
The Use of Viral Vectors for Gene Therapy and Vaccination in Tuberculosis

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Immune Response in Pulmonary TB
3. The Experimental Models of Pulmonary TB
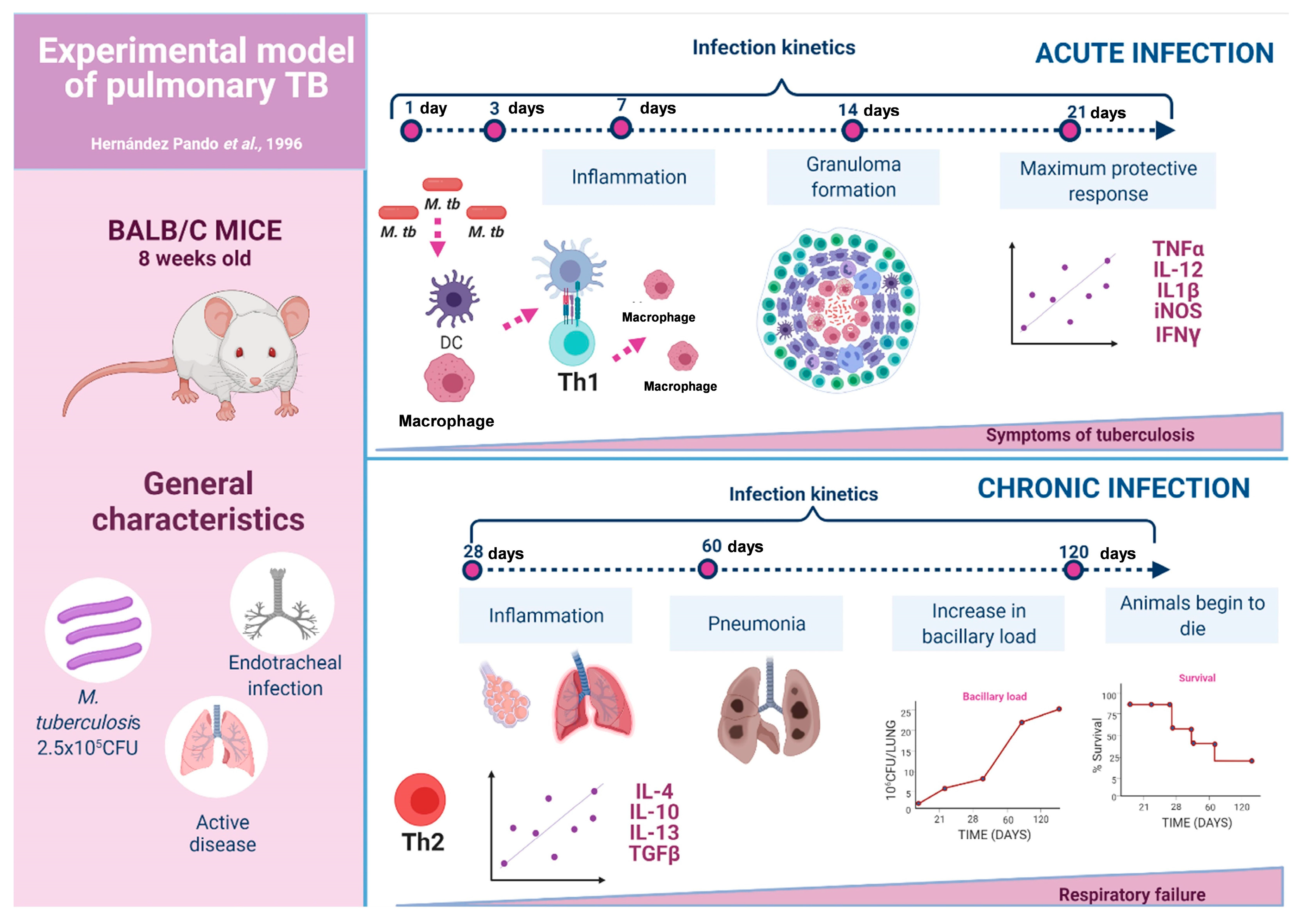
3.1. The Experimental Model of Progressive Pulmonary TB
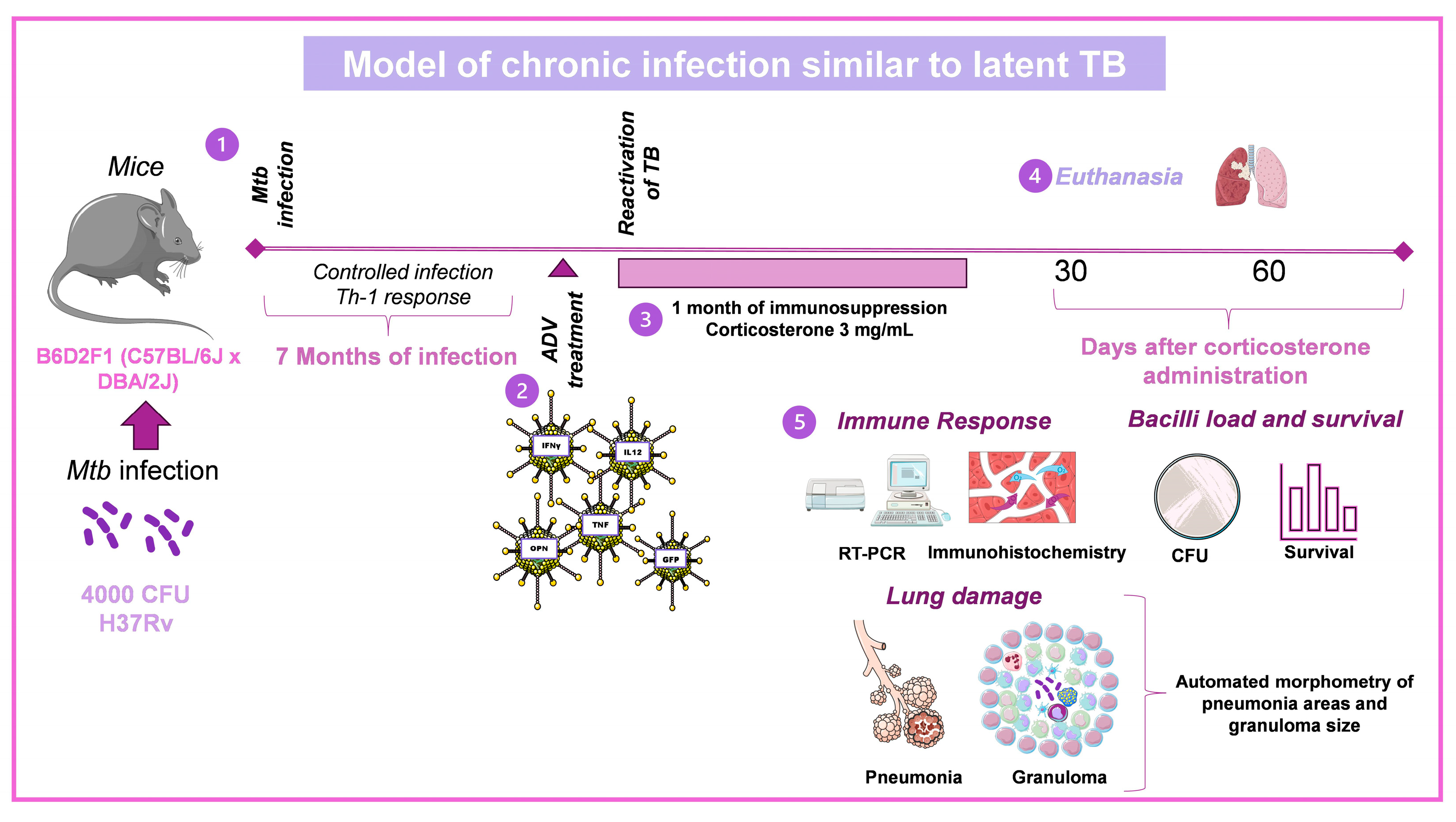
3.2. Model of Chronic Infection Similar to Latent TB
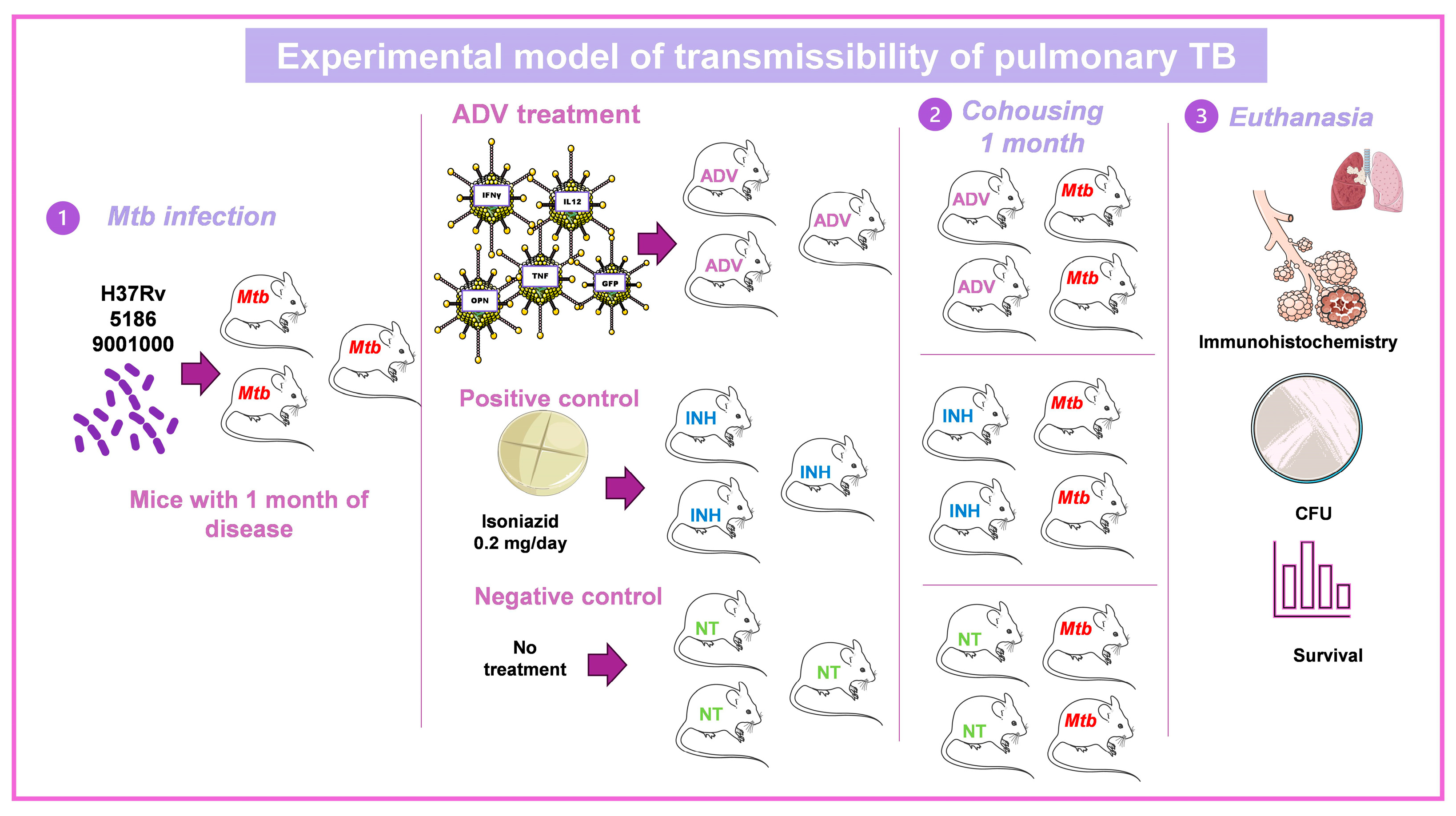
3.3. Murine Model of Mtb Transmissibility
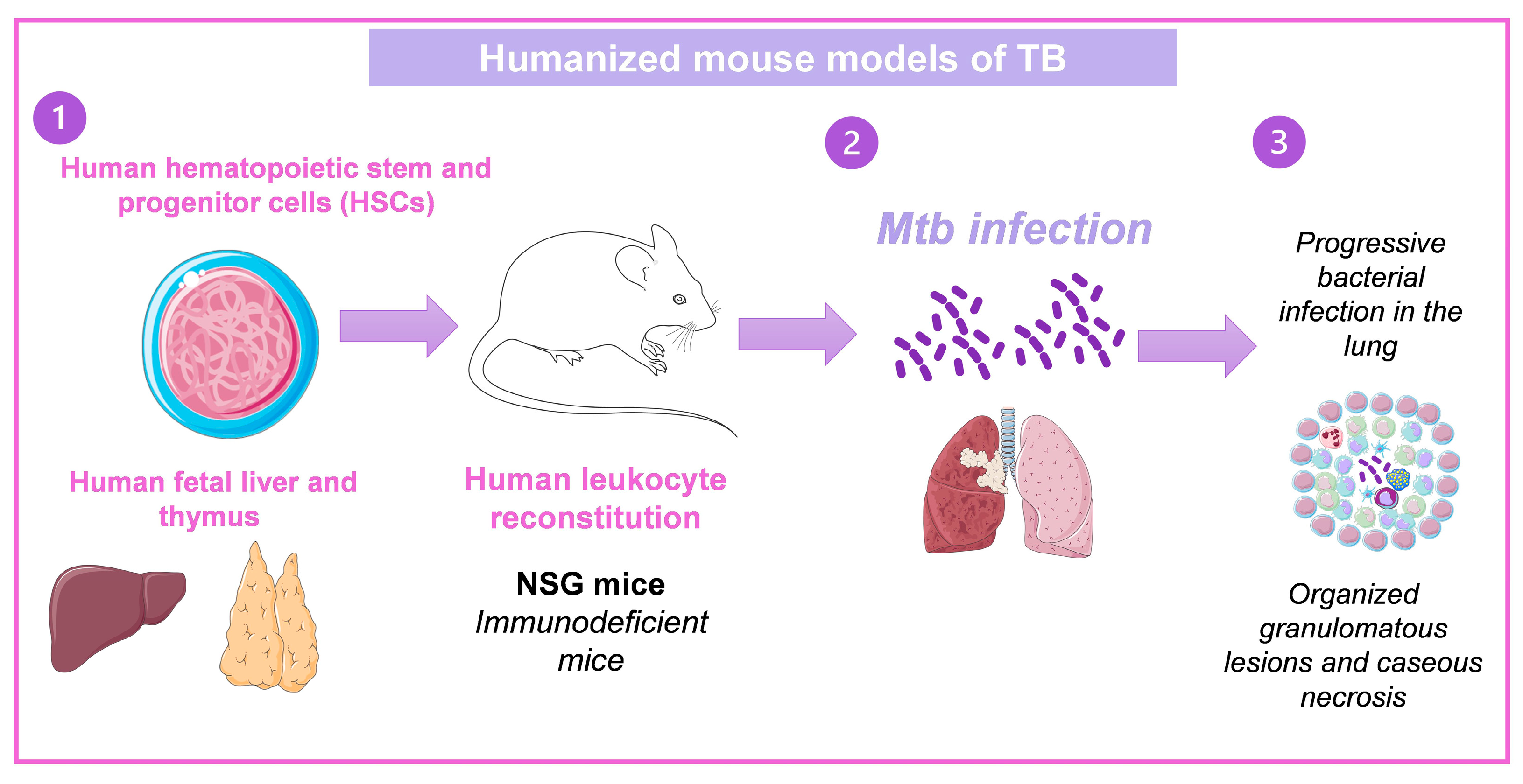
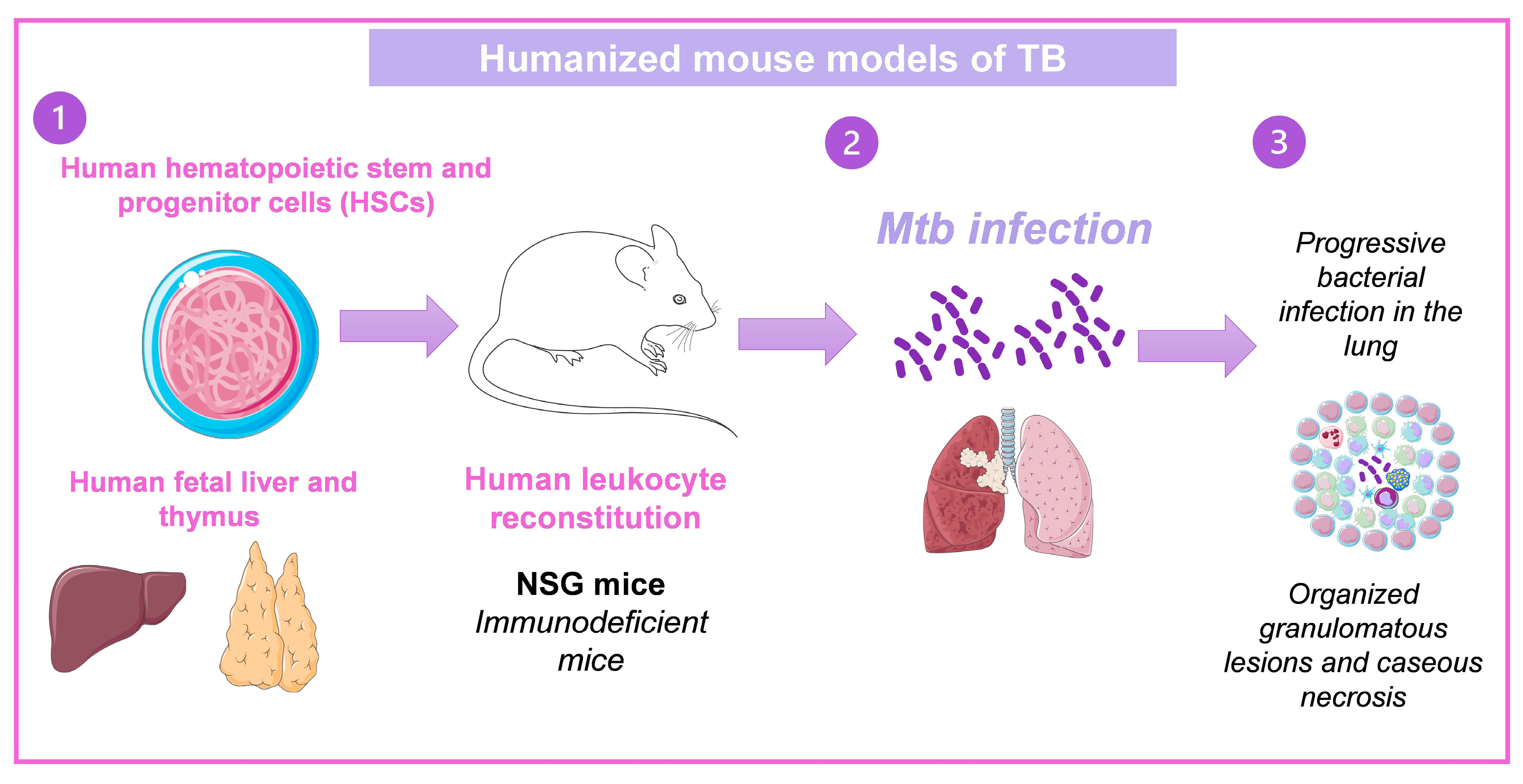
3.4. Humanized Mouse Models of TB
4. Treatment of Pulmonary TB
4.1. Immunotherapy Based on Gene Therapy with Recombinant Adenovirus
4.1.1. The Advantages of Ad as Genetic Vectors
4.1.2. The Drawbacks of Ad as Genetic Vectors
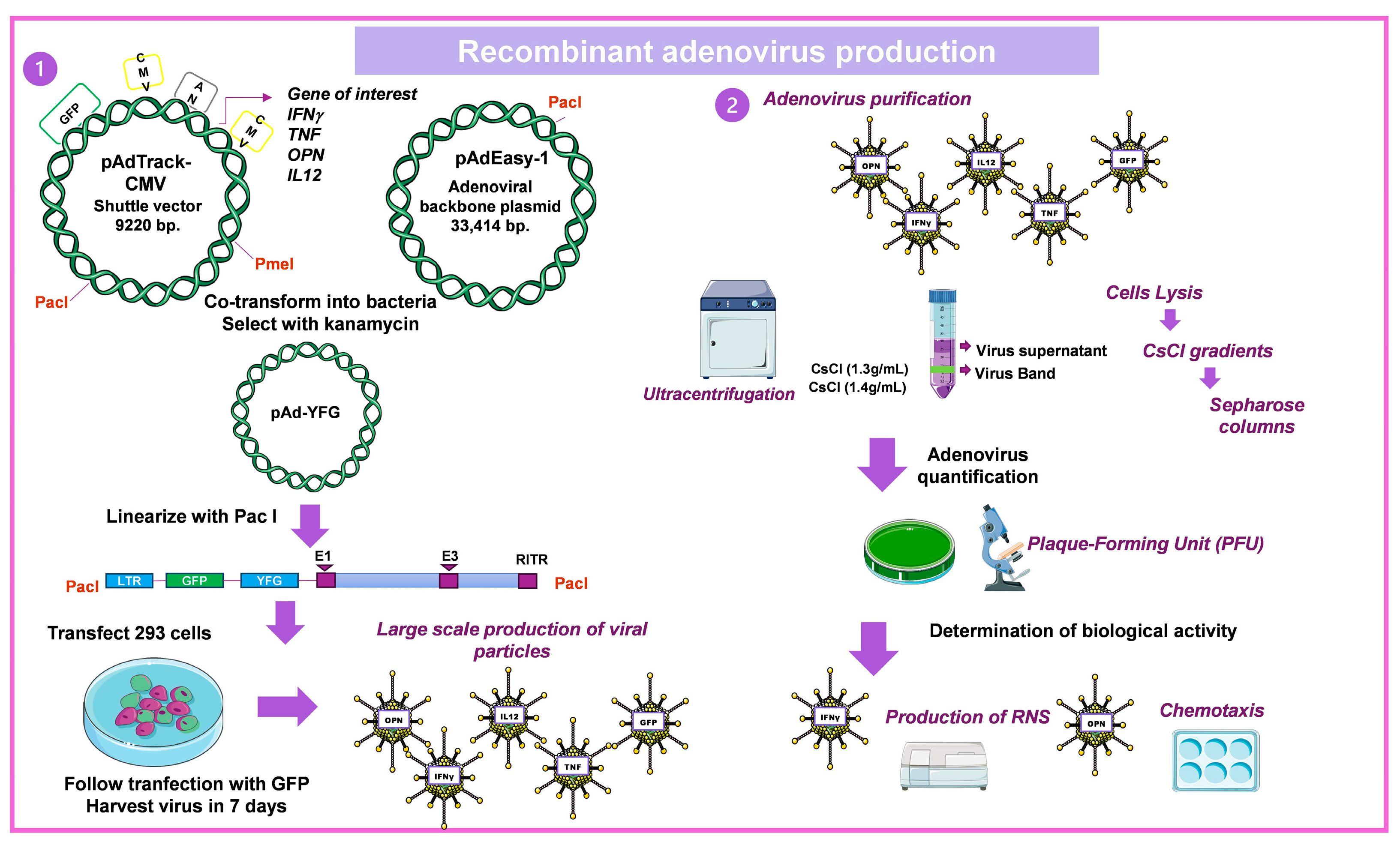
4.1.3. Construction of Recombinant Ad
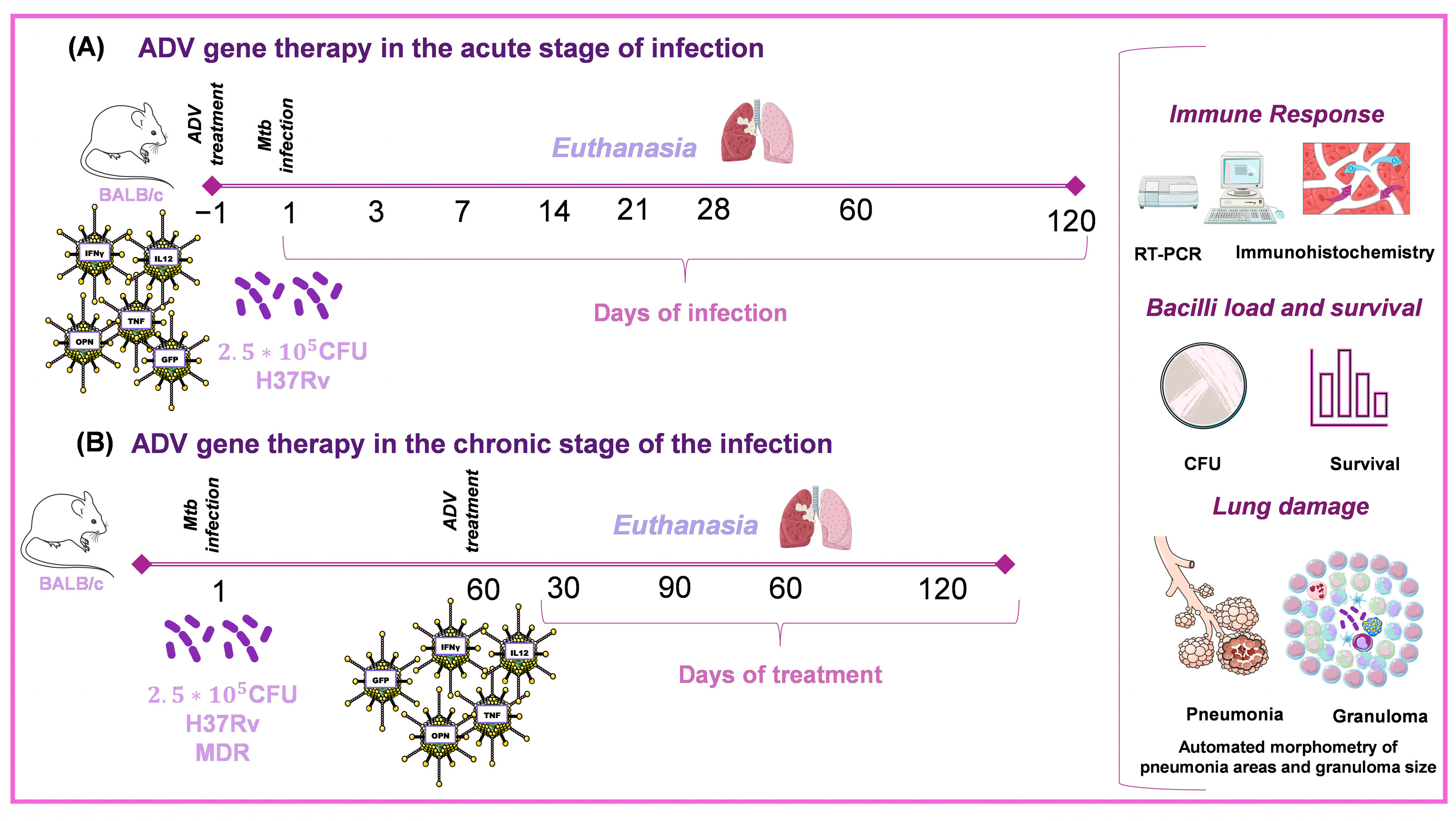
4.2. Recombinant Adenovirus Therapy in Different Experimental Models of TB
4.2.1. AdIFNγ
4.2.2. AdIL12
4.2.3. AdGM/CSF
4.2.4. AdOPN
4.2.5. Ad Antimicrobial Peptides
4.2.6. Ad TNFα
4.2.7. Ad IL-23
4.3. Candidate Vaccine for TB Based on Adenovirus
4.3.1. AdHu5Ag85A
4.3.2. ChAdOx185A-MVA85A
4.3.3. AERAS-402
4.4. Other Virus-Based Therapies in TB
4.4.1. MVA/IL-15/5Mtb
4.4.2. VSVAg85A
4.4.3. Mycobacteriophage D29
4.4.4. TB/FLU-01L
4.4.5. TB/FLU-04L
4.4.6. RhCMV/TB
4.4.7. rLCMV-Based Mtb Vaccine
5. Perspectives
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rahlwes, K.C.; Dias, B.R.S.; Campos, P.C.; Alvarez-Arguedas, S.; Shiloh, M.U. Pathogenicity and virulence of Mycobacterium tuberculosis. Virulence 2023, 14, 2150449. [Google Scholar] [CrossRef] [PubMed]
- Ojo, O.O.; Nadarajah, S.; Kebe, M. Integer time series models for tuberculosis in Africa. Sci. Rep. 2023, 13, 11443. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K. The Pathology of Mycobacterium tuberculosis Infection. Vet. Pathol. 2012, 49, 423–439. [Google Scholar] [CrossRef] [PubMed]
- Poladian, N.; Orujyan, D.; Narinyan, W.; Oganyan, A.K.; Navasardyan, I.; Velpuri, P.; Chorbajian, A.; Venketaraman, V. Role of NF-κB during Mycobacterium tuberculosis Infection. Int. J. Mol. Sci. 2023, 24, 1772. [Google Scholar] [CrossRef] [PubMed]
- O’Garra, A.; Redford, P.S.; McNab, F.W.; Bloom, C.I.; Wilkinson, R.J.; Berry, M.P.R. The Immune Response in Tuberculosis. Annu. Rev. Immunol. 2013, 31, 475–527. [Google Scholar] [CrossRef]
- World Health Organization. Global Tuberculosis Report 2020: Synopsis [Global Tuberculosis Report 2020: Executive Summary]; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Schrager, L.K.; Vekemens, J.; Drager, N.; Lewinsohn, D.M.; Olesen, O.F. The status of tuberculosis vaccine development. Lancet Infect. Dis. 2020, 20, e28–e37. [Google Scholar] [CrossRef]
- Pandit, R.; Singh, P.K.; Kumar, V. Natural Remedies against Multi-Drug Resistant Mycobacterium tuberculosis. J. Tuberc. Res. 2015, 3, 171–183. [Google Scholar] [CrossRef]
- Hameed, H.M.A.; Islam, M.M.; Chhotaray, C.; Wang, C.; Liu, Y.; Tan, Y.; Li, X.; Tan, S.; Delorme, V.; Yew, W.W.; et al. Molecular targets related drug resistance mechanisms in MDR-, XDR-, and TDR-Mycobacterium tuberculosis strains. Front. Cell. Infect. Microbiol. 2018, 8, 114. [Google Scholar] [CrossRef]
- Ramos-Espinosa, O.; Islas-Weinstein, L.; Peralta-Álvarez, M.P.; López-Torres, M.O.; Hernández-Pando, R. The use of immunotherapy for the treatment of tuberculosis. Expert Rev. Respir. Med. 2018, 12, 427–440. [Google Scholar] [CrossRef]
- Wirth, T.; Parker, N.; Ylä-Herttuala, S. History of gene therapy. Gene 2013, 525, 162–169. [Google Scholar] [CrossRef]
- Kreppel, F.; Kochanek, S. Modification of adenovirus gene transfer vectors with synthetic polymers: A scientific review and technical guide. Mol. Ther. 2008, 16, 16–29. [Google Scholar] [CrossRef] [PubMed]
- De Martino, M.; Lodi, L.; Galli, L.; Chiappini, E. Immune Response to Mycobacterium tuberculosis: A Narrative Review. Front. Pediatr. 2019, 7, 350. [Google Scholar] [CrossRef] [PubMed]
- Carabalí-Isajar, M.L.; Rodríguez-Bejarano, O.H.; Amado, T.; Patarroyo, M.A.; Izquierdo, M.A.; Lutz, J.R.; Ocampo, M. Clinical manifestations and immune response to tuberculosis. World J. Microbiol. Biotechnol. 2023, 39, 206. [Google Scholar] [CrossRef]
- Rivas-Santiago, C.E.; Hernández-Pando, R.; Rivas-Santiago, B. Immunotherapy for pulmonary TB: Antimicrobial peptides and their inducers. Immunotherapy 2013, 5, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Pando, R.; Orozco, H.; Aguilar, D. Factors that deregulate the protective immune response in tuberculosis. Arch. Immunol. Ther. Exp. 2009, 57, 355–367. [Google Scholar] [CrossRef]
- Rook, G.A.W. Endocrine and cytokine responses in humans with pulmonary tuberculosis. Brain. Behav. Immun. 2007, 21, 169–170. [Google Scholar] [CrossRef]
- Hernández-Pando, R.; Orozcoe, H.; Sampieri, A.; Pavón, L.; Velasquillo, C.; Larriva-Sahd, J.; Alcocer, J.M.; Madrid, M. V Correlation between the kinetics of Th1, Th2 cells and pathology in a murine model of experimental pulmonary tuberculosis. Immunology 1996, 89, 26–33. [Google Scholar]
- Hernandez-Pando, R.; Orozco, E.H.; Arriaga, K.; Sampieri, A.; Larriva-Sahd, J.; Madrid-Marina, V. Analysis of the local kinetics and localization of interleukin-1α, tumour necrosis factor-α and transforming growth factor-β, during the course of experimental pulmonary tuberculosis. Immunology 1997, 90, 607–617. [Google Scholar] [CrossRef]
- Rivas-Santiago, B.; Sada, E.; Tsutsumi, V.; Aguilar-Léon, D.; Contreras, J.L.; Hernández-Pando, R. Β-Defensin Gene Expression During the Course of Experimental Tuberculosis Infection. J. Infect. Dis. 2006, 194, 697–701. [Google Scholar] [CrossRef]
- Sharma, S.K.; Mohan, A.; Kohli, M. Extrapulmonary tuberculosis. Expert Rev. Respir. Med. 2021, 15, 931–948. [Google Scholar] [CrossRef]
- Rodriguez-Takeuchi, S.Y.; Renjifo, M.E.; Medina, F.J. Extrapulmonary tuberculosis: Pathophysiology and imaging findings. Radiographics 2019, 39, 2023–2037. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Pando, R.; Orozco, H.; Arriaga, K.; Pavön, L.; Rook, G. Treatment with BB-94, A broad spectrum inhibitor of zinc-dependent metalloproteinases, causes deviation of the cytokine profile towards Type-2 in experimental pulmonary tuberculosis in Balb/c mice. Int. J. Exp. Pathol. 2000, 81, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Arriaga, A.K.; Orozco, E.H.; Aguilar, L.D.; Rook, G.A.W.; Hernández Pando, R. Immunological and pathological comparative analysis between experimental latent tuberculous infection and progressive pulmonary tuberculosis. Clin. Exp. Immunol. 2002, 128, 229–237. [Google Scholar] [CrossRef]
- Marquina-Castillo, B.; García-García, L.; Ponce-De-León, A.; Jimenez-Corona, M.E.; Bobadilla-Del Valle, M.; Cano-Arellano, B.; Canizales-Quintero, S.; Martinez-Gamboa, A.; Kato-Maeda, M.; Robertson, B.; et al. Virulence, immunopathology and transmissibility of selected strains of Mycobacterium tuberculosis in a murine model. Immunology 2009, 128, 123–133. [Google Scholar] [CrossRef]
- Walsh, N.C.; Kenney, L.L.; Jangalwe, S.; Aryee, K.E.; Greiner, D.L.; Brehm, M.A.; Shultz, L.D. Humanized Mouse Models of Clinical Disease. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 187–215. [Google Scholar] [CrossRef]
- Calderon, V.E.; Valbuena, G.; Goez, Y.; Judy, B.M.; Huante, M.B.; Sutjita, P.; Johnston, R.K.; Estes, D.M.; Hunter, R.L.; Actor, J.K.; et al. A Humanized Mouse Model of Tuberculosis. PLoS ONE 2013, 8, e63331. [Google Scholar] [CrossRef]
- Nusbaum, R.J.; Calderon, V.E.; Huante, M.B.; Sutjita, P.; Vijayakumar, S.; Lancaster, K.L.; Hunter, R.L.; Actor, J.K.; Cirillo, J.D.; Aronson, J.; et al. Pulmonary Tuberculosis in Humanized Mice Infected with HIV-1. Sci. Rep. 2016, 6, 21522. [Google Scholar] [CrossRef]
- Arrey, F.; Löwe, D.; Kuhlmann, S.; Kaiser, P.; Moura-Alves, P.; Krishnamoorthy, G.; Lozza, L.; Maertzdorf, J.; Skrahina, T.; Skrahina, A.; et al. Humanized mouse model mimicking pathology of human tuberculosis for in vivo evaluation of drug regimens. Front. Immunol. 2019, 10, 89. [Google Scholar] [CrossRef]
- Suárez, I.; Fünger, S.M.; Kröger, S.; Rademacher, J.; Fätkenheuer, G.; Rybniker, J. The Diagnosis and Treatment of Tuberculosis. Dtsch. Arztebl. Int. 2019, 116, 729–735. [Google Scholar] [CrossRef]
- Anderson, L.F.; Tamne, S.; Watson, J.P.; Cohen, T.; Mitnick, C.; Brown, T.; Drobniewski, F.; Abubakar, I. Treatment outcome of multi-drug resistant tuberculosis in the United Kingdom: Retrospective-prospective cohort study from 2004 to 2007. Eurosurveillance 2013, 18, 20601. [Google Scholar] [CrossRef]
- Appaiahgari, M.B.; Vrati, S. Adenoviruses as gene/vaccine delivery vectors: Promises and pitfalls. Expert Opin. Biol. Ther. 2015, 15, 337–351. [Google Scholar] [CrossRef]
- Harrach, B.; Tarján, Z.L.; Benkő, M. Adenoviruses across the animal kingdom: A walk in the zoo. FEBS Lett. 2019, 593, 3660–3673. [Google Scholar] [CrossRef] [PubMed]
- Loustalot, F.; Kremer, E.J.; Salinas, S. Membrane Dynamics and Signaling of the Coxsackievirus and Adenovirus Receptor. Int. Rev. Cell Mol. Biol. 2016, 322, 331–362. [Google Scholar] [CrossRef] [PubMed]
- Hidaka, C.; Milano, E.; Leopold, P.L.; Bergelson, J.M.; Hackett, N.R.; Finberg, R.W.; Wickham, T.J.; Kovesdi, I.; Roelvink, P.; Crystal, R.G. CAR-dependent and CAR-independent pathways of adenovirus vector-mediated gene transfer and expression in human fibroblasts. J. Clin. Investig. 1999, 103, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Kulanayake, S.; Tikoo, S.K. Adenovirus core proteins: Structure and function. Viruses 2021, 13, 388. [Google Scholar] [CrossRef] [PubMed]
- Garbuglia, A.R.; Minosse, C.; Del Porto, P. mRNA- and Adenovirus-Based Vaccines against SARS-CoV-2 in HIV-Positive People. Viruses 2022, 14, 748. [Google Scholar] [CrossRef]
- Srivastava, S.; Dey, S.; Mukhopadhyay, S. Vaccines against Tuberculosis: Where Are We Now? Vaccines 2023, 11, 388. [Google Scholar] [CrossRef]
- Shirley, J.L.; de Jong, Y.P.; Terhorst, C.; Herzog, R.W. Immune Responses to Viral Gene Therapy Vectors. Mol. Ther. 2020, 28, 709–722. [Google Scholar] [CrossRef]
- Young, L.S.; Mautner, V. The promise and potential hazards of adenovirus gene therapy. Gut 2001, 48, 733–736. [Google Scholar] [CrossRef]
- Montes-Galindo, D.A.; Espiritu-Mojarro, A.C.; Melnikov, V.; Moy-López, N.A.; Soriano-Hernandez, A.D.; Galvan-Salazar, H.R.; Guzman-Muñiz, J.; Guzman-Esquivel, J.; Martinez-Fierro, M.L.; Rodriguez-Sanchez, I.P.; et al. Adenovirus 5 produces obesity and adverse metabolic, morphological, and functional changes in the long term in animals fed a balanced diet or a high-fat diet: A study on hamsters. Arch. Virol. 2019, 164, 775–786. [Google Scholar] [CrossRef]
- He, T.C.; Zhou, S.; Da Costa, L.T.; Yu, J.; Kinzler, K.W.; Vogelstein, B. A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. USA 1998, 95, 2509–2514. [Google Scholar] [CrossRef] [PubMed]
- Mata-Espinosa, D.A.; Mendoza-Rodríguez, V.; Aguilar-León, D.; Rosales, R.; López-Casillas, F.; Hernández-Pando, R. Therapeutic effect of recombinant adenovirus encoding interferon-γ in a murine model of progressive pulmonary tuberculosis. Mol. Ther. 2008, 16, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Bazán, S.; Mata-Espinosa, D.; Lozano-Ordaz, V.; Ramos-Espinosa, O.; Barrios-Payán, J.; López-Casillas, F.; Pando, R.H. Immune Regulatory Effect of Osteopontin Gene Therapy in a Murine Model of Multidrug Resistant Pulmonary Tuberculosis. Hum. Gene Ther. 2022, 33, 1037–1051. [Google Scholar] [CrossRef] [PubMed]
- Mata-Espinosa, D.A.; Francisco-Cruz, A.; Marquina-Castillo, B.; Barrios-Payan, J.; Ramos-Espinosa, O.; Bini, E.I.; Xing, Z.; Hernández-Pando, R. Immunotherapeutic effects of recombinant adenovirus encoding interleukin 12 in experimental pulmonary tuberculosis. Scand. J. Immunol. 2019, 89, e12743. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.A.L.; Chan, J.; Triebold, K.J.; Dalton, D.K.; Stewart, T.A.; Bloom, B.R. An essential role for interferon γ in resistance to mycobacterium tuberculosis infection. J. Exp. Med. 1993, 178, 2249–2254. [Google Scholar] [CrossRef]
- Bharti, R.; Srivastava, A.; Roy, T.; Verma, K.; Reddy, D.V.S.; Shafi, H.; Verma, S.; Raman, S.K.; Singh, A.K.; Singh, J.; et al. Transient Transfection of the Respiratory Epithelium with Gamma Interferon for Host-Directed Therapy in Pulmonary Tuberculosis. Mol. Ther. Nucleic Acids 2020, 22, 1121–1128. [Google Scholar] [CrossRef]
- Bharti, R.; Roy, T.; Verma, S.; Reddy, D.V.S.; Shafi, H.; Verma, K.; Raman, S.K.; Pal, S.; Azmi, L.; Singh, A.K.; et al. Transient, inhaled gene therapy with gamma interferon mitigates pathology induced by host response in a mouse model of tuberculosis. Tuberculosis 2022, 134, 102198. [Google Scholar] [CrossRef]
- Cheng, E.M.; Tsarovsky, N.W.; Sondel, P.M.; Rakhmilevich, A.L. Interleukin-12 as an in situ cancer vaccine component: A review. Cancer Immunol. Immunother. 2022, 71, 2057–2065. [Google Scholar] [CrossRef]
- García-Romo, G.S.; Pedroza-González, A.; Aguilar-León, D.; Orozco-Estevez, H.; Lambrecht, B.N.; Estrada-Garcia, I.; Flores-Romo, L.; Hernández-Pando, R. Airways infection with virulent Mycobacterium tuberculosis delays the influx of dendritic cells and the expression of costimulatory molecules in mediastinal lymph nodes. Immunology 2004, 112, 661–668. [Google Scholar] [CrossRef]
- Francisco-Cruz, A.; Mata-Espinosa, D.; Estrada-Parra, S.; Xing, Z.; Hernández-Pando, R. Immunotherapeutic effects of recombinant adenovirus encoding granulocyte-macrophage colony-stimulating factor in experimental pulmonary tuberculosis. Clin. Exp. Immunol. 2013, 171, 283–297. [Google Scholar] [CrossRef]
- Francisco-Cruz, A.; Mata-Espinosa, D.; Ramos-Espinosa, O.; Marquina-Castillo, B.; Estrada-Parra, S.; Xing, Z.; Hernández-Pando, R. Efficacy of gene-therapy based on adenovirus encoding granulocyte-macrophage colony-stimulating factor in drug-sensitive and drug-resistant experimental pulmonary tuberculosis. Tuberculosis 2016, 100, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.X.; Denhardt, D.T. Osteopontin: Role in immune regulation and stress responses. Cytokine Growth Factor Rev. 2008, 19, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Cruz, J.; Ortiz, C.; Guzmán, F.; Fernández-Lafuente, R.; Torres, R. Antimicrobial Peptides: Promising Compounds Against Pathogenic Microorganisms. Curr. Med. Chem. 2014, 21, 2299–2321. [Google Scholar] [CrossRef] [PubMed]
- Rivas-Santiago, B.; Contreras, J.C.L.; Sada, E.; Hernández-Pando, R. The potential role of lung epithelial cells and β-defensins in experimental latent tuberculosis. Scand. J. Immunol. 2008, 67, 448–452. [Google Scholar] [CrossRef]
- Castañeda-Delgado, J.; Hernández-Pando, R.; Serrano, C.J.; Aguilar-León, D.; León-Contreras, J.; Rivas-Santiago, C.; Méndez, R.; González-Curiel, I.; Enciso-Moreno, A.; Rivas-Santiago, B. Kinetics and cellular sources of cathelicidin during the course of experimental latent tuberculous infection and progressive pulmonary tuberculosis. Clin. Exp. Immunol. 2010, 161, 542–550. [Google Scholar] [CrossRef]
- Ramos-Espinosa, O.; Mata-Espinosa, D.; Francisco-Cruz, A.; López-Torres, M.O.; Hernández-Bazán, S.; Barrios-Payán, J.; Marquina-Castillo, B.; Carretero, M.; del Río, M.; Hernández-Pando, R. Immunotherapeutic effect of adenovirus encoding antimicrobial peptides in experimental pulmonary tuberculosis. J. Leukoc. Biol. 2021, 110, 951–963. [Google Scholar] [CrossRef]
- Ramos-Espinosa, O.; Hernández-Bazán, S.; Francisco-Cruz, A.; Mata-Espinosa, D.; Barrios-Payán, J.; Marquina-Castillo, B.; López-Casillas, F.; Carretero, M.; del Río, M.; Hernández-Pando, R. Gene therapy based in antimicrobial peptides and pro-inflammatory cytokine prevents reactivation of experimental latent tuberculosis. Pathog. Dis. 2016, 74, ftw075. [Google Scholar] [CrossRef]
- Keane, J.; Gershon, S.; Wise, R.P.; Mirabile-Levens, E.; Kasznica, J.; Schwieterman, W.D.; Siegel, J.N.; Braun, M.M. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N. Engl. J. Med. 2001, 345, 1098–1104. [Google Scholar] [CrossRef]
- Happel, K.I.; Lockhart, E.A.; Mason, C.M.; Porretta, E.; Keoshkerian, E.; Odden, A.R.; Nelson, S.; Ramsay, A.J. Pulmonary interleukin-23 gene delivery increases local T-cell immunity and controls growth of Mycobacterium tuberculosis in the lungs. Infect. Immun. 2005, 73, 5782–5788. [Google Scholar] [CrossRef]
- Ferreira, R.G.; Gordon, N.F.; Stock, R.; Petrides, D. Adenoviral vector covid-19 vaccines: Process and cost analysis. Processes 2021, 9, 1430. [Google Scholar] [CrossRef]
- Hu, Z.; Lu, S.H.; Lowrie, D.B.; Fan, X.Y. Research Advances for Virus-vectored Tuberculosis Vaccines and Latest Findings on Tuberculosis Vaccine Development. Front. Immunol. 2022, 13, 895020. [Google Scholar] [CrossRef] [PubMed]
- Santosuosso, M.; Zhang, X.; Mccormick, S.; Wang, J.; Hitt, M.; Xing, Z. Protective CD4 and CD8 T Cells within the Airway Lumen 1. J. Immunol. 2005, 174, 7986–7994. [Google Scholar] [CrossRef]
- Xing, Z.; McFarland, C.T.; Sallenave, J.M.; Izzo, A.; Wang, J.; McMurray, D.N. Intranasal mucosal boosting with an adenovirus-vectored vaccine markedly enhances the protection of BCG-primed guinea pigs against pulmonary tuberculosis. PLoS ONE 2009, 4, e5856. [Google Scholar] [CrossRef] [PubMed]
- Pérez De Val, B.; Villarreal-Ramos, B.; Nofrarías, M.; López-Soria, S.; Romera, N.; Singh, M.; Abad, F.X.; Xing, Z.; Vordermeier, H.M.; Domingo, M. Goats primed with Mycobacterium bovis BCG and boosted with a recombinant adenovirus expressing Ag85A show enhanced protection against tuberculosis. Clin. Vaccine Immunol. 2012, 19, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Smaill, F.; Jeyanathan, M.; Smieja, M.; Medina, M.F.; Thanthrige-Don, N.; Zganiacz, A.; Yin, C.; Heriazon, A.; Damjanovic, D.; Puri, L.; et al. A human type 5 adenovirus-based tuberculosis vaccine induces robust T cell responses in humans despite preexisting anti-adenovirus immunity. Sci. Transl. Med. 2013, 5, 205ra134. [Google Scholar] [CrossRef] [PubMed]
- Stylianou, E.; Griffiths, K.L.; Poyntz, H.C.; Harrington-Kandt, R.; Dicks, M.D.; Stockdale, L.; Betts, G.; McShane, H. Improvement of BCG protective efficacy with a novel chimpanzee adenovirus and a modified vaccinia Ankara virus both expressing Ag85A. Vaccine 2015, 33, 6800–6808. [Google Scholar] [CrossRef]
- Wilkie, M.; Satti, I.; Minhinnick, A.; Harris, S.; Riste, M.; Ramon, R.L.; Sheehan, S.; Thomas, Z.R.M.; Wright, D.; Stockdale, L.; et al. A phase I trial evaluating the safety and immunogenicity of a candidate tuberculosis vaccination regimen, ChAdOx1 85A prime—MVA85A boost in healthy UK adults. Vaccine 2020, 38, 779–789. [Google Scholar] [CrossRef]
- Satti, I.; Meyer, J.; Harris, S.A.; Thomas, Z.R.M.; Griffiths, K.; Antrobus, R.D.; Rowland, R.; Ramon, R.L.; Smith, M.; Sheehan, S.; et al. Safety and immunogenicity of a candidate tuberculosis vaccine MVA85A delivered by aerosol in BCG-vaccinated healthy adults: A phase 1, double-blind, randomised controlled trial. Lancet Infect. Dis. 2014, 14, 939–946. [Google Scholar] [CrossRef]
- Sivakumaran, D.; Blatner, G.; Bakken, R.; Hokey, D.; Ritz, C.; Jenum, S.; Grewal, H.M.S. A 2-Dose AERAS-402 Regimen Boosts CD8+ Polyfunctionality in HIV-Negative, BCG-Vaccinated Recipients. Front. Immunol. 2021, 12, 673532. [Google Scholar] [CrossRef]
- Kagina, B.M.N.; Tameris, M.D.; Geldenhuys, H.; Hatherill, M.; Abel, B.; Hussey, G.D.; Scriba, T.J.; Mahomed, H.; Sadoff, J.C.; Hanekom, W.A.; et al. The novel tuberculosis vaccine, AERAS-402, is safe in healthy infants previously vaccinated with BCG, and induces dose-dependent CD4 and CD8T cell responses. Vaccine 2014, 32, 5908–5917. [Google Scholar] [CrossRef]
- Perera, P.Y.; Derrick, S.C.; Kolibab, K.; Momoi, F.; Yamamoto, M.; Morris, S.L.; Waldmann, T.A.; Perera, L.P. A multi-valent vaccinia virus-based tuberculosis vaccine molecularly adjuvanted with interleukin-15 induces robust immune responses in mice. Vaccine 2009, 27, 2121–2127. [Google Scholar] [CrossRef]
- Roediger, E.K.; Kugathasan, K.; Zhang, X.Z.; Lichty, B.D.; Xing, Z. Heterologous boosting of recombinant adenoviral prime immunization with a novel vesicular stomatitis virus-vectored tuberculosis vaccine. Mol. Ther. 2008, 16, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.Y.; Yang, W.H.; Dong, X.K.; Cong, L.M.; Li, N.; Li, Y.; Wen, Z.B.; Yin, Z.; Lan, Z.J.; Li, W.P.; et al. Inhalation Study of Mycobacteriophage D29 Aerosol for Mice by Endotracheal Route and Nose-Only Exposure. J. Aerosol Med. Pulm. Drug Deliv. 2016, 29, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Carrigy, N.B.; Larsen, S.E.; Reese, V.; Pecor, T.; Harrison, M.; Kuehl, P.J.; Hatfull, G.F.; Sauvageau, D.; Baldwin, S.L.; Finlay, W.H.; et al. Prophylaxis of mycobacterium tuberculosis H37Rv infection in a preclinical mouse model via inhalation of nebulized bacteriophage D29. Antimicrob. Agents Chemother. 2019, 63, 101128. [Google Scholar] [CrossRef]
- Stukova, M. Randomized Open Label Phase 1 Clinical Trial of TB/FLU-01L Tuberculosis Vaccine Administered Intranasally or Sublingual in BCG-Vaccinated Healthy Adults. Global Forum on TB Vaccines, New Delhi, India. 2018. Available online: https://tbvaccinesforum.org/wp-content/uploads/2018/03/5GF-Breakout-2-Stukova.pdf (accessed on 23 September 2023).
- Shurygina, A.P.; Zabolotnykh, N.; Vinogradova, T.; Khairullin, B.; Kassenov, M.; Nurpeisova, A.; Sarsenbayeva, G.; Sansyzbay, A.; Vasilyev, K.; Buzitskaya, J.; et al. Preclinical Evaluation of TB/FLU-04L—An Intranasal Influenza Vector-Based Boost Vaccine against Tuberculosis. Int. J. Mol. Sci. 2023, 24, 7439. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.G.; Zak, D.E.; Xu, G.; Ford, J.C.; Marshall, E.E.; Malouli, D.; Gilbride, R.M.; Hughes, C.M.; Ventura, A.B.; Ainslie, E.; et al. Prevention of tuberculosis in rhesus macaques by a cytomegalovirus-based vaccine. Nat. Med. 2018, 24, 130–143. [Google Scholar] [CrossRef]
- Belnoue, E.; Vogelzang, A.; Nieuwenhuizen, N.E.; Krzyzaniak, M.A.; Darbre, S.; Kreutzfeldt, M.; Wagner, I.; Merkler, D.; Lambert, P.H.; Kaufmann, S.H.E.; et al. Replication-Deficient Lymphocytic Choriomeningitis Virus-Vectored Vaccine Candidate for the Induction of T Cell Immunity against Mycobacterium tuberculosis. Int. J. Mol. Sci. 2022, 23, 2700. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mata-Espinosa, D.; Lara-Espinosa, J.V.; Barrios-Payán, J.; Hernández-Pando, R. The Use of Viral Vectors for Gene Therapy and Vaccination in Tuberculosis. Pharmaceuticals 2023, 16, 1475. https://doi.org/10.3390/ph16101475
Mata-Espinosa D, Lara-Espinosa JV, Barrios-Payán J, Hernández-Pando R. The Use of Viral Vectors for Gene Therapy and Vaccination in Tuberculosis. Pharmaceuticals. 2023; 16(10):1475. https://doi.org/10.3390/ph16101475
Chicago/Turabian StyleMata-Espinosa, Dulce, Jacqueline V. Lara-Espinosa, Jorge Barrios-Payán, and Rogelio Hernández-Pando. 2023. "The Use of Viral Vectors for Gene Therapy and Vaccination in Tuberculosis" Pharmaceuticals 16, no. 10: 1475. https://doi.org/10.3390/ph16101475
APA StyleMata-Espinosa, D., Lara-Espinosa, J. V., Barrios-Payán, J., & Hernández-Pando, R. (2023). The Use of Viral Vectors for Gene Therapy and Vaccination in Tuberculosis. Pharmaceuticals, 16(10), 1475. https://doi.org/10.3390/ph16101475