
In Vitro and In Vivo Effects of Synthesis Novel Phenoxyacetamide Derivatives as Potent Apoptotic Inducer against HepG2 Cells through PARP-1 Inhibition

 ,
,  ,
,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
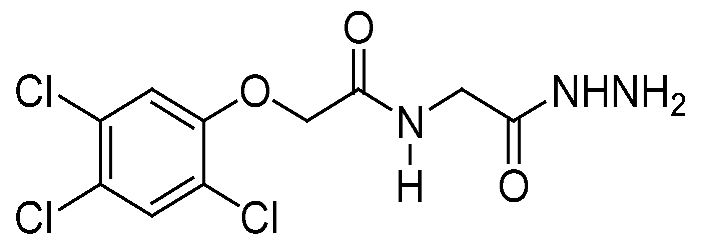
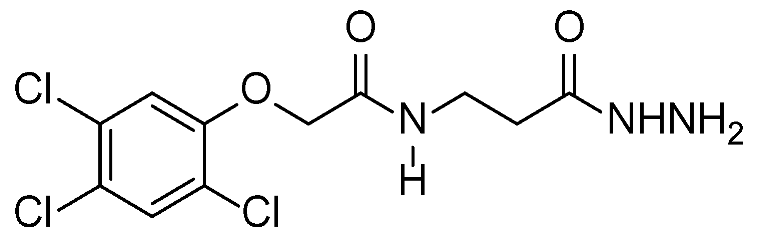
2.2. Cytotoxicity against Different Cell Lines
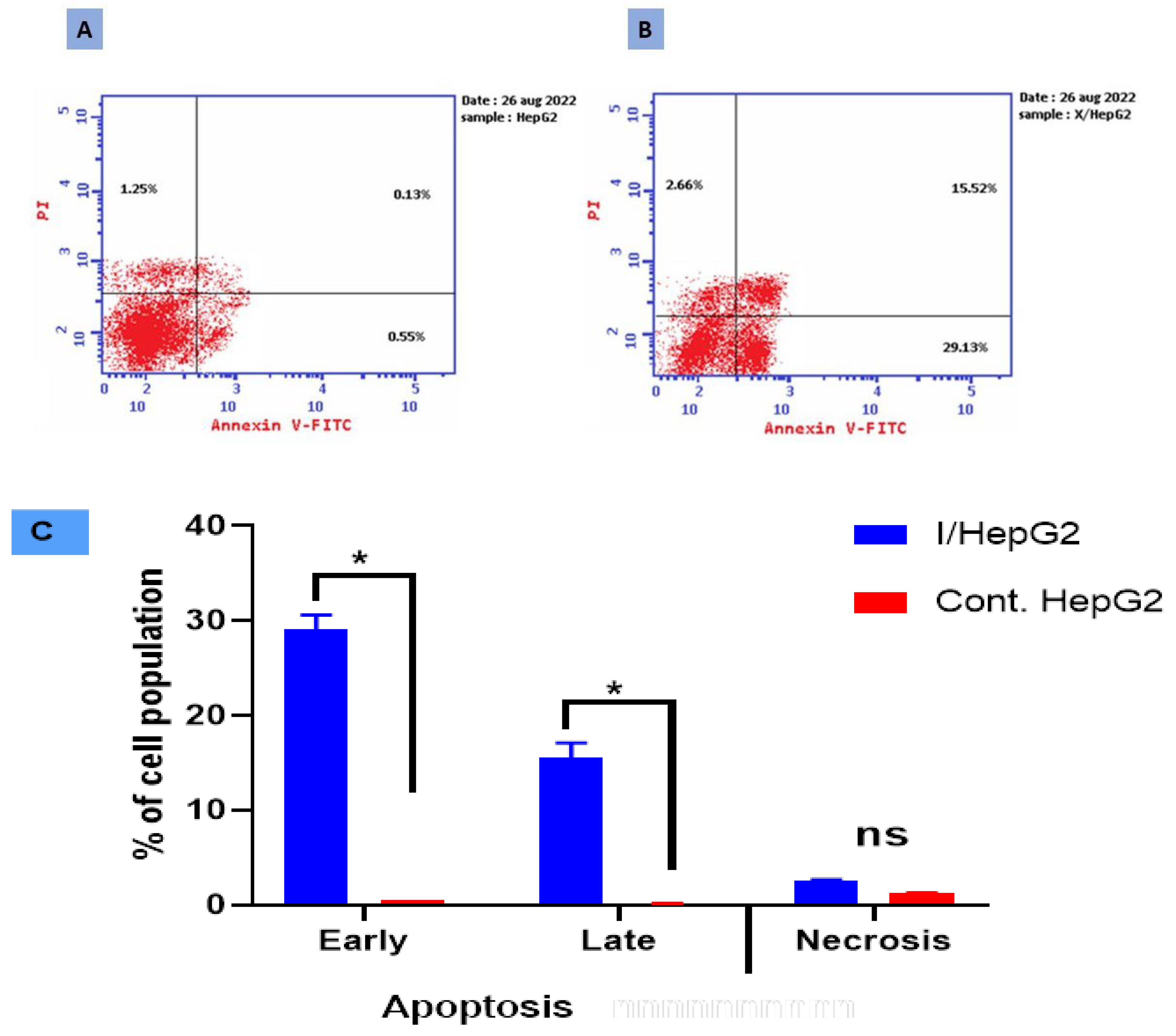
2.3. Apoptotic Evaluation
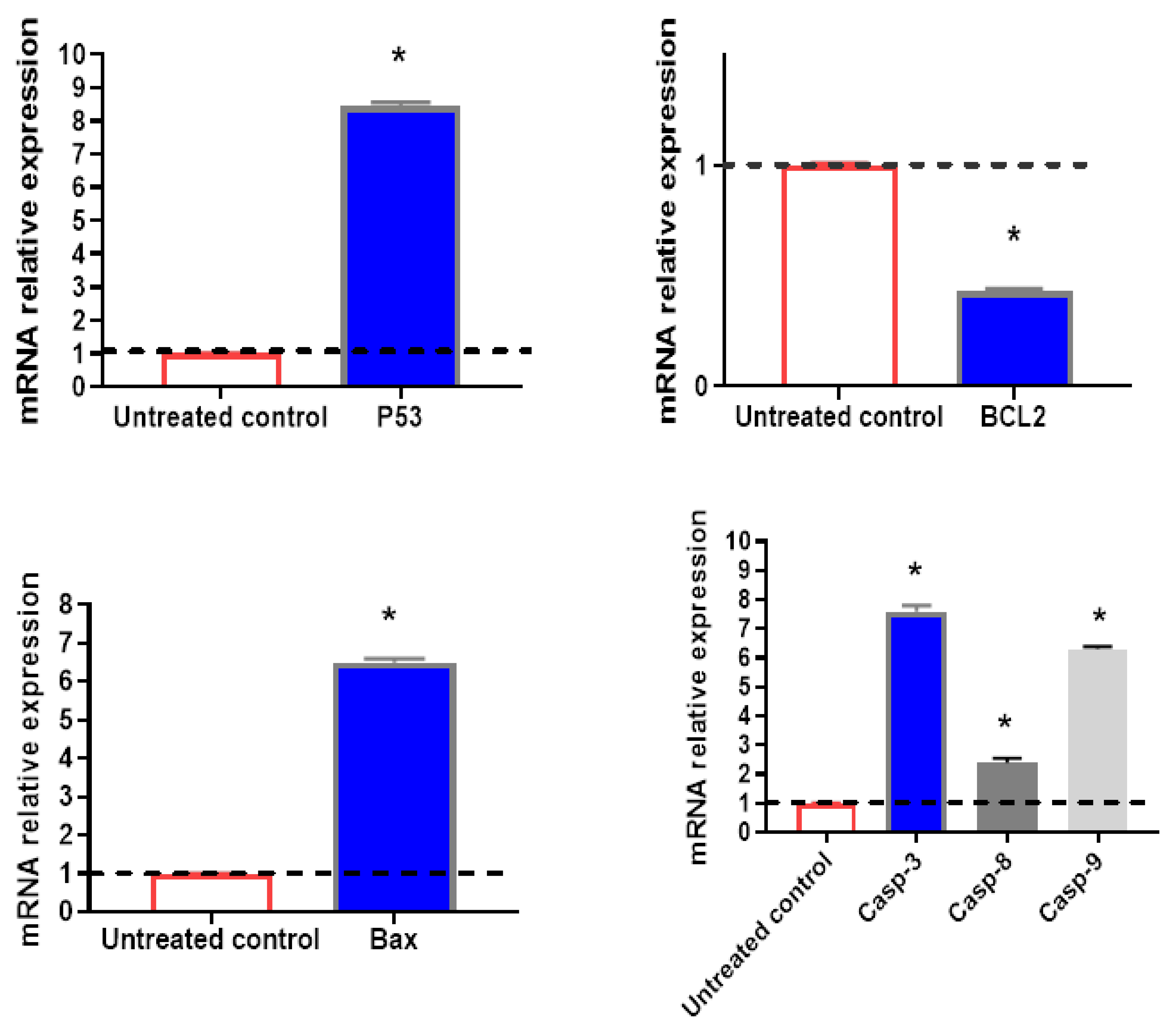
2.4. Pro-Apoptotic and Anti-Apoptotic Genes Using RT-PCR
2.5. In Vivo Results
2.5.1. Antitumor Assays
2.5.2. Hematological Assays
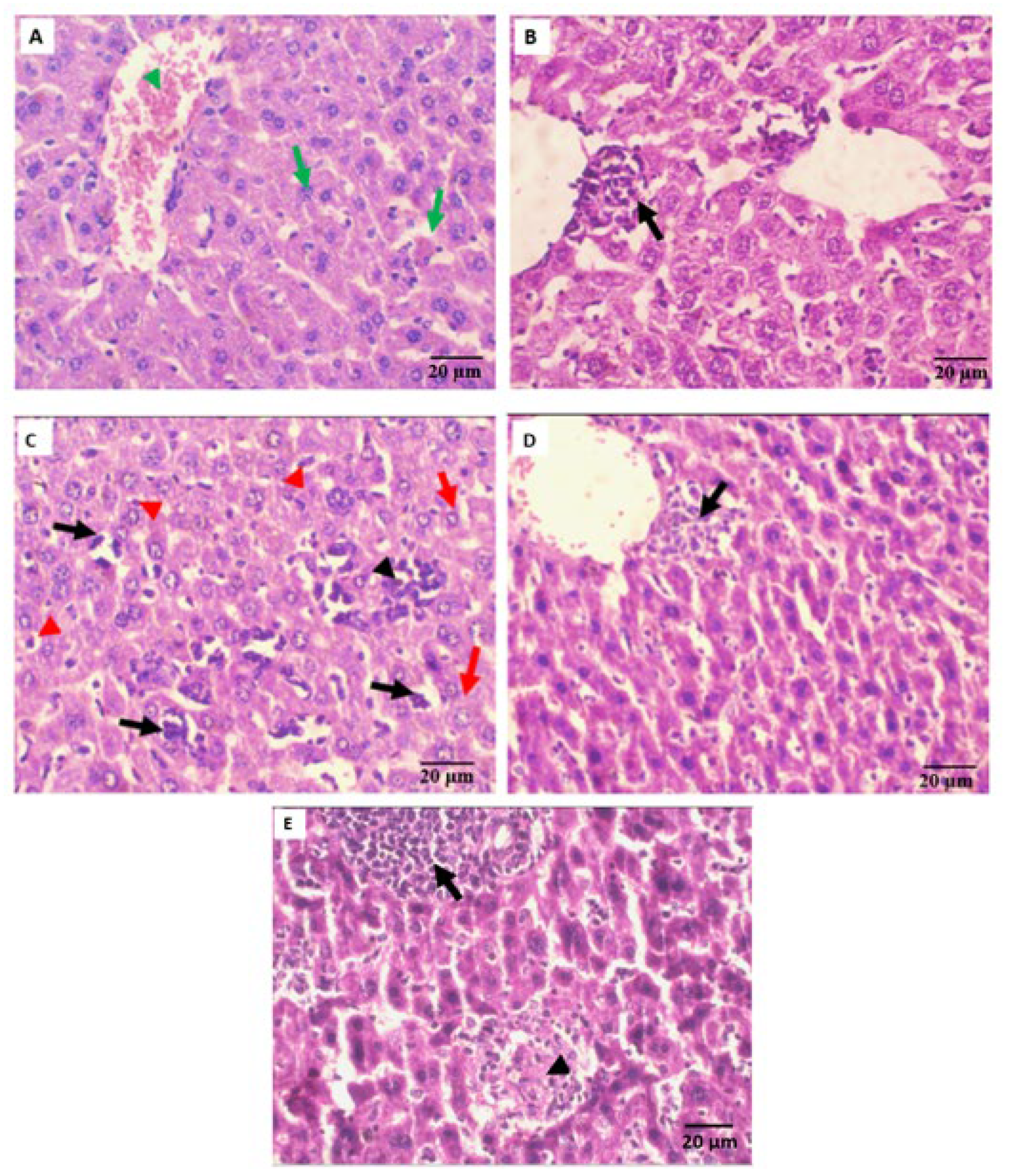
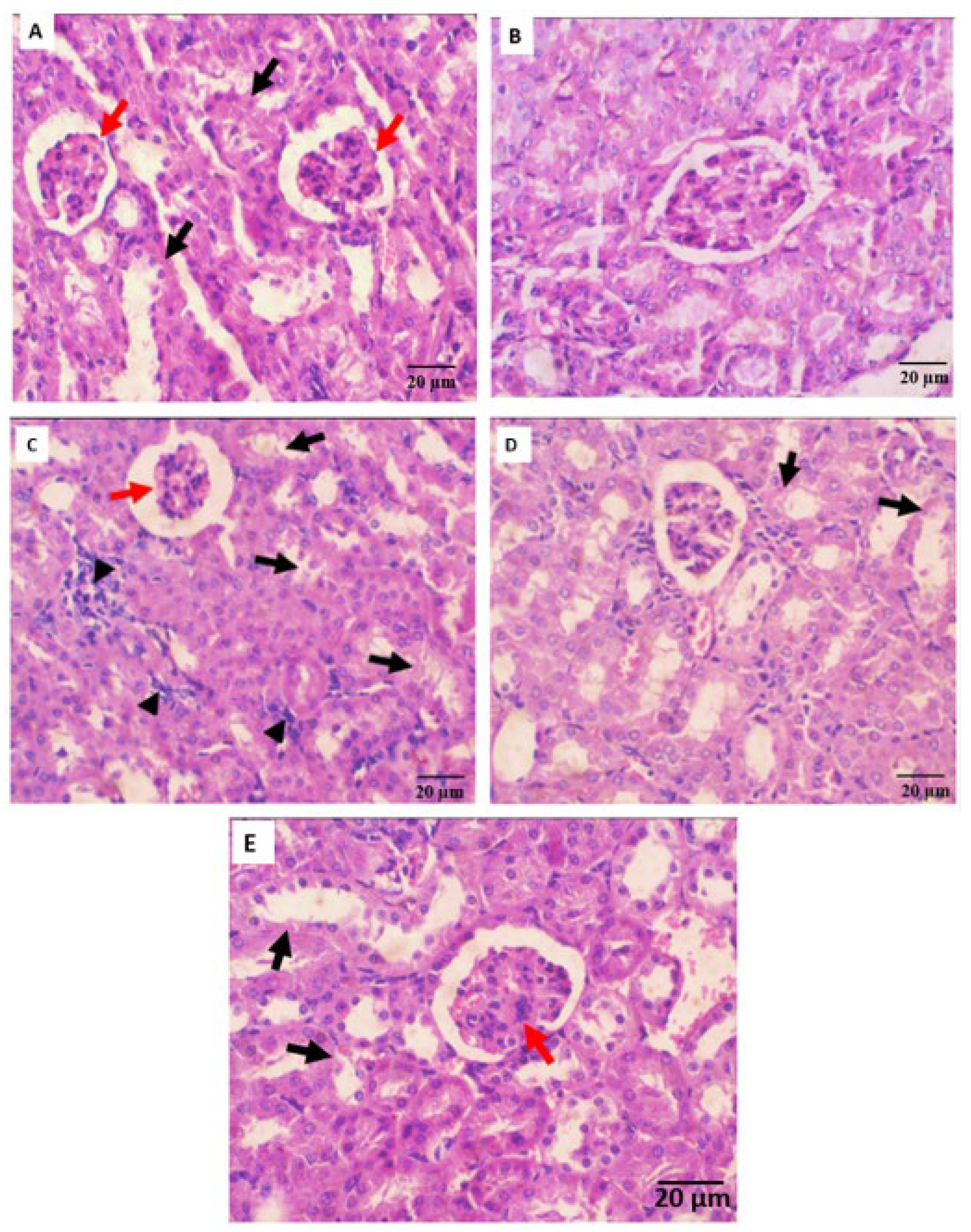
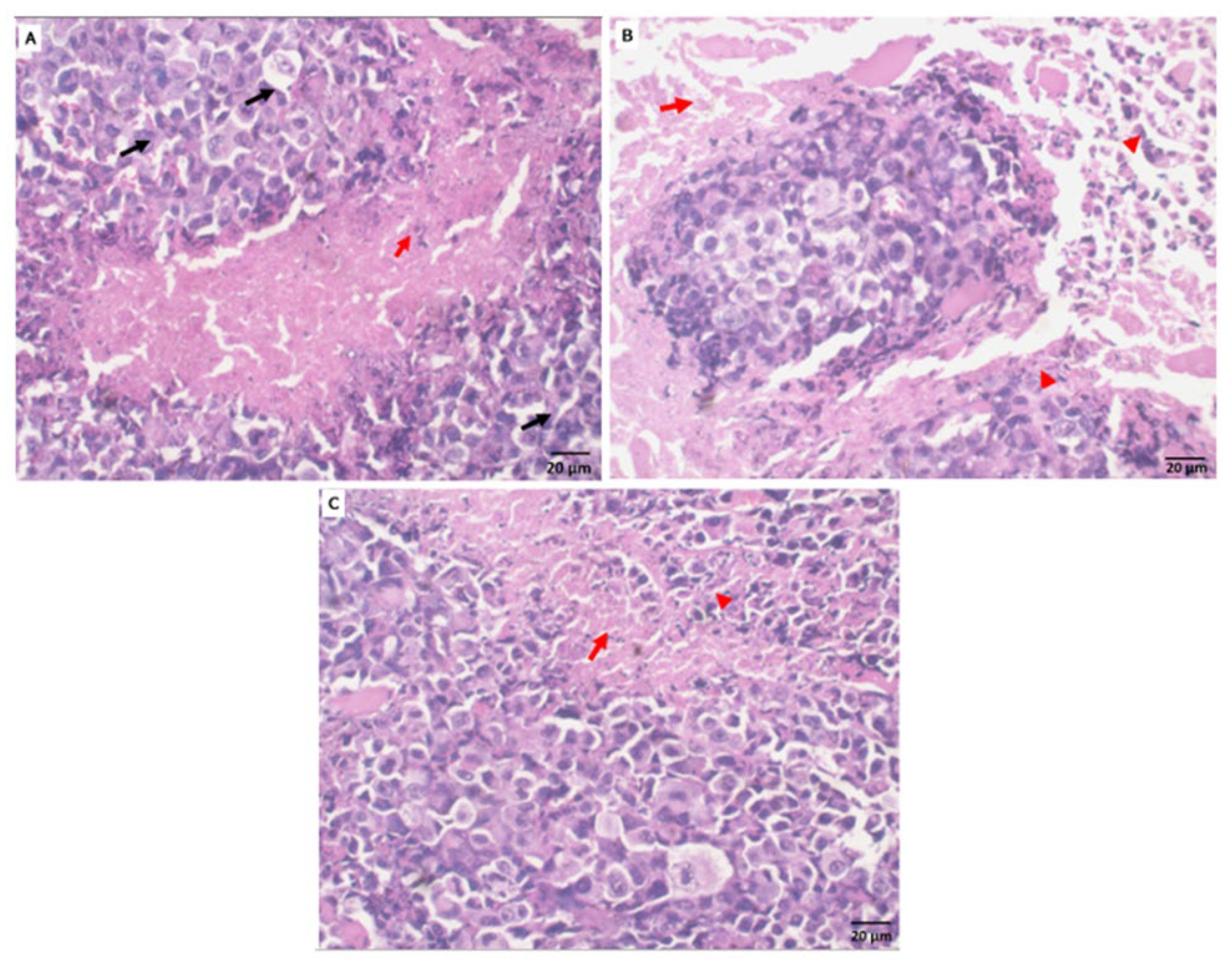
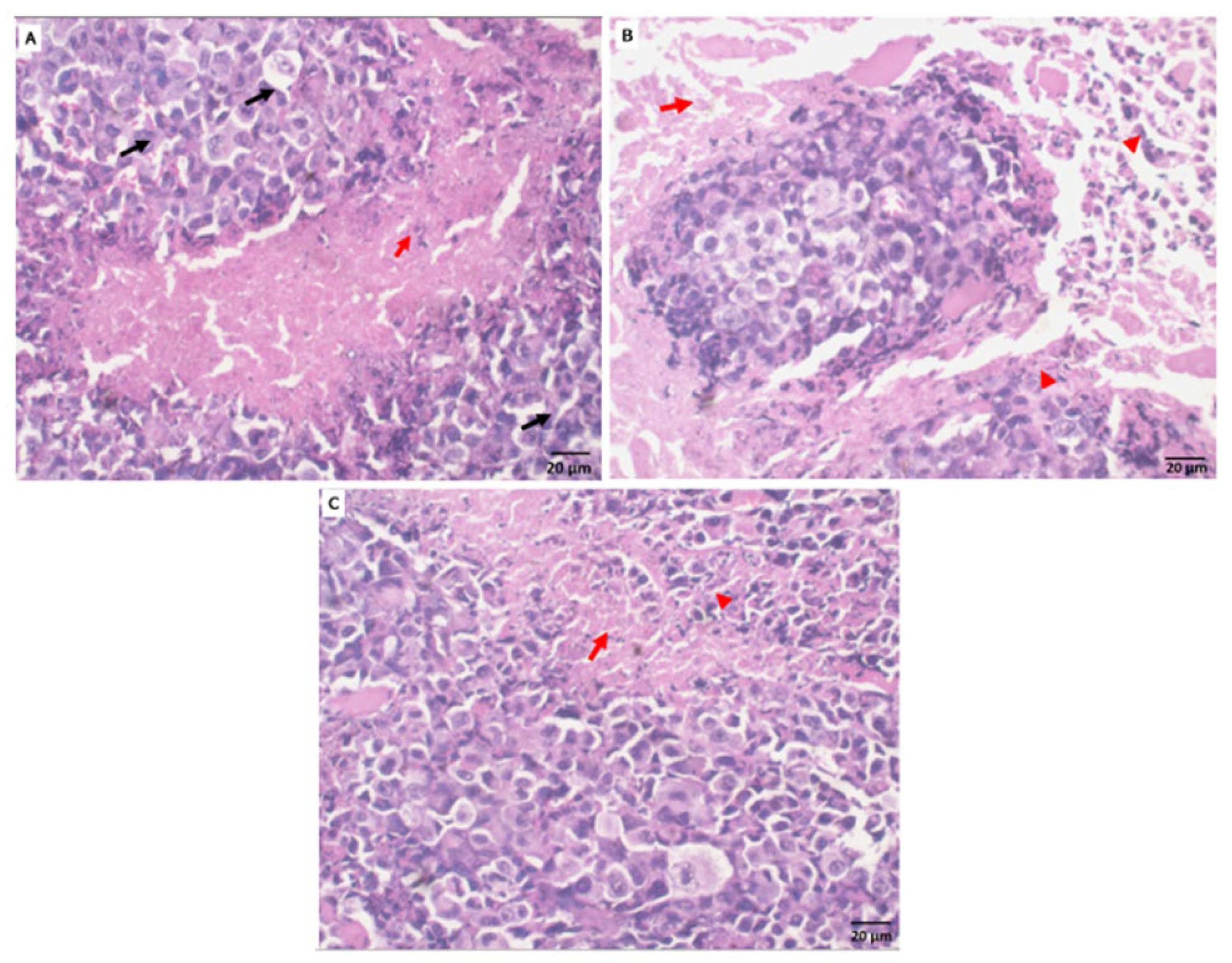
2.5.3. Histopathological Observation
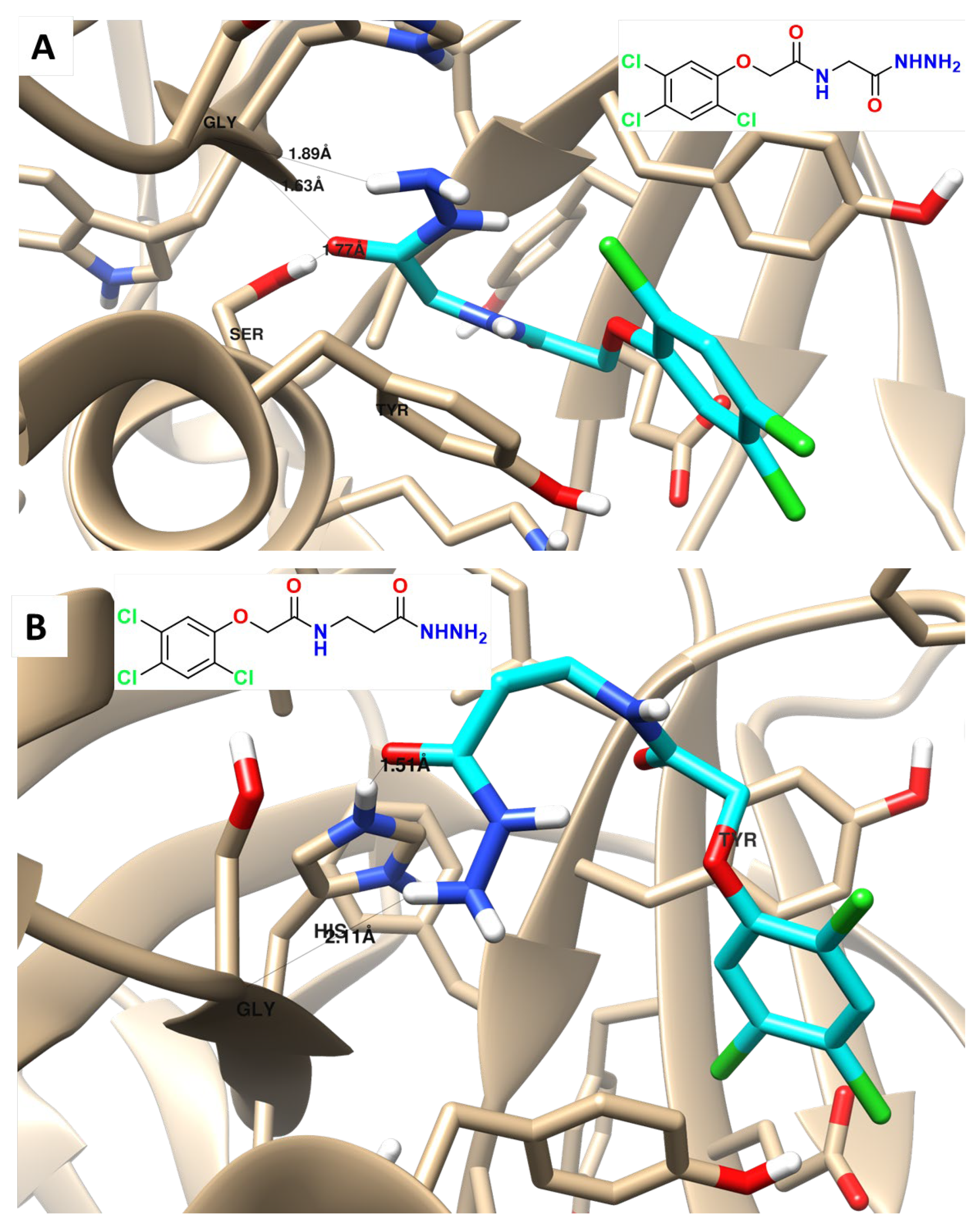
2.6. Molecular Docking
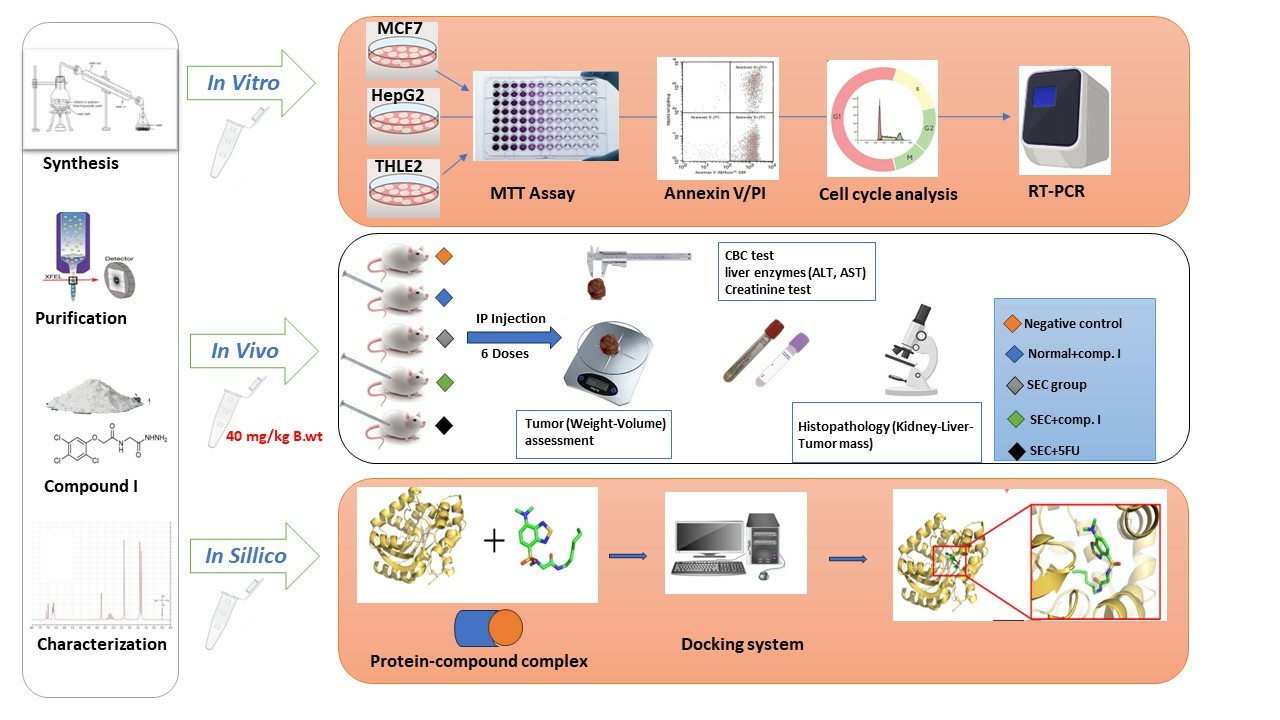
3. Materials and Methods
3.1. Chemistry
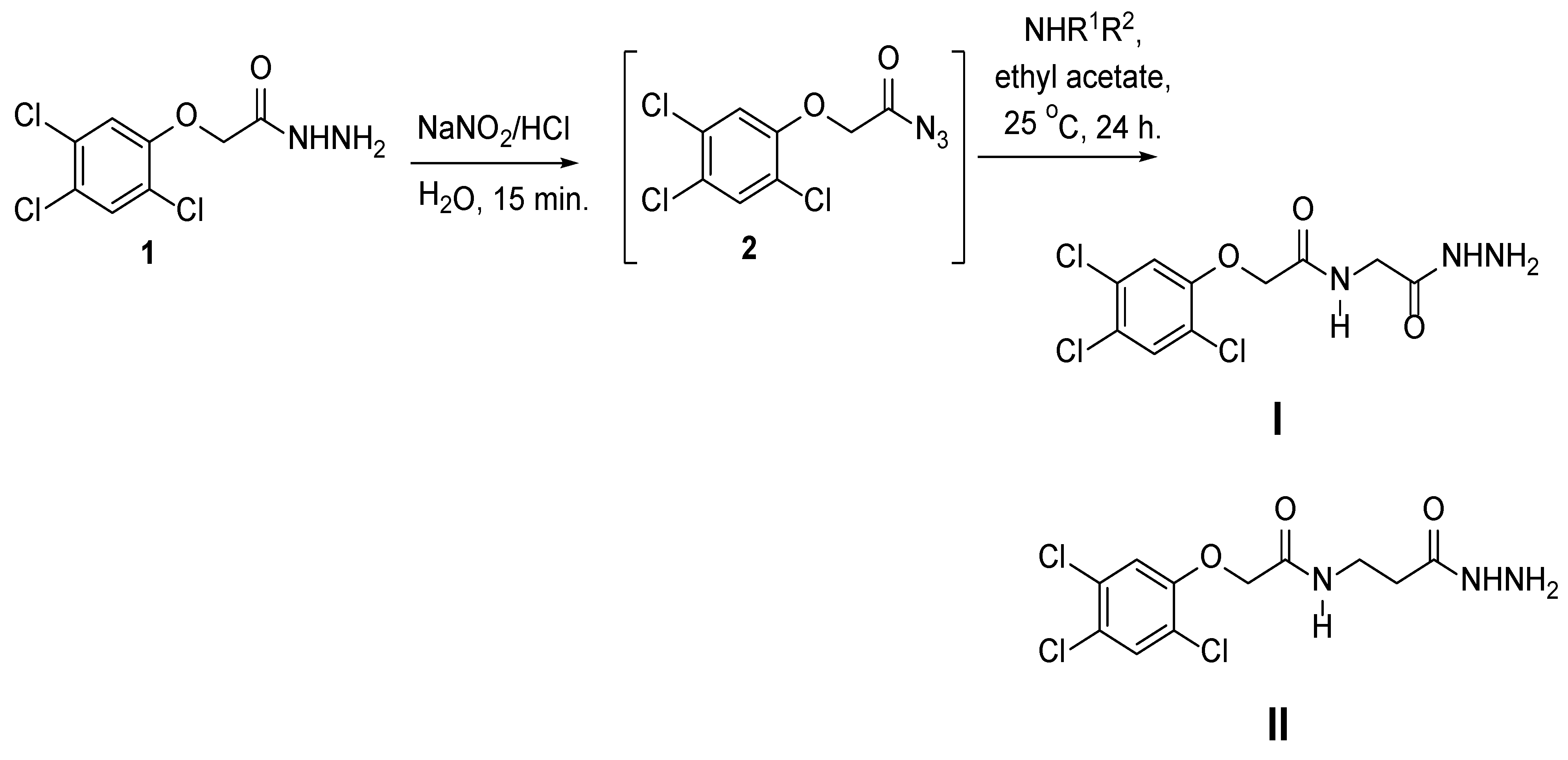
3.1.1. Method for Preparation of Phenoxy Acetamide Derivatives
3.1.2. Synthesis of 2,4,5-Trichloro-Phenoxyacetic Acid Hydrazide (1)
3.2. Cytotoxic Activity
3.3. Apoptotic Evaluation
3.4. Annexin V/PI and Cell Cycle
3.5. Real-Time Polymerase Chain Reaction (RT-PCR)
3.6. In Vivo Anti-Tumor Study
3.6.1. Experimental Design
3.6.2. Hematological, Biochemical, and Histopathological Assessments
3.7. PARP-1 Inhibition and Molecular Docking
3.8. Research Ethics Consideration
3.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Chan, L.K.; Tsui, Y.M.; Ho, D.W.H.; Ng, I.O.L. Cellular heterogeneity and plasticity in liver cancer. Semin. Cancer Biol. 2022, 82, 134–149. [Google Scholar] [CrossRef]
- Hawash, M.; Qaoud, M.T.; Jaradat, N.; Abdallah, S.; Issa, S.; Adnan, N.; Hoshya, M.; Sobuh, S.; Hawash, Z. Anticancer Activity of Thiophene Carboxamide Derivatives as CA-4 Biomimetics: Synthesis, Biological Potency, 3D Spheroid Model, and Molecular Dynamics Simulation. Biomimetics 2022, 7, 247. [Google Scholar] [CrossRef] [PubMed]
- Lazzarotto-da-Silva, G.; Grezzana-Filho, T.J.; Scaffaro, L.A.; Farenzena, M.; Silva, R.K.; de Araujo, A.; Arruda, S.; Feier, F.H.; Prediger, L.; Lazzaretti, G.S.; et al. Percutaneous ethanol injection is an acceptable bridging therapy to hepatocellular carcinoma prior to liver transplantation. Langenbecks Arch. Surg. 2023, 408, 26. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Feng, J.; Liu, W.; Wen, C.; Wu, Y.; Liao, Q.; Zou, L.; Sui, X.; Xie, T.; Zhang, J.; et al. Opportunities and challenges for co-delivery nanomedicines based on combination of phytochemicals with chemotherapeutic drugs in cancer treatment. Adv. Drug Deliv. Rev. 2022, 188, 114445. [Google Scholar] [CrossRef]
- Hawash, M.; Ergun, S.G.; Kahraman, D.C.; Olgac, A.; Hamel, E.; Cetin-Atalay, R.; Baytas, S.N. Novel Indole-Pyrazole Hybrids as Potential Tubulin-Targeting Agents; Synthesis, antiproliferative evaluation, and molecular modeling studies. J. Mol. Struct. 2023, 1285, 135477. [Google Scholar] [CrossRef] [PubMed]
- Al-Ostoot, F.H.; Zabiulla, S.; Khanum, S.A. Recent investigations into synthesis and pharmacological activities of phenoxy acetamide and its derivatives (chalcone, indole, and quinoline) as possible therapeutic candidates. J. Iran. Chem. Soc. 2021, 18, 1839–1875. [Google Scholar] [CrossRef]
- Gull, Y.; Rasool, N.; Noreen, M.; Altaf, A.A.; Musharraf, S.G.; Zubair, M.; Nasim, F.U.H.; Yaqoob, A.; DeFeo, V.; Zia-Ul-Haq, M. Synthesis of N-(6-Arylbenzo [d] thiazole-2-acetamide derivatives and their biological activities: An experimental and computational approach. Molecules 2016, 21, 266. [Google Scholar] [CrossRef] [PubMed]
- Khazir, J.; Mir, B.A.; Chashoo, G.; Maqbool, T.; Riley, D.; Pilcher, L. Design, synthesis, and anticancer evaluation of acetamide and hydrazine analogues of pyrimidine. J. Heterocycl. Chem. 2020, 57, 1306–1318. [Google Scholar] [CrossRef]
- Al-Ostoot, F.H.; Sherapura, A.; Vigneshwaran, V.; Basappa, G.; Vivek, H.K.; Prabhakar, B.T.; Khanum, S.A. Targeting HIF-1α by newly synthesized Indolephenoxyacetamide (IPA) analogs to induce anti-angiogenesis-mediated solid tumor suppression. Pharmacol. Rep. 2021, 73, 1328–1343. [Google Scholar] [CrossRef]
- Arshad, M.F.; Alam, A.; Alshammari, A.A.; Alhazza, M.B.; Alzimam, I.M.; Alam, M.A.; Mustafa, G.; Ansari, M.S.; Alotaibi, A.M.; Alotaibi, A.A.; et al. Thiazole: A versatile standalone moiety contributing to the development of various drugs and biologically active agents. Molecules 2022, 27, 3994. [Google Scholar] [CrossRef]
- Kozyra, P.; Pitucha, M. Terminal phenoxy group as a privileged moiety of the drug scaffold—A short review of most recent studies 2013–2022. Int. J. Mol. Sci. 2022, 23, 8874. [Google Scholar] [CrossRef]
- Palakhachane, S.; Ketkaew, Y.; Chuaypen, N.; Sirirak, J.; Boonsombat, J.; Ruchirawat, S.; Tangkijvanich, P.; Suksamrarn, A.; Limpachayaporn, P. Synthesis of sorafenib analogues incorporating a 1, 2, 3-triazole ring and cytotoxicity towards hepatocellular carcinoma cell lines. Bioorg. Chem. 2021, 112, 104831. [Google Scholar] [CrossRef]
- Čermáková, L.; Hofman, J.; Laštovičková, L.; Havlíčková, L.; Špringrová, I.; Novotná, E.; Wsól, V. Bruton’s Tyrosine Kinase Inhibitor Zanubrutinib Effectively Modulates Cancer Resistance by Inhibiting Anthracycline Metabolism and Efflux. Pharmaceutics 2022, 14, 1994. [Google Scholar] [CrossRef]
- Berglöf, A.; Hamasy, A.; Meinke, S.; Palma, M.; Krstic, A.; Månsson, R.; Kimby, E.; Österborg, A.; Smith, C.I.E. Targets for Ibrutinib Beyond B Cell Malignancies. Scand. J. Immunol. 2015, 82, 208–217. [Google Scholar] [CrossRef]
- Kasonde, L.; Tordrup, D.; Naheed, A.; Zeng, W.; Ahmed, S.; Babar, Z.U.D. Evaluating medicine prices, availability, and affordability in Bangladesh using World Health Organisation and Health Action International methodology. BMC Health Serv. Res. 2019, 19, 383. [Google Scholar] [CrossRef]
- Molica, S.; Tam, C.; Allsup, D.; Polliack, A. Advancements in the Treatment of Cll: The Rise of zanubrutinib as a preferred therapeutic option. Cancers 2023, 15, 3737. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, Y.H.E.; Malojirao, V.H.; Thirusangu, P.; Al-Ghorbani, M.; Prabhakar, B.T.; Khanum, S.A. The Novel 4-Phenyl-2-Phenoxyacetamide Thiazoles modulates the tumor hypoxia leading to the crackdown of neoangiogenesis and evoking the cell death. Eur. J. Med. Chem. 2018, 143, 1826–1839. [Google Scholar] [CrossRef] [PubMed]
- Kesternich, V.; Pérez-Fehrmann, M.; Quezada, V.; Castroagudín, M.; Nelson, R.; Martínez, R. A simple and efficient synthesis of N-[3-chloro-4-(4-chlorophenoxy)-phenyl]-2-hydroxy-3, 5-diiodobenzamide, rafoxanide. Chem. Pap. 2023, 77, 5091–5095. [Google Scholar] [CrossRef]
- Chang, X.; Sun, D.; Shi, D.; Wang, G.; Chen, Y.; Zhang, K.; Tan, H.; Liu, J.; Liu, B.; Ouyang, L. Design, synthesis, and biological evaluation of quinazoline-4 (3H)-one derivatives co-targeting poly (ADP-ribose) polymerase-1 and bromodomain containing protein 4 for breast cancer therapy. Acta Pharm. Sin. B 2021, 11, 156–180. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, Y.H.E.; Thirusangu, P.; Vigneshwaran, V.; Prabhakar, B.T.; Khanum, S.A. The anti-invasive role of novel synthesized pyridazine hydrazide appended phenoxy acetic acid against neoplastic development targeting matrix metalloproteases. Biomed. Pharmacother. 2017, 95, 375–386. [Google Scholar] [CrossRef]
- Thabet, F.M.; Dawood, K.M.; Ragab, E.A.; Nafie, M.S.; Abbas, A.A. Design and synthesis of new bis (1,2,4-triazolo [3,4-b][1,3,4] thiadiazines) and bis ((quinoxalin-2-yl) phenoxy) alkanes as anti-breast cancer agents through dual PARP-1 and EGFR targets inhibition. RSC Adv. 2022, 12, 23644–23660. [Google Scholar] [CrossRef]
- Gołąbek-Grenda, A.; Kaczmarek, M.; Juzwa, W.; Olejnik, A. Natural resveratrol analogs differentially target endometriotic cells into apoptosis pathways. Sci. Rep. 2023, 13, 11468. [Google Scholar] [CrossRef] [PubMed]
- Honrado, C.; Salahi, A.; Adair, S.J.; Moore, J.H.; Bauer, T.W.; Swami, N.S. Automated biophysical classification of apoptotic pancreatic cancer cell subpopulations by using machine learning approaches with impedance cytometry. Lab Chip 2022, 22, 3708–3720. [Google Scholar] [CrossRef]
- Niu, J.; Hao, Q.; Li, T.; Zong, C.; Meng, F.; Fu, J.; Tian, M.; Yu, X. Tiny nuance leads to large difference: Construction of fluorescent probes to visualize early and late apoptotic stages. Sens. Actuators B Chem. 2023, 393, 134200. [Google Scholar] [CrossRef]
- Çıkla-Süzgün, P.; Küçükgüzel, Ş. Recent Progress on Apoptotic Activity of Triazoles. Curr. Drug Targets 2021, 22, 1844–1900. [Google Scholar] [CrossRef] [PubMed]
- Aboelez, M.O.; Belal, A.; Xiang, G.; Ma, X. Design, synthesis, and molecular docking studies of novel pomalidomide-based PROTACs as potential anti-cancer agents targeting EGFR WT and EGFR T790M. J. Enzym. Inhib. Med. Chem. 2022, 37, 1196–1211. [Google Scholar] [CrossRef] [PubMed]
- Mir, M.A.; Sofi, S. Cell Cycle and Cancer. In Therapeutic Potential of Cell Cycle Kinases in Breast Cancer; Springer: Berlin/Heidelberg, Germany, 2023; pp. 83–101. [Google Scholar]
- Xie, R.; Yao, Y.; Tang, P.; Chen, G.; Liu, X.; Yun, F.; Cheng, C.; Wu, X.; Yuan, Q. Design, synthesis, and biological evaluation of novel hydroxamates and 2-aminobenzamides as potent histone deacetylase inhibitors and antitumor agents. Eur. J. Med. Chem. 2017, 134, 1–12. [Google Scholar] [CrossRef]
- Güngör, T.; Ozleyen, A.; Yılmaz, Y.B.; Siyah, P.; Ay, M.; Durdağı, S.; Tumer, T.B. New nimesulide derivatives with amide/sulfonamide moieties: Selective COX-2 inhibition and antitumor effects. Eur. J. Med. Chem. 2021, 221, 113566. [Google Scholar] [CrossRef]
- Green, D.R. The mitochondrial pathway of apoptosis Part I: MOMP and beyond. Cold Spring Harb. Perspect. Biol. 2022, 14, a041038. [Google Scholar] [CrossRef]
- D’arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis, and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Ahmad, A.; Tiwari, R.K.; Saeed, M.; Al-Amrah, H.; Han, I.; Choi, E.-H.; Yadav, D.K.; Ansari, I.A. Carvacrol instigates intrinsic and extrinsic apoptosis with abrogation of cell cycle progression in cervical cancer cells: Inhibition of Hedgehog/GLI signaling cascade. Front. Chem. 2023, 10, 1064191. [Google Scholar] [CrossRef] [PubMed]
- Loreto, C.; La Rocca, G.; Anzalone, R.; Caltabiano, R.; Vespasiani, G.; Castorina, S.; Ralph, D.J.; Cellek, S.; Musumeci, G.; Giunta, S. The role of intrinsic pathway in apoptosis activation and progression in Peyronie’s disease. BioMed Res. Int. 2014, 2014, 616149. [Google Scholar] [CrossRef] [PubMed]
- Changizi, Z.; Moslehi, A.; Rohani, A.H.; Eidi, A. Chlorogenic acid induces 4T1 breast cancer tumor’s apoptosis via p53, Bax, Bcl-2, and caspase-3 signaling pathways in BALB/c mice. J. Biochem. Mol. Toxicol. 2021, 35, e22642. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, W.; Zhang, B.; Gao, Z.; Zhang, Q.; Deng, H.; Han, L.; Shen, X.L. Perfluorooctanoic acid induces hepatocellular endoplasmic reticulum stress and mitochondrial-mediated apoptosis in vitro via endoplasmic reticulum-mitochondria communication. Chem. Biol. Interact. 2022, 354, 109844. [Google Scholar] [CrossRef]
- Zhang, C.; Ni, J.; Fan, W.; Hou, J. Nutlin-3 Promotes TRAIL-Induced Liver Cancer Cells Apoptosis by Activating p53 to Inhibit bcl-2 and Surviving Expression. Ann. Clin. Lab. Sci. 2022, 52, 601–610. [Google Scholar]
- Dawood, K.M.; Raslan, M.A.; Abbas, A.A.; Mohamed, B.E.; Nafie, M.S. Novel bis-amide-based bis-thiazoles as anti-colorectal cancer agents through Bcl-2 inhibition: Synthesis, in vitro, and in vivo studies. Anti-Cancer Agents Med. Chem. (Former Curr. Med. Chem.-Anti-Cancer Agents) 2023, 23, 328–345. [Google Scholar] [CrossRef]
- Das, M.; Mondal, S.; Ghosh, R.; Biswas, P.; Moussa, Z.; Darbar, S.; Ahmed, S.A.; Das, A.K.; Bhattacharya, S.S.; Pal, D.; et al. A nano erythropoiesis-stimulating agent for the treatment of anemia and associated disorders. iScience 2022, 25, 105021. [Google Scholar] [CrossRef]
- Liu, Z.; Li, S.; Ling, Y.; Jiang, H.; Kuang, L.; Bao, J.; Li, J.; Zhang, P.; Jin, H. Interventional Effects of Grape Skin Extract against Lung Injury Induced by Artificial Fine Particulate Matter in a Rat Model. Future Integr. Med. 2022, 1, 2–12. [Google Scholar] [CrossRef]
- Courneya, K.S.; Booth, C.M. Exercise as cancer treatment: A clinical oncology framework for exercise oncology research. Front. Oncol. 2022, 12, 957135. [Google Scholar] [CrossRef] [PubMed]
- Sakthivel, K.M.; Guruvayoorappan, C. Acacia ferruginea inhibits tumor progression by regulating inflammatory mediators-(TNF-α, iNOS, COX-2, IL-1β, IL-6, IFN-γ, IL-2, GM-CSF) and pro-angiogenic growth factor-VEGF. Asian Pac. J. Cancer Prev. 2013, 14, 3909–3919. [Google Scholar] [CrossRef]
- Ansari, A.; Seth, A.; Dutta, M.; Qamar, T.; Katiyar, S.; Jaiswal, A.K.; Rani, A.; Majhi, S.; Kumar, M.; Bhatta, R.S.; et al. Discovery, SAR and mechanistic studies of quinazolinone-based acetamide derivatives in experimental visceral leishmaniasis. Eur. J. Med. Chem. 2023, 257, 115524. [Google Scholar] [CrossRef]
- Rowbottom, M.W.; Bain, G.; Calderon, I.; Lasof, T.; Lonergan, D.; Lai, A.; Huang, F.; Darlington, J.; Prodanovich, P.; Santini, A.M.; et al. Identification of 4-(aminomethyl)-6-(trifluoromethyl)-2-(phenoxy) pyridine derivatives as potent, selective, and orally efficacious inhibitors of the copper-dependent amine oxidase, lysyl oxidase-like 2 (LOXL2). J. Med. Chem. 2017, 60, 4403–4423. [Google Scholar] [CrossRef]
- Liu, J.; Cui, X.; Guo, F.; Li, X.; Li, L.; Pan, J.; Tao, S.; Huang, R.; Feng, Y.; Ma, L.; et al. 2-methylquinazoline derivative F7 as a potent and selective HDAC6 inhibitor protected against rhabdomyolysis-induced acute kidney injury. PLoS ONE 2019, 14, e0224158. [Google Scholar] [CrossRef]
- Poulou, E.; Hackenberger, C.P. Staudinger Ligation and Reactions–From Bioorthogonal Labeling to Next-Generation Biopharmaceuticals. Isr. J. Chem. 2023, 63, e202200057. [Google Scholar] [CrossRef]
- Patil, S.S.; Rathod, V.K. Extraction and purification of curcuminoids from Curcuma longa using microwave-assisted deep eutectic solvent-based system and cost estimation. Process Biochem. 2023, 126, 61–71. [Google Scholar] [CrossRef]
- Savithri, K.; Kumar, B.V.; Vivek, H.K.; Revanasiddappa, H.D. Synthesis and Characterization of Cobalt (III) and Copper (II) Complexes of 2-((E)-(6-Fluorobenzo [d] thiazol-2-ylimino) methyl)-4-chlorophenol: DNA Binding and Nuclease Studies—SOD and Antimicrobial Activities. Int. J. Spectrosc. 2018, 2018, 8759372. [Google Scholar] [CrossRef]
- Freshney, R.I. Culture of Animal Cells: A Manual of Basic Technique and Specialized Applications; John Wiley & Sons: Hoboken, NJ, USA, 2015. [Google Scholar]
- Liu, T.; Krysiak, K.; Shirai, C.L.; Kim, S.; Shao, J.; Ndonwi, M.; Walter, M.J. Knockdown of HSPA9 induces TP53-dependent apoptosis in human hematopoietic progenitor cells. PLoS ONE 2017, 12, e0170470. [Google Scholar] [CrossRef] [PubMed]
- Gad, E.M.; Nafie, M.S.; Eltamany, E.H.; Hammad, M.S.; Barakat, A.; Boraei, A.T. Discovery of new apoptosis-inducing agents for breast cancer based on ethyl 2-amino-4, 5, 6, 7-tetra hydrobenzo [b] thiophene-3-carboxylate: Synthesis, in vitro, and in vivo activity evaluation. Molecules 2020, 25, 2523. [Google Scholar] [CrossRef]
- Sarhan, A.A.; Boraei, A.T.; Barakat, A.; Nafie, M.S. Discovery of hydrazide-based pyridazino [4,5-b] indole scaffold as a new phosphoinositide 3-kinase (PI3K) inhibitor for breast cancer therapy. RSC Adv. 2020, 10, 19534–19541. [Google Scholar] [CrossRef]
- Meier, J.; Theakston, R.D.G. Approximate LD50 determinations of snake venoms using eight to ten experimental animals. Toxicon 1986, 24, 395–401. [Google Scholar] [CrossRef]
- Bincoletto, C.; Eberlin, S.; Figueiredo, C.A.; Luengo, M.B.; Queiroz, M.L. Effects produced by Royal Jelly on hematopoiesis: Relation with host resistance against Ehrlich ascites tumor challenge. Int. Immunopharmacol. 2005, 5, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Kendall, R.; Benoit, E.; Bogiages, J.; Bordenkircher, R.; Caple, K.; Chen, L.L.; Cheng, T.; Hoshino, T.; Kelley, J.; Ngo, N.; et al. Performance evaluation of the Abbott Cell-Dyn 1800 automated hematology analyzer. Lab. Hematol. Off. Publ. Int. Soc. Lab. Hematol. 2003, 9, 143–152. [Google Scholar]
- Dufour, D.R.; Lott, J.A.; Nolte, F.S.; Gretch, D.R.; Koff, R.S.; Seeff, L.B. Diagnosis and monitoring of hepatic injury. I. Performance characteristics of laboratory tests. Clin. Chem. 2000, 46, 2027–2049. [Google Scholar] [CrossRef] [PubMed]
- Hafkenscheid, J.C.; Dijt, C.C. Determination of serum aminotransferases: Activation by pyridoxal-5′-phosphate in relation to substrate concentration. Clin. Chem. 1979, 25, 55–59. [Google Scholar] [CrossRef]
- Larsen, K. Creatinine assay in the presence of protein with LKB 8600 Reaction Rate Analyser. Clin. Chim. Acta Int. J. Clin. Chem. 1972, 38, 475–476. [Google Scholar]
- Banchroft, J.D.; Stevens, A.; Turner, D.R. Theory and Practice of Histological Techniques, 4th ed.; Churchill Livingstone: London, UK, 1996. [Google Scholar]
- Nafie, M.S.; Khodair, A.I.; Hassan, H.A.Y.; El-Fadeal, N.M.A.; Bogari, H.A.; Elhady, S.S.; Ahmed, S.A. Evaluation of 2-thioxoimadazolidin-4-one derivatives as potent anti-cancer agents through apoptosis induction and antioxidant activation: In vitro and in vivo Approaches. Molecules 2021, 27, 83. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration (µM) | 100 | 25 | 6.30 | 1.6 | 0.4 | IC50 (µM) |
|---|---|---|---|---|---|---|
| Compound I/MCF-7 | 30.12 ± 2.30 * | 38.18 ± 2.25 * | 51.41 ± 2.50 * | 63.92 ± 3.30 * | 73.41 ± 2.90 * | 7.43 ± 0.44 |
| Compound II/MCF-7 | 37 ± 2.39 * | 46.37 ± 2.28 * | 55.93 ± 2.58 * | 68.43 ± 3.28 * | 79.44 ± 3.10 * | 16.37 ± 0.97 |
| 5-FU/MCF-7 | 33.81 ± 2.26 * | 44.01 ± 2.31 * | 53.50 ± 2.66 * | 62.46 ± 3.36 * | 73.46 ± 3.30 * | 10.53 ± 0.58 |
| Concentration (µM) | 100 | 25 | 6.30 | 1.6 | 0.4 | IC50 (µM) |
|---|---|---|---|---|---|---|
| Compound I/HepG2 | 29.78 ± 3.62 * | 37.93 ± 2.91 * | 50.88 ± 3.10 * | 63.11 ± 2.85 * | 73.17 ± 3.20 * | 1.43 ± 0.31 |
| Compound II/HepG2 | 36.62 ± 3.25 * | 46.21 ± 3.26 * | 55.55 ± 3.25 * | 68.18 ± 2.93 * | 79.42 ± 3.66 * | 6.52 ± 0.86 |
| 5-FU/HepG2 | 33.38 ± 2.50 * | 44.64 ± 3.32 * | 52.83 ± 3.71 * | 62.25 ± 2.90 * | 73.11 ± 3.62 * | 5.32 ± 0.34 |
| Concentration (µM) | 100 | 25 | 6.30 | 1.6 | 0.4 | IC50 (µM) |
|---|---|---|---|---|---|---|
| Compound I/THLE-2 | 40.29 ± 2.70 * | 54.12 ± 2.25 * | 64.52 ± 4.10 * | 80.04 ± 3.33 * | 88.03 ± 4.25 * | 36.27 ± 2.03 |
| Compound I/HepG2 | 29.78 ± 3.62 * | 37.93 ± 2.91 * | 50.88 ± 3.10 * | 63.11 ± 2.85 * | 73.17 ± 3.20 * | 1.43 ± 0.31 |
| Groups | Negative Control | Normal + Comp. I | Positive Control (SEC) | SEC + 5-FU | SEC + Comp. I | ||
|---|---|---|---|---|---|---|---|
| Parameters | |||||||
| Mice’s body weight. (g) | 1st day | 21.85 ± 0.45 | 22.34 ± 0.20 | 23.35 ± 0.24 | 23.90 ± 0.22 | 24.20 ± 0.46 | |
| 15th day | 24.25 ± 0.49 | 25.34 ± 0.22 | 32.93 ± 0.58 | 29.97 ± 0.28 * | 30.02 ± 0.41 * | ||
| Tumor weight (mg) | ----- | --- | 6770 ± 0.20 | 4320 ± 0.22 * | 3890 ± 0.46 *,# | ||
| Tumor volume (mm3) | ----- | --- | 4960 ± 0.30 | 1760 ± 0.36 * | 1260 ± 0.11 *,# | ||
| Parameters Treatments | Hematological Parameters | Liver Parameters | Kidney Parameters | |||||
|---|---|---|---|---|---|---|---|---|
| RBCs (×106/µL) | Hb (g/dl) | WBC (×103/μL) | Lymphocyte (×103/μL) | Granulocyte (×103/μL) | ALT (U/L) | AST (U/L) | Creatinine (mg/dL) | |
| Negative control | 8.33 ± 0.28 | 13.20 ± 0.61 | 5.26 ± 0.30 | 2.20 ± 0.17 | 2.8 ± 0.2 | 40 ± 2.10 | 71 ± 4.04 | 0.58 ± 0.05 |
| Normal + comp. I | 7.97 ± 0.23 | 13.30 ± 0.28 | 5.73 ± 0.50 | 1.88 ± 0.27 | 3.46 ± 0.41 | 43 ± 2.88 | 77 ± 2.10 | 0.61 ± 0.07 |
| Positive control (SEC) | 5.97 ± 0.17 | 10.03 ± 0.74 | 10.86 ± 0.73 | 5.40 ± 0.52 | 4.26 ± 0.20 | 62 ± 2.64 | 139 ± 3.60 | 0.86 ± 0.15 |
| SEC + comp. I | 6.66 ± 0.08 * | 12.76 ± 0.75 * | 9.90 ± 0.45 | 4.20 ± 0.10 * | 3.50 ± 0.10 * | 47.66 ± 2.51 * | 111 ± 2.64 *,# | 0.85 ± 0.10 |
| SEC + 5-FU | 6.90 ± 0.36 * | 12.45 ± 0.58 * | 9.80 ± 0.43 | 4.34 ± 0.15 * | 3.7 ± 0.26 * | 53.33 ± 2.88 * | 98 ± 4.10 * | 0.81 ± 0.06 |
| Gene | Forward Primer (5′-3′) | Reverse Primer (3′-5′) |
|---|---|---|
| Bax | ATGGACGGGTCCGGGGAG | ATCCAGCCCAACAGCCGC |
| Bcl2 | AAG CCG GCG ACGACT TCT | GGT GCC GGT TCA GGTACT CA |
| p53 | ATGTTTTGCCAACTGGCCAAG | TGAGCAGCGCTCATGGTG |
| Casp3 | TGTTTGTGTGCTTCTGAGCC | CACGCCATGTCATCATCAAC |
| Casp8 | AGAAGAGGGTCATCCTGGGAGA | TCAGGACTTCCTTCAAGGCTGC |
| Casp9 | CATTTCATGGTGGAGGTGAAG | GGGAACTGCAGGTGGCTG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sayed, M.M.; Nabil, Z.I.; El-Shenawy, N.S.; Al-Eisa, R.A.; Nafie, M.S. In Vitro and In Vivo Effects of Synthesis Novel Phenoxyacetamide Derivatives as Potent Apoptotic Inducer against HepG2 Cells through PARP-1 Inhibition. Pharmaceuticals 2023, 16, 1524. https://doi.org/10.3390/ph16111524
Sayed MM, Nabil ZI, El-Shenawy NS, Al-Eisa RA, Nafie MS. In Vitro and In Vivo Effects of Synthesis Novel Phenoxyacetamide Derivatives as Potent Apoptotic Inducer against HepG2 Cells through PARP-1 Inhibition. Pharmaceuticals. 2023; 16(11):1524. https://doi.org/10.3390/ph16111524
Chicago/Turabian StyleSayed, Mai M., Zohour I. Nabil, Nahla S. El-Shenawy, Rasha A. Al-Eisa, and Mohamed S. Nafie. 2023. "In Vitro and In Vivo Effects of Synthesis Novel Phenoxyacetamide Derivatives as Potent Apoptotic Inducer against HepG2 Cells through PARP-1 Inhibition" Pharmaceuticals 16, no. 11: 1524. https://doi.org/10.3390/ph16111524
APA StyleSayed, M. M., Nabil, Z. I., El-Shenawy, N. S., Al-Eisa, R. A., & Nafie, M. S. (2023). In Vitro and In Vivo Effects of Synthesis Novel Phenoxyacetamide Derivatives as Potent Apoptotic Inducer against HepG2 Cells through PARP-1 Inhibition. Pharmaceuticals, 16(11), 1524. https://doi.org/10.3390/ph16111524