
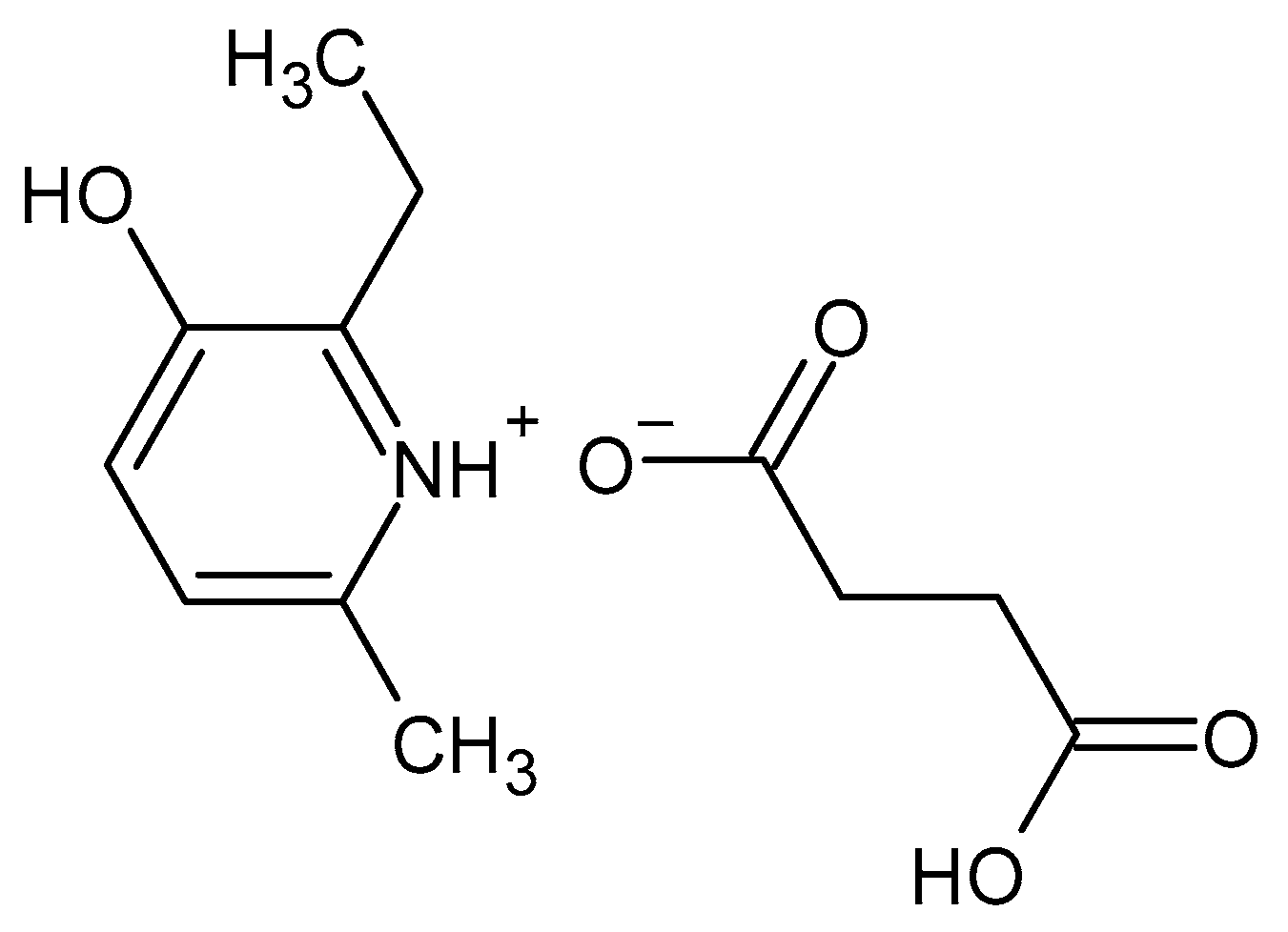
Ethylmethylhydroxypyridine Succinate Is an Inhibitor but Not a Substrate of ABCB1 and SLCO1B1

 ,
,
Abstract
:
1. Introduction
2. Results
2.1. MTT Assay
2.2. EMHPS Transport through the Caco-2 Cells Monolayer
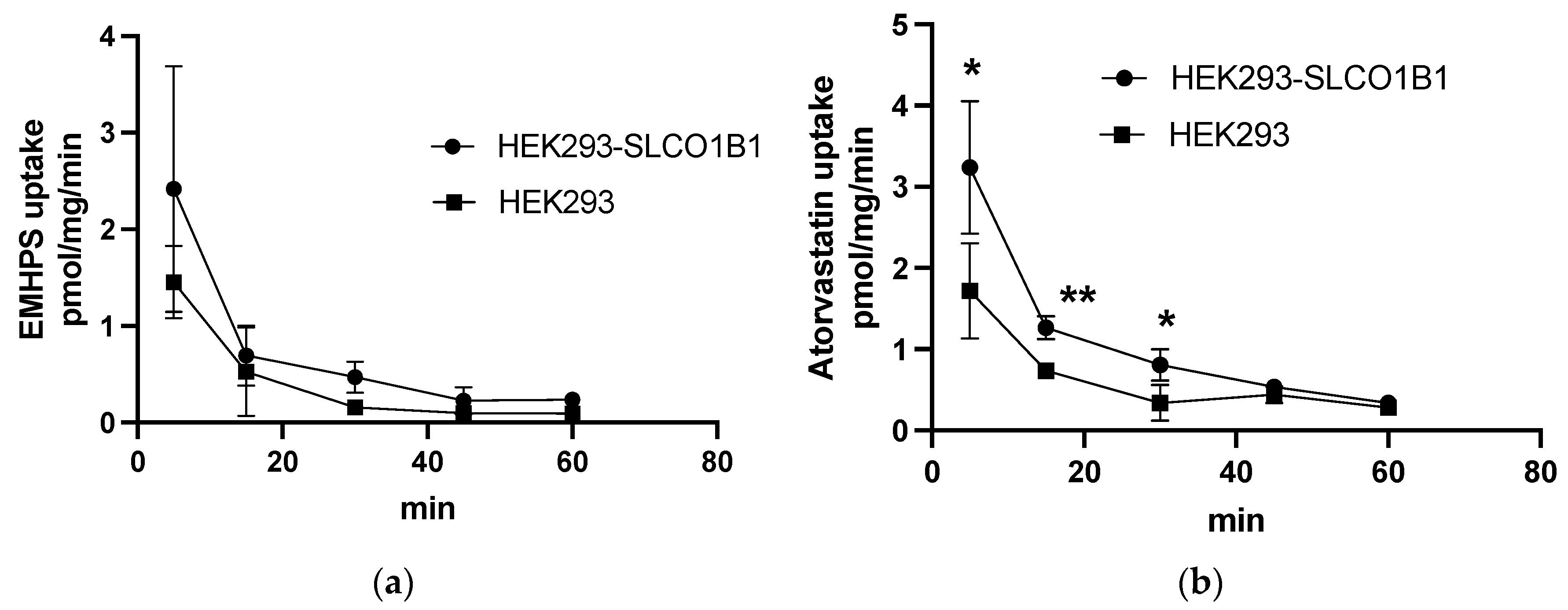
2.3. EMHPS Transport in HEK293-SLCO1B1 Cells
2.4. Effect of EMHPS on the ABCB1-Mediated Transport of Fexofenadine
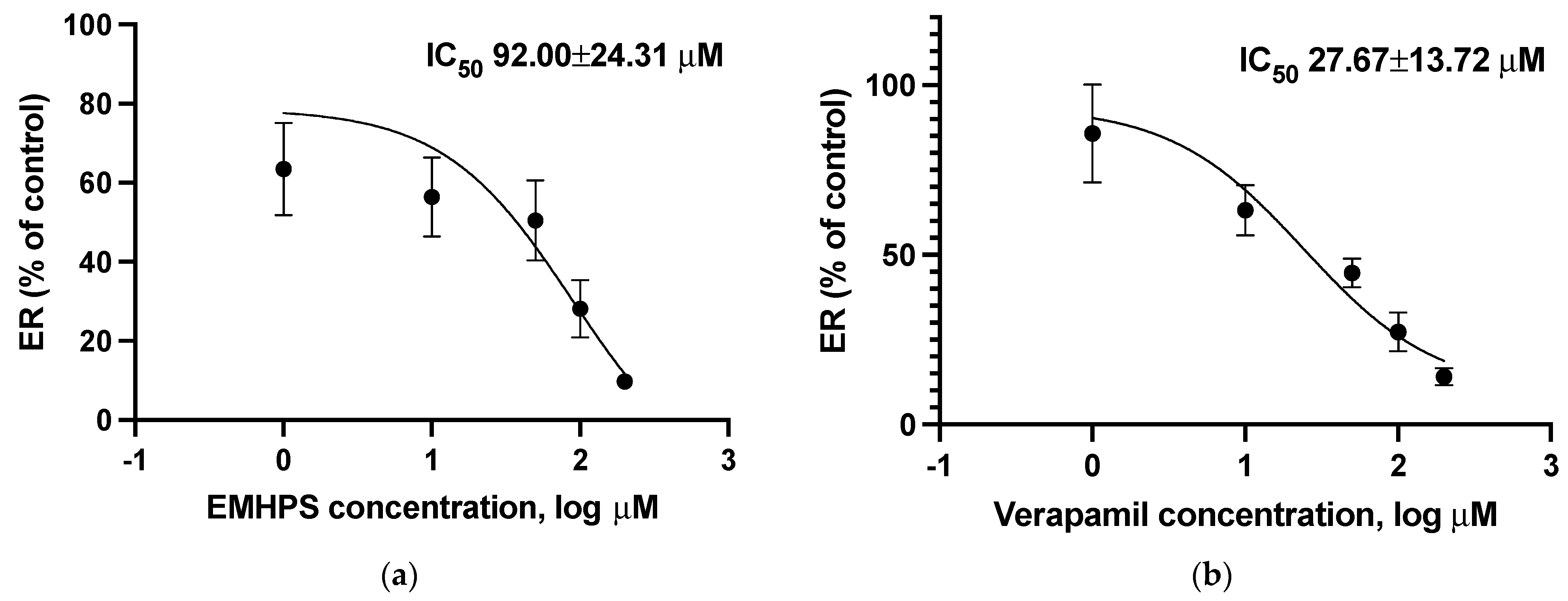
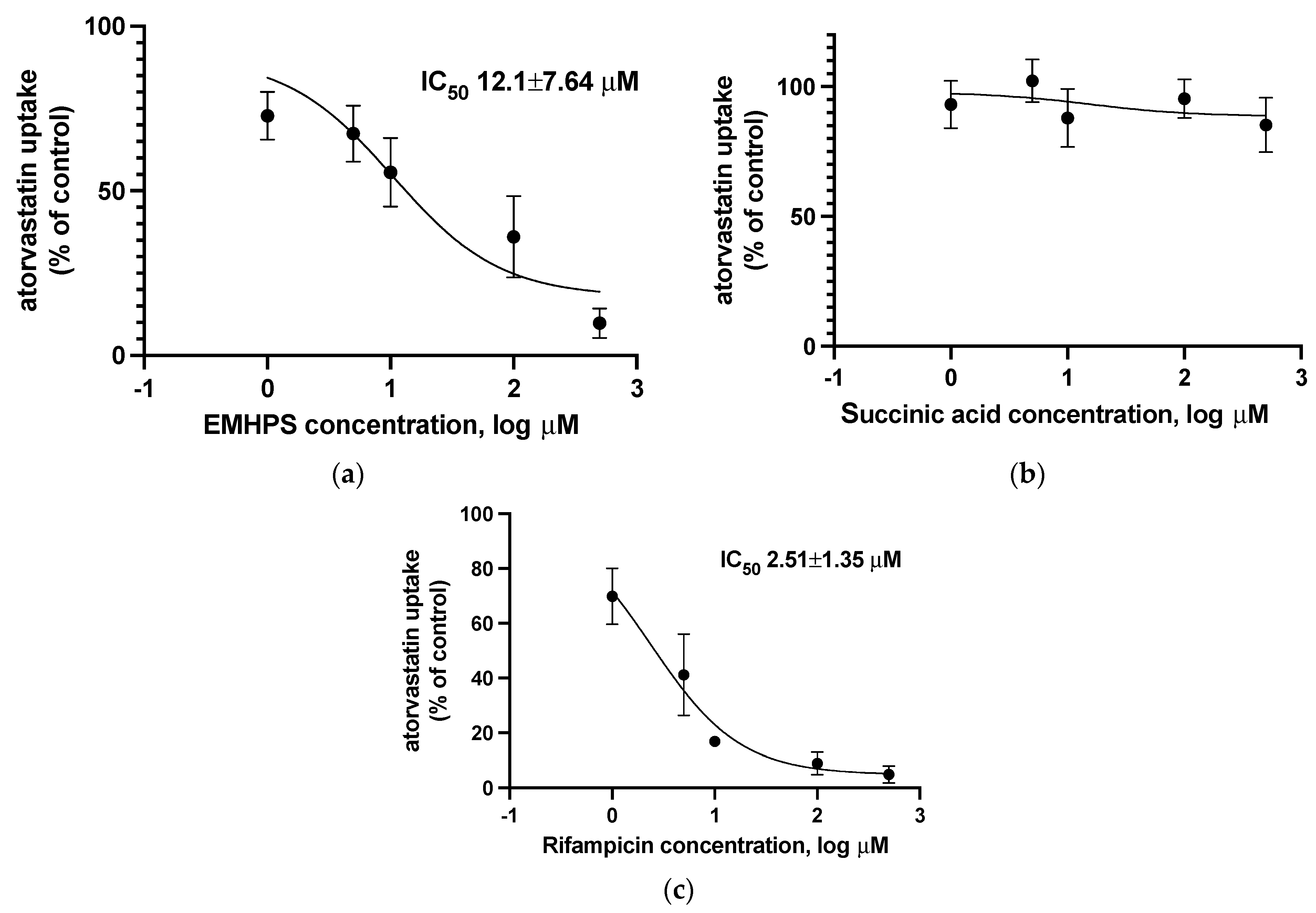
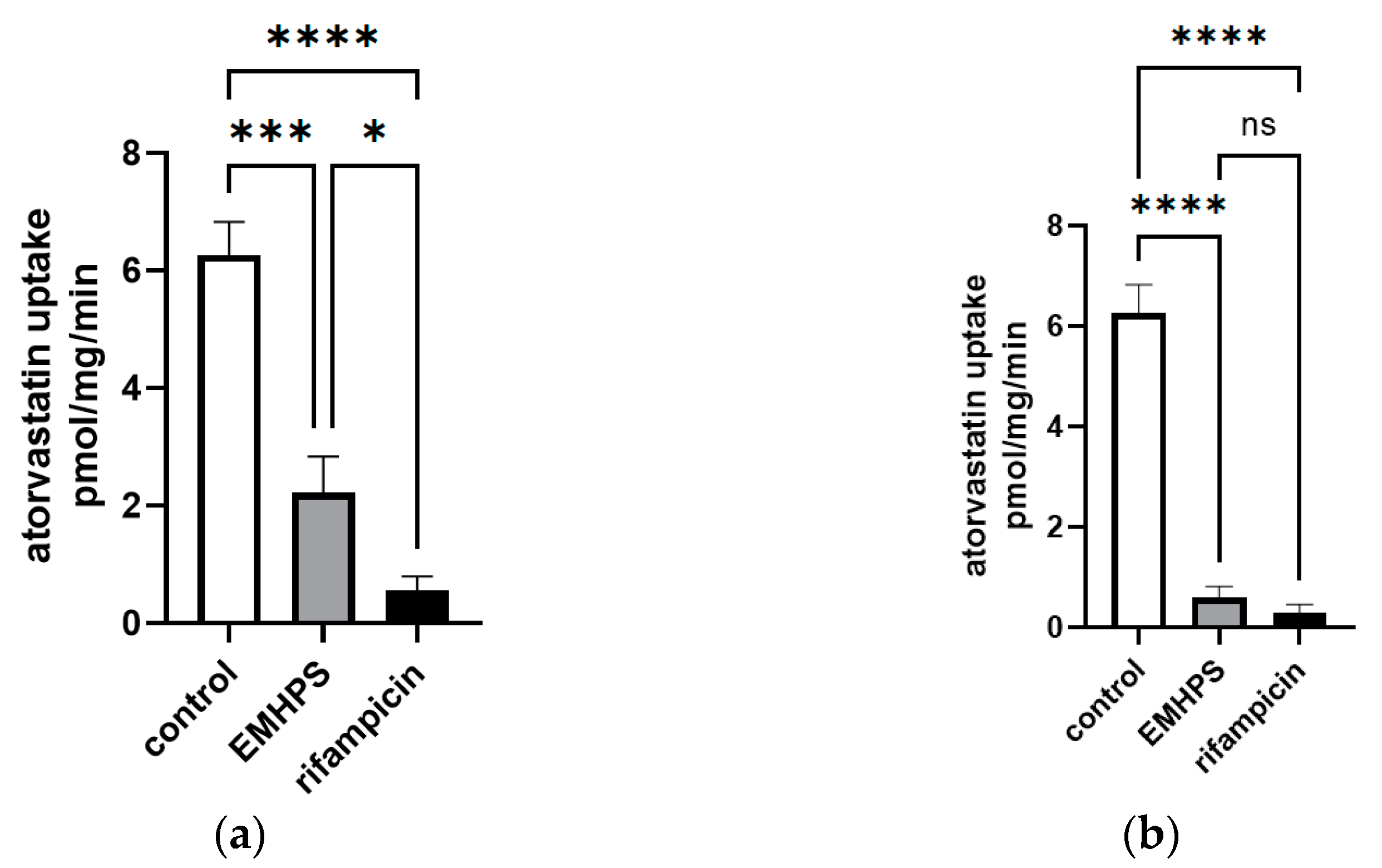
2.5. Effect of EMHPS on SLCO1B1 Activity
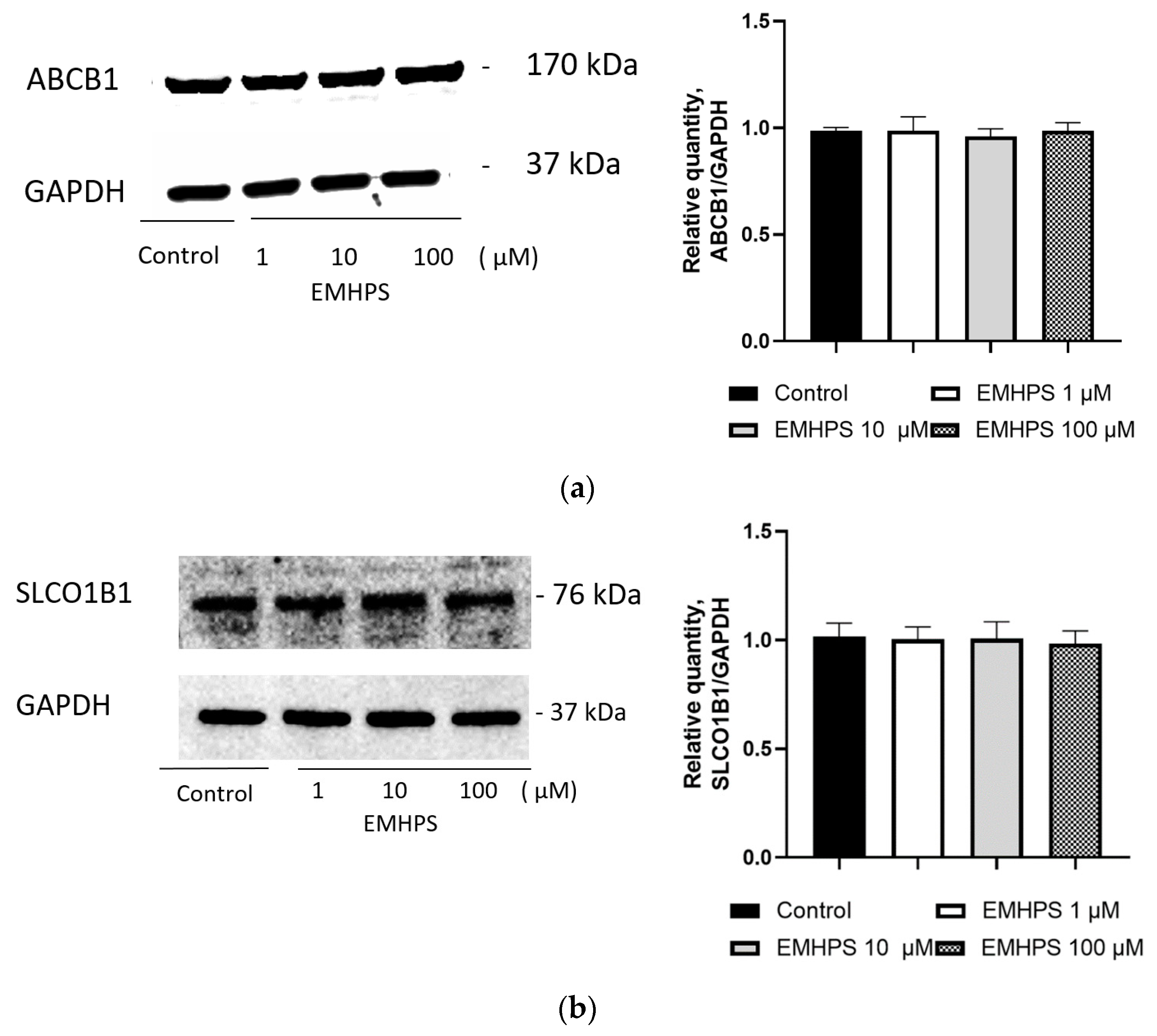
2.6. Effect of EMHPS on Expression of ABCB1 and SLCO1B1
3. Discussion
4. Materials and Methods
4.1. Caco-2 Cell Culture
4.2. HEK293-SLCO1B1 Cell Line
4.3. HepG2 Cell Line
4.4. Cytotoxicity (MTT) Assay
4.5. Transport Experiments on Caco-2 Cells
4.6. Uptake Experiments
4.7. Fexofenadine HPLC-UV Analysis
4.8. Atorvastatin LC-MS/MS Analysis
4.9. EMHPS HPLC-UV Analysis
4.10. EMHPS LC-MS/MS Analysis
4.11. Effect of EMHPS on the Expression of ABCB1 and SLCO1B1
4.12. Western Blot
4.13. Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Voronina, T.A. Antioxidant Mexidol. The Main Neuropsychotropic Effects and the Mechanism of Action; Institute of Pharmacology, Russian Academy of Medical Sciences: Moscow, Russia, 2013. [Google Scholar]
- Gupta, D.S.; Parab, S.B.; Kaur, G. Promising effects of emoxypine and its succinate derivative in the management of various diseases-with insights on recent patent applications. Curr. Res. Pharmacol. Drug Discov. 2022, 3, 100121. [Google Scholar] [CrossRef]
- Hoa, N.T.; Van Bay, M.; Mechler, A.; Vo, Q.V. Theoretical insights into the antiradical activity and copper-catalysed oxidative damage of mexidol in the physiological environment. R Soc. Open Sci. 2022, 9, e211239. [Google Scholar] [CrossRef]
- Semikasheva, O.V.; Yakupova, L.R.; Borisov, I.M.; Safiullin, R.L. Quantitative Analysis of the Antioxidant Activity of Mexidol. Pharm. Chem. J. 2021, 54, 1282–1285. [Google Scholar] [CrossRef]
- Karmanova, E.E.; Chernikov, A.V.; Usacheva, A.M.; Bruskov, V.I. Antioxidant and Gene-Protective Properties of Ethylmethylhydroxypyridine Succinate (Mexidol) in X-Ray Irradiation. Pharm. Chem. J. 2020, 54, 673–677. [Google Scholar] [CrossRef]
- Lukyanova, L.D.; Germanova, E.L.; Tsybina, T.A.; Chernobaeva, G.N. Energotropic effect of succinate-containing derivatives of 3-hydroxypyridine. Bull. Exp. Biol. Med. 2009, 148, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Hamel, D.; Sanchez, M.; Duhamel, F.; Roy, O.; Honoré, J.-C.; Noueihed, B.; Zhou, T.; Nadeau-Vallée, M.; Hou, X.; Lavoie, J.-C.; et al. G-Protein–Coupled Receptor 91 and Succinate Are Key Contributors in Neonatal Postcerebral Hypoxia-Ischemia Recovery. Arter. Thromb. Vasc. Biol. 2014, 34, 285–293. [Google Scholar] [CrossRef]
- Stakhovskaya, L.V.; Shamalov, N.A.; Khasanova, D.R.; Mel’nikova, E.V.; Agaf’ina, A.S.; Golikov, K.V.; Bogdanov, E.I.; Yakupova, A.A.; Roshkovskaya, L.V.; Lukinykh, L.V.; et al. Results of a Randomized, Double-Blind, Multicenter, Placebo-Controlled, Parallel-Group Study of the Efficacy and Safety of Mexidol in Prolonged Sequential Therapy of Patients in the Acute and Early Recovery Stages of Hemispheric Stroke (the EPICA study). Neurosci. Behav. Physiol. 2018, 48, 929–938. [Google Scholar] [CrossRef]
- Fedin, A.; Zakharov, V.; Tanashyan, M.; Chukanova, E.; Madzhidova, E.; Shchepankevich, L.; Ostroumova, O. Results of an international multicenter, randomized, double-blind, placebo-controlled study assessing the efficacy and safety of sequential therapy with Mexidol and Mexidol FORTE 250 in patients with chronic brain ischemia (MEMO). Zhurnal Nevrol. I Psikhiatrii Im. SS Korsakova 2021, 121, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Shchulkin, A.; Kazakhmedov, E.; Galochkin, S.; Tolkacheva, V.; Kobalava, Z. Effects of mexidol in patients with chronic brain ischemia and chronic heart failure (II-III functional class). Kardiol. I Serdechno-Sosud. Khirurgiya 2020, 13, 427–434. [Google Scholar] [CrossRef]
- Baranov, P.A.; Appolonova, S.A.; Rodchenkov, G.M.; Sariev, A.K.; Zherdev, V.P. Effect of mexidol on 6beta/free hydroxycortisol ratio. Possibility of CYP3A4 activation. Eksperimental’naia I Klin. Farmakol. 2010, 73, 39–40. [Google Scholar]
- Schulkin, A.V. Mexidol: Modern aspects of the pharmacokinetics and pharmacodynamics. Farmateka 2016, 4–16, 65–71. [Google Scholar]
- Nigam, S.K. What do drug transporters really do? Nat. Rev. Drug Discov. 2014, 14, 29–44. [Google Scholar] [CrossRef]
- Vasiliou, V.; Vasiliou, K.; Nebert, D.W. Human ATP-binding cassette (ABC) transporter family. Hum. Genom. 2008, 3, 281. [Google Scholar] [CrossRef]
- Sauna, Z.E.; Ambudkar, S.V. About a switch: How P-glycoprotein (ABCB1) harnesses the energy of ATP binding and hydrolysis to do mechanical work. Mol. Cancer Ther. 2007, 6, 13–23. [Google Scholar] [CrossRef] [PubMed]
- König, J.; Müller, F.; Fromm, M.F. Transporters and Drug-Drug Interactions: Important Determinants of Drug Disposition and Effects. Pharmacol. Rev. 2013, 65, 944–966. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.H. P-Glycoprotein, a gatekeeper in the blood–brain barrier. Adv. Drug Deliv. Rev. 1999, 36, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Amin, L. P-glycoprotein Inhibition for Optimal Drug Delivery. Drug Target Insights 2013, 7, DTI-S12519. [Google Scholar] [CrossRef]
- Funk, C. The role of hepatic transporters in drug elimination. Expert Opin. Drug Metab. Toxicol. 2008, 4, 363–379. [Google Scholar] [CrossRef]
- Simonson, S.G.; Raza, A.; Martin, P.D.; Mitchell, P.D.; Jarcho, J.A.; Brown, C.D.A.; Windass, A.S.; Schneck, D.W. Rosuvastatin pharmacokinetics in heart transplant recipients administered an antirejection regimen including cyclosporine. Clin. Pharmacol. Ther. 2004, 76, 167–177. [Google Scholar] [CrossRef]
- Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER). In Vitro Drug Interaction Studies—Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry. Clin. Pharmacol. 2020, 1–43. Available online: https://www.fda.gov/media/134582/download (accessed on 10 May 2023).
- Voronina, T.A.; Ivanova, E.A. Combined administration of mexidol with known medicines. Zhurnal Nevrol. I Psikhiatrii Im. S.S. Korsakova 2019, 119, 115–124. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Chufan, E.E.; Sim, H.M.; Ambudkar, S.V. Molecular Basis of the Polyspecificity of P-Glycoprotein (ABCB1). Adv. Cancer Res. 2015, 125, 71–96. [Google Scholar] [CrossRef] [PubMed]
- Garrison, D.A.; Talebi, Z.; Eisenmann, E.D.; Sparreboom, A.; Baker, S.D. Role of OATP1B1 and OATP1B3 in Drug-Drug Interactions Mediated by Tyrosine Kinase Inhibitors. Pharmaceutics 2020, 12, 856. [Google Scholar] [CrossRef] [PubMed]
- Nagayasu, M.; Ozeki, K.; Sakurai, Y.; Tsutsui, H.; Onoue, S. Simplified Method to Determine the Efflux Ratio on P-Glycoprotein Substrates Using Three-Compartment Model Analysis for Caco-2 Cell Assay Data. Pharm Res. 2020, 37, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Scialis, R.J.; Feng, B.; Leach, K. Detection of Statin Cytotoxicity Is Increased in Cells Expressing the OATP1B1 Transporter. Toxicol. Sci. 2013, 134, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Danielson, M.L.; Sawada, G.A.; Raub, T.J.; Desai, P.V. In Silico and in Vitro Assessment of OATP1B1 Inhibition in Drug Discovery. Mol Pharm. 2018, 15, 3060–3068. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Y.D.; Strong, J.M.; Reynolds, K.S.; Huang, S.M. A regulatory viewpoint on transporter-based drug interactions. Xenobiotica 2008, 38, 709–724. [Google Scholar] [CrossRef]
- Wessler, J.D.; Grip, L.T.; Mendell, J.; Giugliano, R.P. The P-glycoprotein transport system and cardiovascular drugs. J Am Coll Cardiol. 2013, 61, 2495–2502. [Google Scholar] [CrossRef]
- Ferry, D.R.; Russell, M.A.; Cullen, M.H. P-glycoprotein possesses A 1,4-dihydropyridine-selective drug acceptor site which is alloserically coupled to a vinca-alkaloid-selective binding site. Biochem. Biophys. Res. Commun. 1992, 188, 440–445. [Google Scholar] [CrossRef]
- Ferreira, R.J.; Ferreira, M.J.U.; dos Santos, D.J.V.A. Molecular Docking Characterizes Substrate-Binding Sites and Efflux Modulation Mechanisms within P-Glycoprotein. J. Chem. Inf. Model 2013, 53, 1747–1760. [Google Scholar] [CrossRef]
- Dastvan, R.; Mishra, S.; Peskova, Y.B.; Nakamoto, R.K.; Mchaourab, H.S. Mechanism of allosteric modulation of P-glycoprotein by transport substrates and inhibitors. Science 2019, 364, 689–692. [Google Scholar] [CrossRef]
- Artursson, P.; Palm, K.; Luthman, K. Caco-2 monolayers in experimental and theoretical predictions of drug transport. Adv. Drug Deliv. Rev. 2001, 46, 27–43. [Google Scholar] [CrossRef]
- Tolosa, L.; Donato, M.T.; Gómez-Lechón, M.J. General cytotoxicity assessment by means of the MTT assay. Methods Mol. Biol. 2015, 1250, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Van Breemen, R.B.; Li, Y. Caco-2 cell permeability assays to measure drug absorption. Expert Opin. Drug Metab. Toxicol. 2005, 1, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Yakusheva, E.N.; Chernyh, I.V.; Shchulkin, A.V.; Gatsanoga, M.V. Design of HPLC methods of fexofenadine quantitative analysis in blood plasma. Pharmacokinet. Pharmacodyn. 2017, 2, 35–38. [Google Scholar]
- Erokhina, P.D.; Myl’nikov, P.Y.; Ganina, S.O.; Konyakhin, E.A.; Shchulkin, A.V.; Slepnev, A.A.; Yakusheva, E.N. Development and Validation of the Quantitative Determination of Atorvastatin in HepG2 Cell Line Using High-Performance Liquid Chromatography with Mass-Spectrometric Detection. IP Pavlov Rus. Med. Biol. Her. 2022, 30, 149–158. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | 10 µM | 100 µM | 250 µM |
|---|---|---|---|
| Papp b-a | 7.56 × 10−6 ± 2.98 × 10−6 | 6.63 × 10−6 ± 2.15 × 10−6 | 1.65 × 10−5 ± 4.16 × 10−6 |
| Papp a-b | 7.66 × 10−6 ± 1.71 × 10−6 | 7.05 × 10−6 ± 2.15 × 10−6 | 2.23 × 10−5 ± 1.99 × 10−6 |
| ER | 0.99 ± 0.323 | 0.94 ± 0.039 | 0.75 ± 0.26 |
| Concentration of EMHPS | Papp b-a, cm/s | Papp a-b, cm/s | ER |
|---|---|---|---|
| Control | 3.64 × 10−6 ± 0.21 × 10−6 | 0.61 × 10−6 ± 0.21 × 10−6 | 6.39 ± 2.04 |
| 1 µM | 4.2 × 10−6 ± 0.3 × 10−6 | 1.1 × 10−6 ± 0.27 × 10−6 | 3.94 ± 0.75 |
| 10 µM | 4.15 × 10−6 ± 0.67 × 10−6 | 1.22 × 10−6 ± 0.29 × 10−6 | 3.64 ± 1.50 |
| 50 µM | 3.61 × 10−6 ± 1.19 × 10−6 | 1.29 × 10−6 ± 0.87 × 10−6 | 3.18 ± 0.89 * |
| 100 µM | 2.57 × 10−6 ± 0.27 × 10−6 | 1.54 × 10−6 ± 0.4 × 10−6 | 1.73 ± 0.37 ** |
| 200 µM | 1.73 × 10−6 ± 0.18 × 10−6 * | 2.9 × 10−6 ± 0.29 × 10−6* | 0.6 ± 0.12 *** |
| Concentration of EMHPS | Papp b-a, cm/s | Papp a-b, cm/s | ER |
|---|---|---|---|
| Control | 3.64 × 10−6 ± 0.21 × 10−6 | 0.61 × 10−6 ± 0.21⋅10−6 | 6.39 ± 2.04 |
| 1 µM | 3.45 × 10−6 ± 0.43 × 10−6 | 0.65 × 10−6 ± 0.13 × 10−6 | 5.55 ± 1.86 |
| 10 µM | 3.75 × 10−6 ± 0.19 × 10−6 | 0.66 × 10−6 ± 0.12 × 10−6 | 5.79 ± 0.97 |
| 50 µM | 4.06 × 10−6 ± 0.21 × 10−6 | 0.73 × 10−6 ± 0.04 × 10−6 | 5.5 ± 0.35 |
| 100 µM | 3.39 × 10−6 ± 0.38 × 10−6 | 0.52 × 10−6 ± 0.11 × 10−6 | 6.69 ± 1.53 |
| 200 µM | 3.98 × 10−6 ± 0.65 × 10−6 | 0.64 × 10−6 ± 0.11 × 10−6 | 6.41 ± 1.89 |
| Concentration of Verapamil | Papp b-a, cm/s | Papp a-b, cm/s | ER |
|---|---|---|---|
| Control | 3.64 × 10−6 ± 0.21 × 10−6 | 0.61 × 10−6 ± 0.21 × 10−6 | 6.39 ± 2.04 |
| 1 µM | 3.89 × 10−6 ± 0.80 × 10−6 | 0.76 × 10−6 ± 0.26 × 10−6 | 5.29 ± 0.77 |
| 10 µM | 3.2 × 10−6 ± 0.5 × 10−6 | 0.85 × 10−6 ± 0.31 × 10−6 | 4.01 ± 1.56 |
| 50 µM | 1.47 × 10−6 ± 0.25 × 10−6 *** | 0.55 × 10−6 ± 0.2 × 10−6 | 2.81 ± 0.69 * |
| 100 µM | 1.15 × 10−6 ± 0.4 × 10−6 *** | 0.67 × 10−6 ± 0.15 × 10−6 | 1.82 ± 0.98 ** |
| 200 µM | 1.23 × 10−6 ± 0.19 × 10−6 *** | 1.46 × 10−6 ± 0.43 × 10−6 * | 0.89 ± 0.26 ** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shchulkin, A.V.; Erokhina, P.D.; Goncharenko, A.V.; Mylnikov, P.Y.; Chernykh, I.V.; Abalenikhina, Y.V.; Kotliarova, M.S.; Yakusheva, E.N. Ethylmethylhydroxypyridine Succinate Is an Inhibitor but Not a Substrate of ABCB1 and SLCO1B1. Pharmaceuticals 2023, 16, 1529. https://doi.org/10.3390/ph16111529
Shchulkin AV, Erokhina PD, Goncharenko AV, Mylnikov PY, Chernykh IV, Abalenikhina YV, Kotliarova MS, Yakusheva EN. Ethylmethylhydroxypyridine Succinate Is an Inhibitor but Not a Substrate of ABCB1 and SLCO1B1. Pharmaceuticals. 2023; 16(11):1529. https://doi.org/10.3390/ph16111529
Chicago/Turabian StyleShchulkin, Aleksey V., Pelageya D. Erokhina, Anna V. Goncharenko, Pavel Yu. Mylnikov, Ivan V. Chernykh, Yulia V. Abalenikhina, Maria S. Kotliarova, and Elena N. Yakusheva. 2023. "Ethylmethylhydroxypyridine Succinate Is an Inhibitor but Not a Substrate of ABCB1 and SLCO1B1" Pharmaceuticals 16, no. 11: 1529. https://doi.org/10.3390/ph16111529
APA StyleShchulkin, A. V., Erokhina, P. D., Goncharenko, A. V., Mylnikov, P. Y., Chernykh, I. V., Abalenikhina, Y. V., Kotliarova, M. S., & Yakusheva, E. N. (2023). Ethylmethylhydroxypyridine Succinate Is an Inhibitor but Not a Substrate of ABCB1 and SLCO1B1. Pharmaceuticals, 16(11), 1529. https://doi.org/10.3390/ph16111529