRef]
- Al-Soud, Y.A.; Al-Masoudi, I.A.; Saeed, B.; Beifuß, U.; Al-Masoudi, N.A. Synthesis of New 1H-1,2,4-Triazolylcoumarins and Their Antitumor and Anti-HIV Activities. Chem. Heterocycl. Compd. 2006, 42, 583–590. [Google Scholar] [CrossRef]
- Kumar, S.; Khokra, S.L.; Yadav, A. Triazole Analogues as Potential Pharmacological Agents: A Brief Review. Future J. Pharm. Sci. 2021, 7, 106. [Google Scholar] [CrossRef]
- Jebir, R.M.; Mustafa, Y.F. Natural Coumarin-Lead Compounds: A Review of Their Medicinal Potentials. Iraqi J. Pharm. 2022, 18, 139–161. [Google Scholar] [CrossRef]
- Trivedi, J.C.; Bariwal, J.B.; Upadhyay, K.D.; Naliapara, Y.T.; Joshi, S.K.; Pannecouque, C.C.; de Clercq, E.; Shah, A.K. Improved and Rapid Synthesis of New Coumarinyl Chalcone Derivatives and Their Antiviral Activity. Tetrahedron Lett. 2007, 48, 8472–8474. [Google Scholar] [CrossRef]
- Patel, R.B.; Chikhalia, K.H.; Pannecouque, C.; de Clercq, E. Synthesis of Novel PETT Analogues: 3,4-Dimethoxy Phenyl Ethyl 1,3,5-Triazinyl Thiourea Derivatives and Their Antibacterial and Anti-HIV Studies. J. Braz. Chem. Soc. 2007, 18, 312–321. [Google Scholar] [CrossRef]
- Reddy, Y.T.; Reddy, P.N.; Crooks, P.A.; De Clercq, E.; Rao, G.V.P.; Rajitha, B. Synthesis and biological evaluation of novel substituted n1-[1-benzyl-3-(3-tert-butylcarbamoyl-octahydroisoquinolin-2yl)-2-hydroxy-propyl]-2-[(2-oxo-2h-chromene-3-carbonyl) amino] succinamide analogs as αντι-viral and αντι-hiv agents. Heterocycl. Commun. 2008, 14, 419–426. [Google Scholar] [CrossRef]
- Al-Soud, Y.A.; Al-Sa’doni, H.H.; Amajaour, H.A.S.; Salih, K.S.M.; Mubarak, M.S.; Al-Masoudi, N.A.; Jaber, I.H. Synthesis, Characterization and Anti-HIV and Antitumor Activities of New Coumarin Derivatives. Z. Naturforschung B 2008, 63, 83–89. [Google Scholar] [CrossRef]
- Al-Masoudi, N.A.; Pfleiderer, W.; Pannecouque, C. Nitroimidazoles Part 7. Synthesis and Anti-HIV Activity of New 4-Nitroimidazole Derivatives. Z. Naturforschung-Sect. B J. Chem. Sci. 2012, 67, 835–842. [Google Scholar] [CrossRef]
- Al-Soud, Y.A.; Al-Masoudi, N.A.; de Clercq, E.; Paneccoque, C. Nitroimidazoles, Part 4: Synthesis and Anti-HIV Activity of New 5-Alkylsulfanyl and 5-(4′-Arylsulfonyl)Piperazinyl-4-Nitroimidazole Derivatives. Heteroat. Chem. 2007, 18, 333–340. [Google Scholar] [CrossRef]
- Jorgensen, W.L. Efficient Drug Lead Discovery and Optimization. Acc. Chem. Res. 2009, 42, 724–733. [Google Scholar] [CrossRef]
- Mahajan, D.H.; Pannecouque, C.; de Clercq, E.; Chikhalia, K.H. Synthesis and Studies of New 2-(Coumarin-4-Yloxy)-4,6-(Substituted)-s- Triazine Derivatives as Potential Anti-HIV Agents. Arch. Pharm. 2009, 342, 281–290. [Google Scholar] [CrossRef]
- Olomola, T.O.; Klein, R.; Lobb, K.A.; Sayed, Y.; Kaye, P.T. Towards the Synthesis of Coumarin Derivatives as Potential Dual-Action HIV-1 Protease and Reverse Transcriptase Inhibitors. Tetrahedron Lett. 2010, 51, 6325–6328. [Google Scholar] [CrossRef]
- Livani, Z.A.; Safakish, M.; Hajimahdi, Z.; Soleymani, S.; Zabihollahi, R.; Aghasadeghi, M.R.; Alipour, E.; Zarghi, A. Design, Synthesis, Molecular Modeling, In Silico ADME Studies and Anti-HIV-1 Assay of New Diazocoumarin Derivatives. Iran J. Pharm. Res. 2018, 17, 65–77. [Google Scholar] [CrossRef]
- Jesumoroti, O.J.; Faridoon; Mnkandhla, D.; Isaacs, M.; Hoppe, H.C.; Klein, R. Evaluation of Novel N′-(3-Hydroxybenzoyl)-2-Oxo-2H-Chromene-3-Carbohydrazide Derivatives as Potential HIV-1 Integrase Inhibitors. Medchemcomm 2019, 10, 80–88. [Google Scholar] [CrossRef]
- Esposito, F.; Ambrosio, F.A.; Maleddu, R.; Costa, G.; Rocca, R.; Maccioni, E.; Catalano, R.; Romeo, I.; Eleftheriou, P.; Karia, D.C.; et al. Chromenone Derivatives as a Versatile Scaffold with Dual Mode of Inhibition of HIV-1 Reverse Transcriptase-Associated Ribonuclease H Function and Integrase Activity. Eur. J. Med. Chem. 2019, 182, 111617. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Ma, L.; Wen, J.; Dong, B.; Wang, Y.; Wang, Z.; Zhou, J.; Zhang, G.; Wang, J.; Guo, Y.; et al. Rational Design and Structure−Activity Relationship of Coumarin Derivatives Effective on HIV-1 Protease and Partially on HIV-1 Reverse Transcriptase. Eur. J. Med. Chem. 2020, 186, 111900. [Google Scholar] [CrossRef] [PubMed]
- Maly, D.J.; Leonetti, F.; Backes, B.J.; Dauber, D.S.; Harris, J.L.; Craik, C.S.; Ellman, J.A. Expedient Solid-Phase Synthesis of Fluorogenic Protease Substrates Using the 7-Amino-4-Carbamoylmethylcoumarin (ACC) Fluorophore. J. Org. Chem. 2002, 67, 910–915. [Google Scholar] [CrossRef]
- Drzewiecka, A.; Koziol, A.E.; Borowski, P.; Sanna, G.; Giliberti, G.; La Colla, P.; Zawadowski, T.; Struga, M. Structural and Antivirial Studies of Dipetalactone and Its Methyl Derivative. J. Mol. Struct. 2013, 1054–1055, 150–156. [Google Scholar] [CrossRef]
- Kasralikar, H.M.; Jadhavar, S.C.; Bhusare, S.R. Synthesis and Molecular Docking Study of Novel Chromeno-Chromenones as Anti-HIV-1 NNRT Inhibitors. Synlett 2015, 26, 1969–1972. [Google Scholar] [CrossRef]
- Kasralikar, H.M.; Jadhavar, S.C.; Bhusare, S.R. Synthesis and Molecular Docking Studies of Oxochromenyl Xanthenone and Indolyl Xanthenone Derivatives as Anti-HIV-1 RT Inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 3882–3886. [Google Scholar] [CrossRef]
- Rashamuse, T.J.; Klein, R.; Kaye, P.T. Synthesis of Baylis-Hillman-Derived Phosphonated 3-(Benzylaminomethyl) Coumarins. Synth. Commun. 2010, 40, 3683–3690. [Google Scholar] [CrossRef]
- Chalkha, M.; Nakkabi, A.; Ben Hadda, T.; Berredjem, M.; El Moussaoui, A.; Bakhouch, M.; Saadi, M.; El Ammari, L.; Almalki, F.A.; Laaroussi, H.; et al. Crystallographic Study, Biological Assessment and POM/Docking Studies of Pyrazoles-Sulfonamide Hybrids (PSH): Identification of a Combined Antibacterial/Antiviral Pharmacophore Sites Leading to in-Silico Screening the Anti-COVID-19 Activity. J. Mol. Struct. 2022, 1267, 133605. [Google Scholar] [CrossRef]
- Lakhrissi, Y.; Rbaa, M.; Tuzun, B.; Hichar, A.; Anouar, E.H.; Ounine, K.; Almalki, F.; Ben Hadda, T.; Zarrouk, A.; Lakhrissi, B. Synthesis, Structural Confirmation, Antibacterial Properties and Bio-Informatics Computational Analyses of New Pyrrole Based on 8-Hydroxyquinoline. J. Mol. Struct. 2022, 1259, 132683. [Google Scholar] [CrossRef]
- Rbaa, M.; Haida, S.; Tuzun, B.; Hichar, A.; El Hassane, A.; Kribii, A.; Lakhrissi, Y.; Ben Hadda, T.; Zarrouk, A.; Lakhrissi, B.; et al. Synthesis, Characterization and Bioactivity of Novel 8-Hydroxyquinoline Derivatives: Experimental, Molecular Docking, DFT and POM Analyses. J. Mol. Struct. 2022, 1258, 132688. [Google Scholar] [CrossRef]
- Esharkawy, E.R.; Almalki, F.; Ben Hadda, T. In Vitro Potential Antiviral SARS-CoV-19- Activity of Natural Product Thymohydroquinone and Dithymoquinone from Nigella Sativa. Bioorg. Chem. 2022, 120, 105587. [Google Scholar] [CrossRef] [PubMed]
- Titi, A.; Messali, M.; Alqurashy, B.A.; Touzani, R.; Shiga, T.; Oshio, H.; Fettouhi, M.; Rajabi, M.; Almalki, F.A.; Ben Hadda, T. Synthesis, Characterization, X-Ray Crystal Study and Bioctivities of Pyrazole Derivatives: Identification of Antitumor, Antifungal and Antibacterial Pharmacophore Sites. J. Mol. Struct. 2020, 1205, 127625. [Google Scholar] [CrossRef]
- Alawadi, D.Y.; Saadeh, H.A.; Kaur, H.; Goyal, K.; Sehgal, R.; Ben Hadda, T.; Elsawy, N.A.; Mubarak, M.S. Metronidazole Derivatives as a New Class of Antiparasitic Agents: Synthesis, Prediction of Biological Activity, and Molecular Properties. Med. Chem. Res. 2015, 24, 1196–1209. [Google Scholar] [CrossRef]
- Lahsasni, S.; Ben Hadda, T.; Masand, V.; Pathan, N.B.; Parvez, A.; Warad, I.; Shaheen, U.; Bader, A.; Aljofan, M. POM Analyses of Raltegravir Derivatives: A New Reflection Enlightening the Mechanism of HIV-Integrase Inhibition. Res. Chem. Intermed. 2015, 41, 5121–5136. [Google Scholar] [CrossRef]
- Hakkou, Z.; Maciuk, A.; Leblais, V.; Bouanani, N.E.; Mekhfi, H.; Bnouham, M.; Aziz, M.; Ziyyat, A.; Rauf, A.; Ben Hadda, T.; et al. Antihypertensive and Vasodilator Effects of Methanolic Extract of Inula Viscosa: Biological Evaluation and POM Analysis of Cynarin, Chlorogenic Acid as Potential Hypertensive. Biomed. Pharmacother. 2017, 93, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Ben Hadda, T.; Khardli, F.Z.; Mimouni, M.; Daoudi, M.; Kerbal, A.; Salgado-Zamora, H.; Gandhare, N.; Parvez, A.; Lahsasni, S. Impact of Geometric Parameters, Charge, and Lipophilicity on Bioactivity of Armed Quinoxaline, Benzothiaole, and Benzothiazine: Pom Analyses of Antibacterial and Antifungal Activity. Phosphorus Sulfur Silicon Relat. Elem. 2014, 189, 753–761. [Google Scholar] [CrossRef]
- Rachedi, K.O.; Ouk, T.S.; Bahadi, R.; Bouzina, A.; Djouad, S.E.; Bechlem, K.; Zerrouki, R.; Ben Hadda, T.; Almalki, F.; Berredjem, M. Synthesis, DFT and POM Analyses of Cytotoxicity Activity of α-Amidophosphonates Derivatives: Identification of Potential Antiviral O,O-Pharmacophore Site. J. Mol. Struct. 2019, 1197, 196–203. [Google Scholar] [CrossRef]
- Berredjem, M.; Bouzina, A.; Bahadi, R.; Bouacida, S.; Rastija, V.; Djouad, S.E.; Sothea, T.O.; Almalki, F.A.; Ben Hadda, T.; Aissaoui, M. Antitumor Activity, X-Ray Crystallography, in Silico Study of Some-Sulfamido-Phosphonates. Identification of Pharmacophore Sites. J. Mol. Struct. 2022, 1250, 131886. [Google Scholar] [CrossRef]
- Ben Hadda, T.; Berredjem, M.; Almalki, F.A.; Rastija, V.; Jamalis, J.; Bin Emran, T.; Abu-Izneid, T.; Esharkawy, E.; Rodriguez, L.C.; Alqahtani, A.M. How to Face COVID-19: Proposed Treatments Based on Remdesivir and Hydroxychloroquine in the Presence of Zinc Sulfate. Docking/DFT/POM Structural Analysis. J. Biomol. Struct. Dyn. 2022, 40, 9429–9442. [Google Scholar] [CrossRef]
- Mabkhot, Y.N.; Alatibi, F.; El-Sayed, N.N.E.; Al-Showiman, S.; Kheder, N.A.; Wadood, A.; Rauf, A.; Bawazeer, S.; Ben Hadda, T. Antimicrobial Activity of Some Novel Armed Thiophene Derivatives and Petra/Osiris/Molinspiration (POM) Analyses. Molecules 2016, 21, 222. [Google Scholar] [CrossRef]
- Bhat, A.R.; Dongre, R.S.; Almalki, F.A.; Berredjem, M.; Aissaoui, M.; Touzani, R.; Ben Hadda, T.; Akhter, M.S. Synthesis, Biological Activity and POM/DFT/Docking Analyses of Annulated Pyrano[2,3-d]Pyrimidine Derivatives: Identification of Antibacterial and Antitumor Pharmacophore Sites. Bioorg. Chem. 2021, 106, 104480. [Google Scholar] [CrossRef] [PubMed]
- Chalkha, M.; Bakhouch, M.; Akhazzane, M.; Bourass, M.; Nicolas, Y.; Al Houari, G.; El Yazidi, M. Design, Synthesis and Characterization of Functionalized Pyrazole Derivatives Bearing Amide and Sulfonamide Moieties from Aza-Aurones. J. Chem. Sci. 2020, 132, 86. [Google Scholar] [CrossRef]
- Taneja, S.C.; Uhar, K.L.; Atal, C.K. A Novel Ring Opening of Coumarins. Synth. Commun. 1980, 10, 37–42. [Google Scholar] [CrossRef]
- Available online: https://Www.maroc.ma/fr/actualites/research-excellence-awards-une-vingtaine-de-travaux-de-recherche-et-dinnovation-primes (accessed on 4 July 2023).
- Ben Hadda, T. Ranking 2023- Taibi BEN HADDA. 2022. Available online: https://www.adscientificindex.com/scientist/taibi-ben-hadda/91200 (accessed on 4 July 2023).






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suleiman, M.; Almalki, F.A.; Ben Hadda, T.; Kawsar, S.M.A.; Chander, S.; Murugesan, S.; Bhat, A.R.; Bogoyavlenskiy, A.; Jamalis, J. Recent Progress in Synthesis, POM Analyses and SAR of Coumarin-Hybrids as Potential Anti-HIV Agents—A Mini Review. Pharmaceuticals 2023, 16, 1538. https://doi.org/10.3390/ph16111538
Suleiman M, Almalki FA, Ben Hadda T, Kawsar SMA, Chander S, Murugesan S, Bhat AR, Bogoyavlenskiy A, Jamalis J. Recent Progress in Synthesis, POM Analyses and SAR of Coumarin-Hybrids as Potential Anti-HIV Agents—A Mini Review. Pharmaceuticals. 2023; 16(11):1538. https://doi.org/10.3390/ph16111538
Chicago/Turabian StyleSuleiman, Mustapha, Faisal A. Almalki, Taibi Ben Hadda, Sarkar M. A. Kawsar, Subhash Chander, Sankaranarayanan Murugesan, Ajmal R. Bhat, Andrey Bogoyavlenskiy, and Joazaizulfazli Jamalis. 2023. "Recent Progress in Synthesis, POM Analyses and SAR of Coumarin-Hybrids as Potential Anti-HIV Agents—A Mini Review" Pharmaceuticals 16, no. 11: 1538. https://doi.org/10.3390/ph16111538
APA StyleSuleiman, M., Almalki, F. A., Ben Hadda, T., Kawsar, S. M. A., Chander, S., Murugesan, S., Bhat, A. R., Bogoyavlenskiy, A., & Jamalis, J. (2023). Recent Progress in Synthesis, POM Analyses and SAR of Coumarin-Hybrids as Potential Anti-HIV Agents—A Mini Review. Pharmaceuticals, 16(11), 1538. https://doi.org/10.3390/ph16111538