
A Pharmacological Investigation of Eph-Ephrin Antagonism in Prostate Cancer: UniPR1331 Efficacy Evidence

 , , ,
, , , 
 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
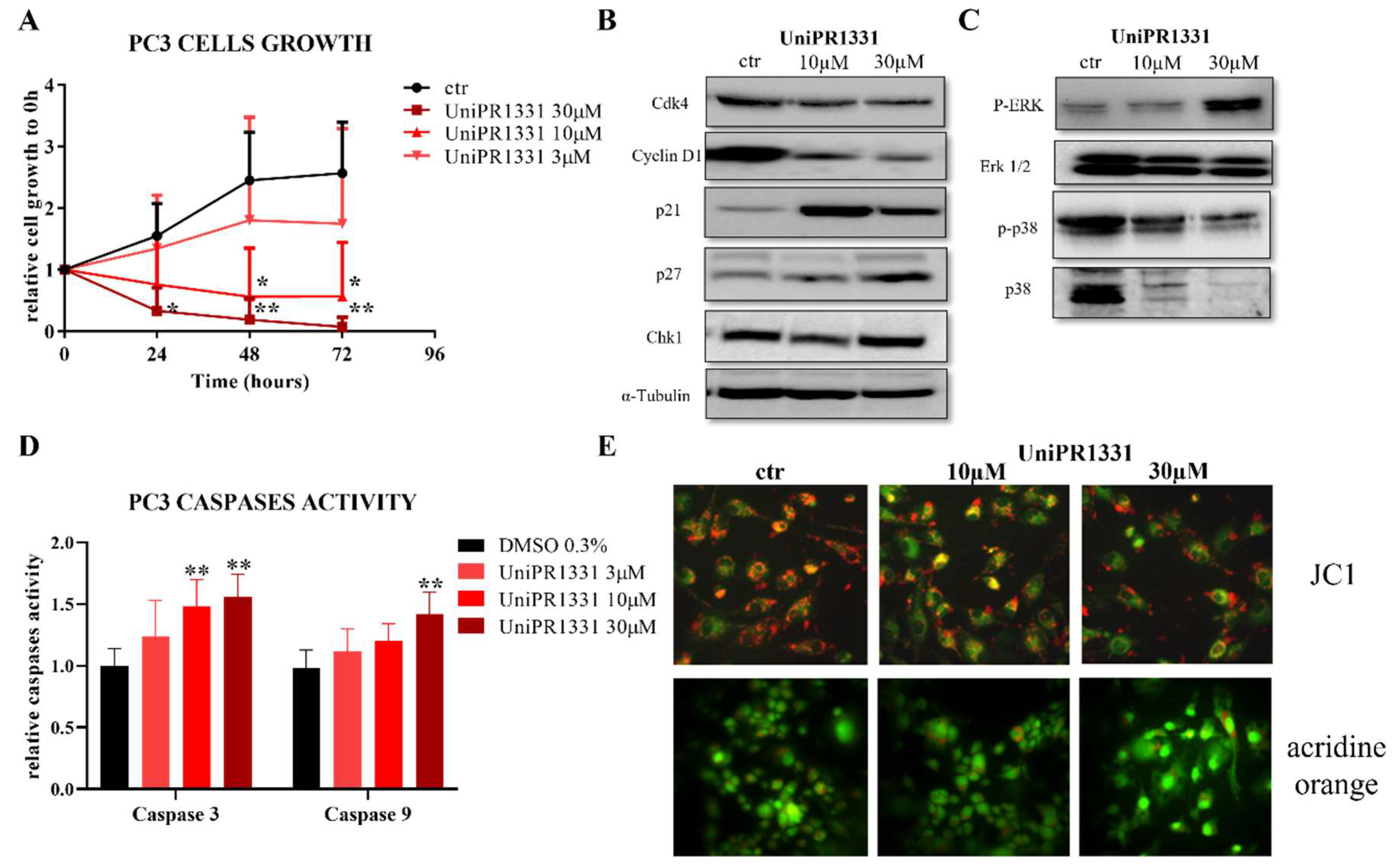
2.1. UniPR1331 Inhibited PC3 Cells’ Growth In Vitro
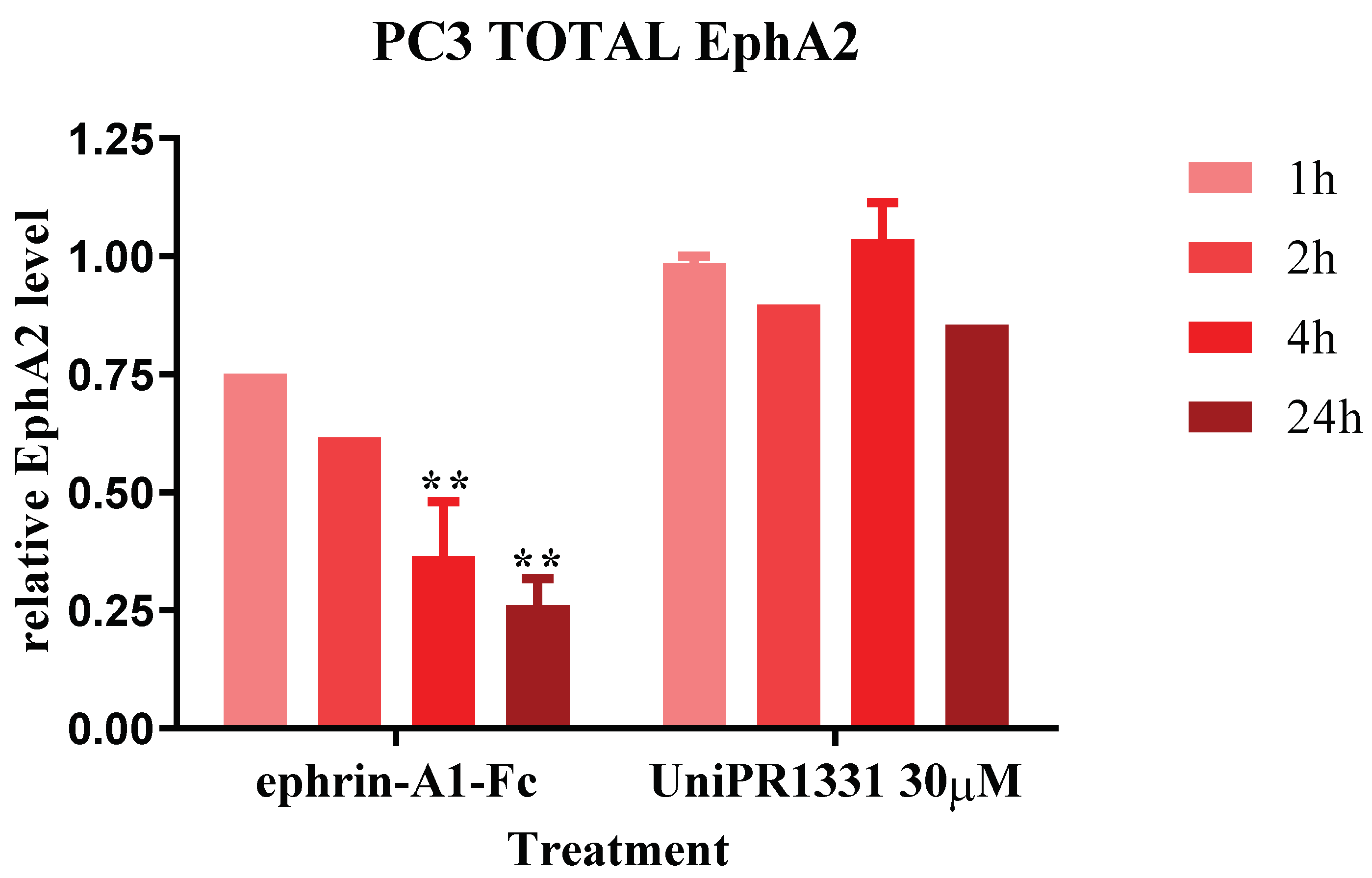
2.2. UniPR1331 Did Not Cause the EphA2 Internalization in PC3 Cells
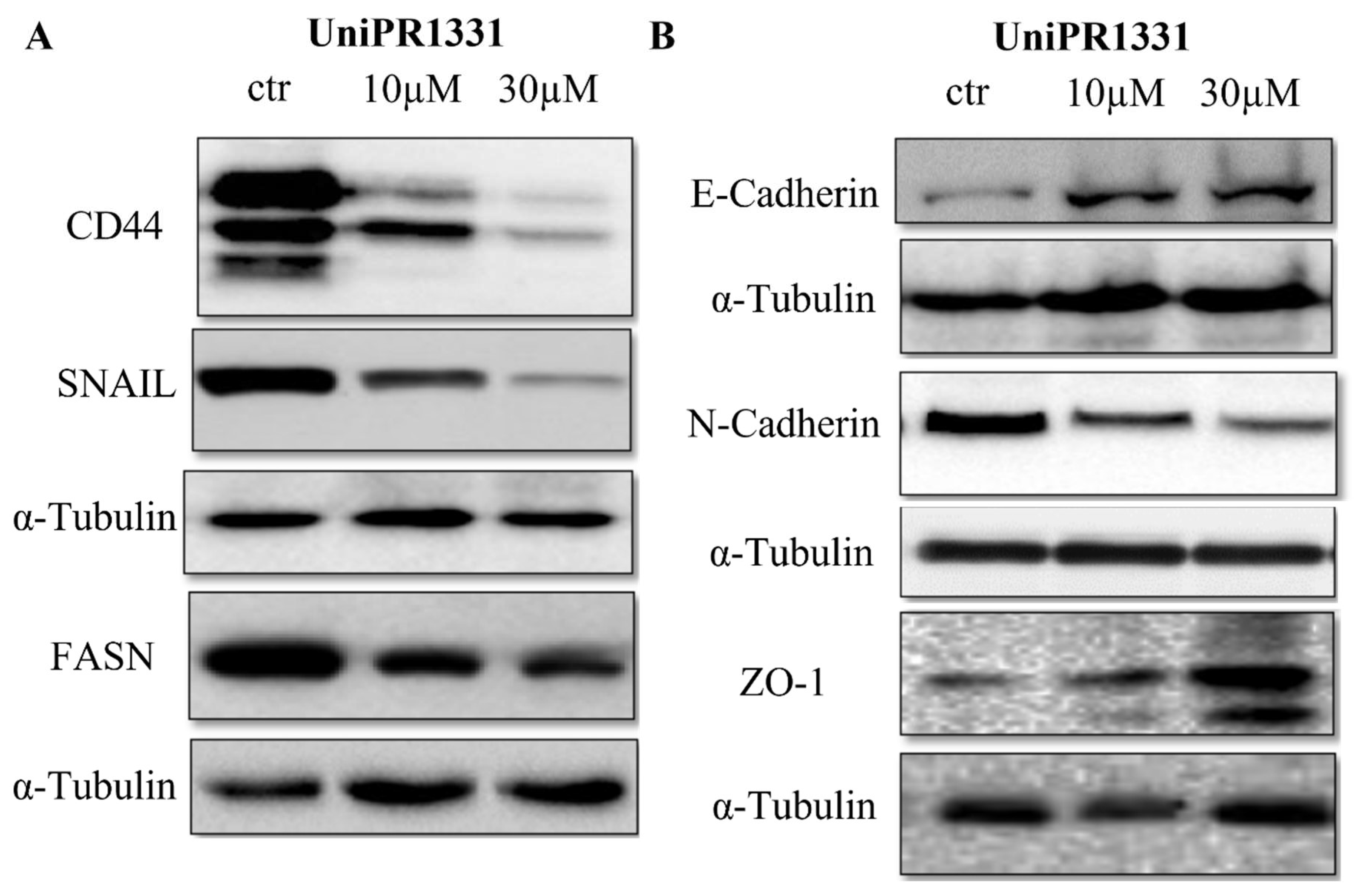
2.3. UniPR1331 Inhibited PC3 Epithelial–Mesenchymal Transition (EMT) and Promoted Epithelial Phenotype
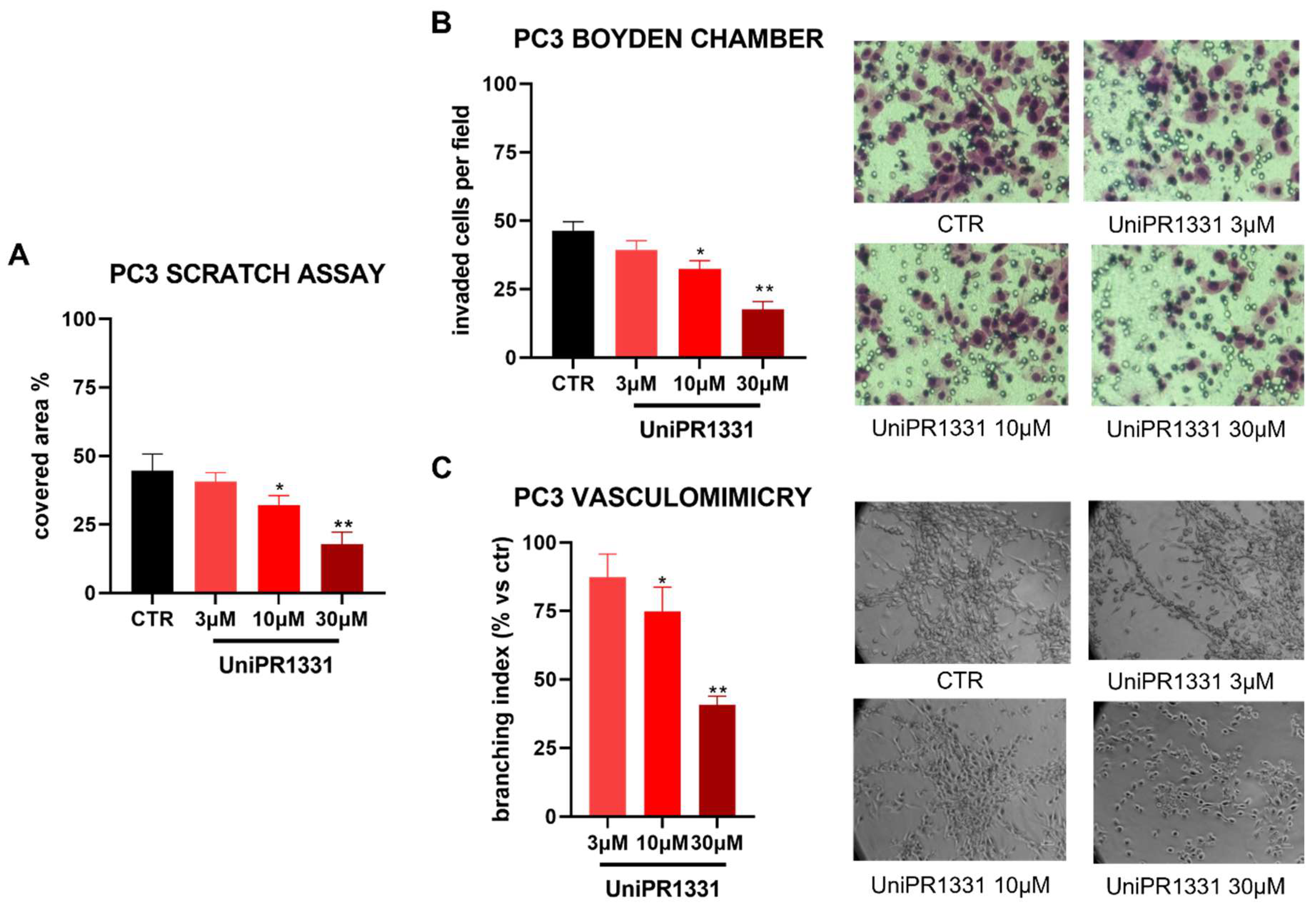
2.4. UniPR1331 Inhibited PC3 Migration, Invasion, and Vasculomimicry Capabilities
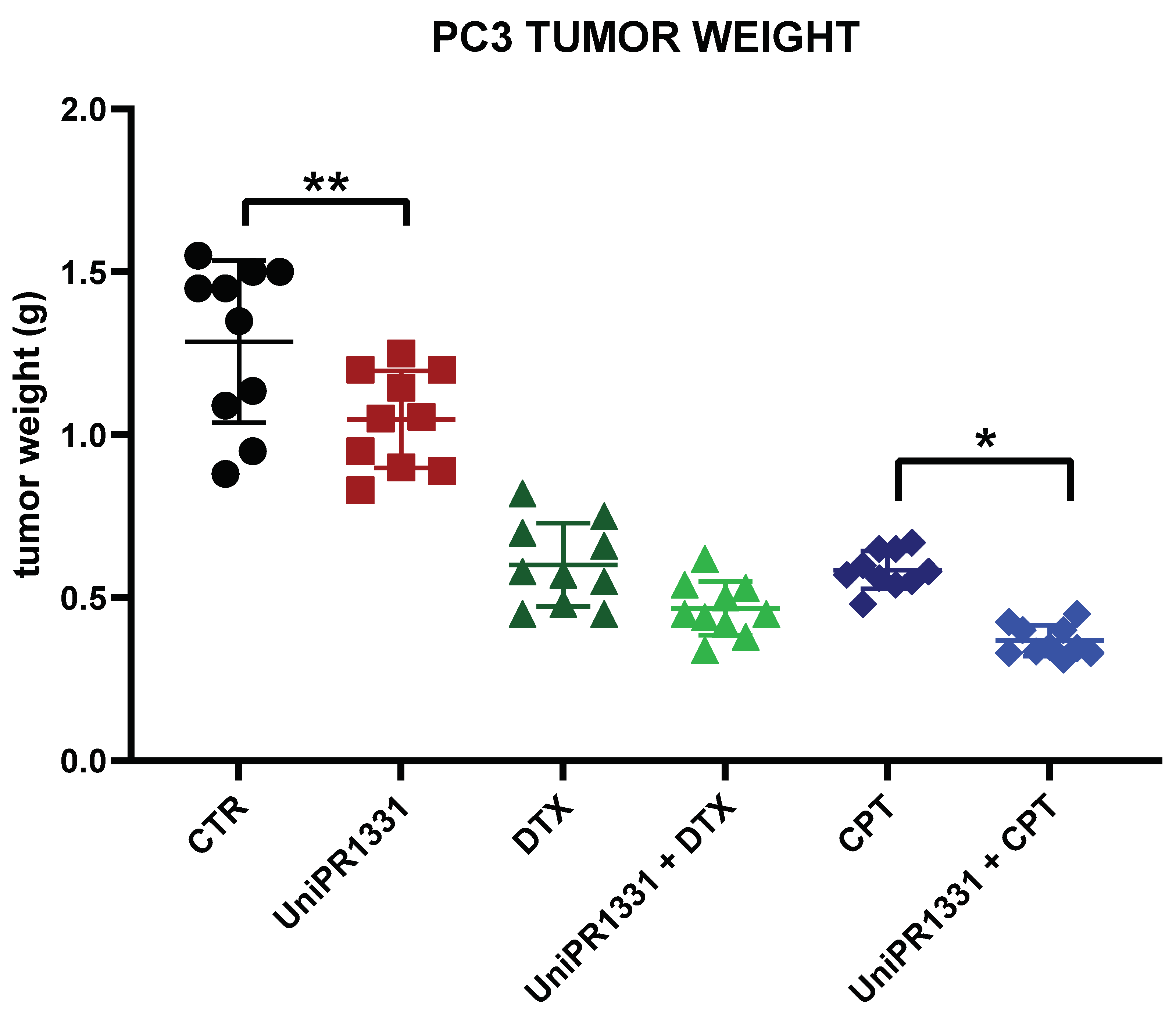
2.5. UniPR1331 and the Associations with Cytotoxic Drugs Reduced the Cancer Growth in PC3-Xenograft Mice
3. Discussion
4. Materials and Methods
4.1. PC3 Culture
4.2. MTT Assay
4.3. Human Total EphA2 ELISA
4.4. Preparation of Cell Lysates and Western Blot Analysis
4.5. JC1 Staining and Acridin Orange Staining
4.6. Scratch Assay
4.7. Boyden Chamber
4.8. Vasculomimicry (VM)
4.9. Xenograft Model
4.10. Treatments for In Vivo Experiments
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kania, A.; Klein, R. Mechanisms of Ephrin-Eph Signalling in Development, Physiology and Disease. Nat. Rev. Mol. Cell. Biol. 2016, 17, 240–256. [Google Scholar] [CrossRef]
- Giorgio, C.; Zanotti, I.; Lodola, A.; Tognolini, M. Ephrin or Not? Six Tough Questions on Eph Targeting. Expert Opin. Ther. Targets 2020, 24, 403–415. [Google Scholar] [CrossRef]
- Kosinski, C.; Li, V.S.W.; Chan, A.S.Y.; Zhang, J.; Ho, C.; Tsui, W.Y.; Chan, T.L.; Mifflin, R.C.; Powell, D.W.; Yuen, S.T.; et al. Gene Expression Patterns of Human Colon Tops and Basal Crypts and BMP Antagonists as Intestinal Stem Cell Niche Factors. Proc. Natl. Acad. Sci. USA 2007, 104, 15418–15423. [Google Scholar] [CrossRef]
- Nomura, T.; Göritz, C.; Catchpole, T.; Henkemeyer, M.; Frisén, J. EphB Signaling Controls Lineage Plasticity of Adult Neural Stem Cell Niche Cells. Cell Stem Cell 2010, 7, 730–743. [Google Scholar] [CrossRef]
- Ji, H.; Goode, R.J.A.; Vaillant, F.; Mathivanan, S.; Kapp, E.A.; Mathias, R.A.; Lindeman, G.J.; Visvader, J.E.; Simpson, R.J. Proteomic Profiling of Secretome and Adherent Plasma Membranes from Distinct Mammary Epithelial Cell Subpopulations. Proteomics 2011, 11, 4029–4039. [Google Scholar] [CrossRef]
- Genander, M.; Holmberg, J.; Frisén, J. Ephrins Negatively Regulate Cell Proliferation in the Epidermis and Hair Follicle. Stem Cells 2010, 28, 1196–1205. [Google Scholar] [CrossRef]
- Konstantinova, I.; Nikolova, G.; Ohara-Imaizumi, M.; Meda, P.; Kucera, T.; Zarbalis, K.; Wurst, W.; Nagamatsu, S.; Lammert, E. EphA-Ephrin-A-Mediated Beta Cell Communication Regulates Insulin Secretion from Pancreatic Islets. Cell 2007, 129, 359–370. [Google Scholar] [CrossRef]
- Prevost, N.; Woulfe, D.; Tanaka, T.; Brass, L.F. Interactions between Eph Kinases and Ephrins Provide a Mechanism to Support Platelet Aggregation Once Cell-to-Cell Contact Has Occurred. Proc. Natl. Acad. Sci. USA 2002, 99, 9219–9224. [Google Scholar] [CrossRef] [PubMed]
- Janes, P.W.; Vail, M.E.; Ernst, M.; Scott, A.M. Eph Receptors in the Immunosuppressive Tumor Microenvironment. Cancer Res. 2021, 81, 801–805. [Google Scholar] [CrossRef]
- Arora, S.; Scott, A.M.; Janes, P.W. Eph Receptors in Cancer. Biomedicines 2023, 11, 315. [Google Scholar] [CrossRef]
- Pergaris, A.; Danas, E.; Goutas, D.; Sykaras, A.G.; Soranidis, A.; Theocharis, S. The Clinical Impact of the EPH/Ephrin System in Cancer: Unwinding the Thread. Int. J. Mol. Sci. 2021, 22, 8412. [Google Scholar] [CrossRef]
- Anderton, M.; van der Meulen, E.; Blumenthal, M.J.; Schäfer, G. The Role of the Eph Receptor Family in Tumorigenesis. Cancers 2021, 13, 206. [Google Scholar] [CrossRef]
- Lodola, A.; Giorgio, C.; Incerti, M.; Zanotti, I.; Tognolini, M. Targeting Eph/Ephrin System in Cancer Therapy. Eur. J. Med. Chem. 2017, 142, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Tröster, A.; Jores, N.; Mineev, K.S.; Sreeramulu, S.; DiPrima, M.; Tosato, G.; Schwalbe, H. Targeting EPHA2 with Kinase Inhibitors in Colorectal Cancer. ChemMedChem 2023, e202300420. [Google Scholar] [CrossRef] [PubMed]
- Psilopatis, I.; Karniadakis, I.; Danos, K.S.; Vrettou, K.; Michaelidou, K.; Mavridis, K.; Agelaki, S.; Theocharis, S. May EPH/Ephrin Targeting Revolutionize Lung Cancer Treatment? IJMS 2022, 24, 93. [Google Scholar] [CrossRef] [PubMed]
- Buckens, O.J.; El Hassouni, B.; Giovannetti, E.; Peters, G.J. The Role of Eph Receptors in Cancer and How to Target Them: Novel Approaches in Cancer Treatment. Expert Opin. Investig. Drugs 2020, 29, 567–582. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, C.; Hassan Mohamed, I.; Flammini, L.; Barocelli, E.; Incerti, M.; Lodola, A.; Tognolini, M. Lithocholic Acid Is an Eph-Ephrin Ligand Interfering with Eph-Kinase Activation. PLoS ONE 2011, 6, e18128. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, C.; Incerti, M.; Corrado, M.; Rusnati, M.; Chiodelli, P.; Russo, S.; Callegari, D.; Ferlenghi, F.; Ballabeni, V.; Barocelli, E.; et al. Pharmacological Evaluation of New Bioavailable Small Molecules Targeting Eph/Ephrin Interaction. Biochem. Pharmacol. 2018, 147, 21–29. [Google Scholar] [CrossRef]
- Tognolini, M.; Incerti, M.; Pala, D.; Russo, S.; Castelli, R.; Hassan-Mohamed, I.; Giorgio, C.; Lodola, A. Target Hopping as a Useful Tool for the Identification of Novel EphA2 Protein-Protein Antagonists. ChemMedChem 2014, 9, 67–72. [Google Scholar] [CrossRef]
- Castelli, R.; Tognolini, M.; Vacondio, F.; Incerti, M.; Pala, D.; Callegari, D.; Bertoni, S.; Giorgio, C.; Hassan-Mohamed, I.; Zanotti, I.; et al. Δ(5)-Cholenoyl-Amino Acids as Selective and Orally Available Antagonists of the Eph-Ephrin System. Eur. J. Med. Chem. 2015, 103, 312–324. [Google Scholar] [CrossRef]
- Miao, H.; Burnett, E.; Kinch, M.; Simon, E.; Wang, B. Activation of EphA2 Kinase Suppresses Integrin Function and Causes Focal-Adhesion-Kinase Dephosphorylation. Nat. Cell Biol. 2000, 2, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Petty, A.; Myshkin, E.; Qin, H.; Guo, H.; Miao, H.; Tochtrop, G.P.; Hsieh, J.-T.; Page, P.; Liu, L.; Lindner, D.J.; et al. A Small Molecule Agonist of EphA2 Receptor Tyrosine Kinase Inhibits Tumor Cell Migration in Vitro and Prostate Cancer Metastasis in Vivo. PLoS ONE 2012, 7, e42120. [Google Scholar] [CrossRef] [PubMed]
- Salem, A.F.; Gambini, L.; Billet, S.; Sun, Y.; Oshiro, H.; Zhao, M.; Hoffman, R.M.; Bhowmick, N.A.; Pellecchia, M. Prostate Cancer Metastases Are Strongly Inhibited by Agonistic Epha2 Ligands in an Orthotopic Mouse Model. Cancers 2020, 12, 2854. [Google Scholar] [CrossRef]
- Festuccia, C.; Gravina, G.L.; Giorgio, C.; Mancini, A.; Pellegrini, C.; Colapietro, A.; Delle Monache, S.; Maturo, M.G.; Sferra, R.; Chiodelli, P.; et al. UniPR1331, a Small Molecule Targeting Eph/Ephrin Interaction, Prolongs Survival in Glioblastoma and Potentiates the Effect of Antiangiogenic Therapy in Mice. Oncotarget 2018, 9, 24347–24363. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.; Gale, N.W.; Aldrich, T.H.; Maisonpierre, P.C.; Lhotak, V.; Pawson, T.; Goldfarb, M.; Yancopoulos, G.D. Ligands for EPH-Related Receptor Tyrosine Kinases That Require Membrane Attachment or Clustering for Activity. Science 1994, 266, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Serttas, R.; Erdogan, S. Pretreatment of Prostate Cancer Cells with Salinomycin and Wnt Inhibitor Increases the Efficacy of Cabazitaxel by Inducing Apoptosis and Decreasing Cancer Stem Cells. Med. Oncol. 2023, 40, 194. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Liu, H.; Qiu, G.-P.; Gao, F. Silencing Akt1 Enhances the Resistance of Prostate Cancer Cells to Starvation and Inhibits Starvation-Induced Lung Metastasis through Epithelial-Mesenchymal Transition in Prostate Cancer. Med. Oncol. 2021, 39, 8. [Google Scholar] [CrossRef] [PubMed]
- Zadra, G.; Ribeiro, C.F.; Chetta, P.; Ho, Y.; Cacciatore, S.; Gao, X.; Syamala, S.; Bango, C.; Photopoulos, C.; Huang, Y.; et al. Inhibition of de Novo Lipogenesis Targets Androgen Receptor Signaling in Castration-Resistant Prostate Cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 631–640. [Google Scholar] [CrossRef]
- Sun, Y.; Schaar, A.; Sukumaran, P.; Dhasarathy, A.; Singh, B.B. TGFβ-Induced Epithelial-to-Mesenchymal Transition in Prostate Cancer Cells Is Mediated via TRPM7 Expression. Mol. Carcinog. 2018, 57, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.A.; Peixoto, A.; Neves, M.; Gaiteiro, C.; Reis, C.A.; Assaraf, Y.G.; Santos, L.L. Mechanisms of Cisplatin Resistance and Targeting of Cancer Stem Cells: Adding Glycosylation to the Equation. Drug Resist. Updates 2016, 24, 34–54. [Google Scholar] [CrossRef]
- Fox, B.P.; Tabone, C.J.; Kandpal, R.P. Potential Clinical Relevance of Eph Receptors and Ephrin Ligands Expressed in Prostate Carcinoma Cell Lines. Biochem. Biophys. Res. Commun. 2006, 342, 1263–1272. [Google Scholar] [CrossRef]
- Hassan-Mohamed, I.; Giorgio, C.; Incerti, M.; Russo, S.; Pala, D.; Pasquale, E.B.; Zanotti, I.; Vicini, P.; Barocelli, E.; Rivara, S.; et al. UniPR129 Is a Competitive Small Molecule Eph-Ephrin Antagonist Blocking in Vitro Angiogenesis at Low Micromolar Concentrations. Br. J. Pharmacol. 2014, 171, 5195–5208. [Google Scholar] [CrossRef]
- Yang, N.-Y.; Fernandez, C.; Richter, M.; Xiao, Z.; Valencia, F.; Tice, D.A.; Pasquale, E.B. Crosstalk of the EphA2 Receptor with a Serine/Threonine Phosphatase Suppresses the Akt-MTORC1 Pathway in Cancer Cells. Cell. Signal. 2011, 23, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Tawadros, T.; Brown, M.D.; Hart, C.A.; Clarke, N.W. Ligand-Independent Activation of EphA2 by Arachidonic Acid Induces Metastasis-like Behaviour in Prostate Cancer Cells. Br. J. Cancer 2012, 107, 1737–1744. [Google Scholar] [CrossRef]
- Shukla, S.; Gupta, S. Apigenin-Induced Cell Cycle Arrest Is Mediated by Modulation of MAPK, PI3K-Akt, and Loss of Cyclin D1 Associated Retinoblastoma Dephosphorylation in Human Prostate Cancer Cells. Cell Cycle 2007, 6, 1102–1114. [Google Scholar] [CrossRef]
- Lin, J.-F.; Tsai, T.-F.; Liao, P.-C.; Lin, Y.-H.; Lin, Y.-C.; Chen, H.-E.; Chou, K.-Y.; Hwang, T.I.-S. Benzyl Isothiocyanate Induces Protective Autophagy in Human Prostate Cancer Cells via Inhibition of MTOR Signaling. Carcinogenesis 2013, 34, 406–414. [Google Scholar] [CrossRef]
- Kim, S.-H.; Kim, K.-Y.; Yu, S.-N.; Park, S.-K.; Choi, H.-D.; Ji, J.-H.; Ahn, S.-C. Autophagy Inhibition Enhances Silibinin-Induced Apoptosis by Regulating Reactive Oxygen Species Production in Human Prostate Cancer PC-3 Cells. Biochem. Biophys. Res. Commun. 2015, 468, 151–156. [Google Scholar] [CrossRef]
- Tai, S.; Sun, Y.; Squires, J.M.; Zhang, H.; Oh, W.K.; Liang, C.-Z.; Huang, J. PC3 Is a Cell Line Characteristic of Prostatic Small Cell Carcinoma. Prostate 2011, 71, 1668–1679. [Google Scholar] [CrossRef]
- Yu, M.; Wang, J.; Muller, D.J.; Helenius, J. In PC3 Prostate Cancer Cells Ephrin Receptors Crosstalk to Β1-Integrins to Strengthen Adhesion to Collagen Type I. Sci. Rep. 2015, 5, 8206. [Google Scholar] [CrossRef]
- Kaighn, M.E.; Narayan, K.S.; Ohnuki, Y.; Lechner, J.F.; Jones, L.W. Establishment and Characterization of a Human Prostatic Carcinoma Cell Line (PC-3). Invest. Urol. 1979, 17, 16–23. [Google Scholar]
- Tae, J.H.; Chang, I.H. Animal Models of Bone Metastatic Prostate Cancer. Investig. Clin. Urol. 2023, 64, 219. [Google Scholar] [CrossRef]
- Mollica, V.; Nuvola, G.; Tassinari, E.; Nigro, M.C.; Marchetti, A.; Rosellini, M.; Rizzo, A.; Errani, C.; Massari, F. Bone Targeting Agents in Patients with Prostate Cancer: General Toxicities and Osteonecrosis of the Jaw. Curr. Oncol. 2022, 29, 1709–1722. [Google Scholar] [CrossRef]
- Ferrari, F.R.; Giorgio, C.; Zappia, A.; Ballabeni, V.; Bertoni, S.; Barocelli, E.; Scalvini, L.; Galvani, F.; Mor, M.; Lodola, A.; et al. Pharmacological Characterization of Second Generation FXR Agonists as Effective EphA2 Antagonists: A Successful Application of Target Hopping Approach. Biochem. Pharmacol. 2023, 209, 115452. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Festuccia, C.; Corrado, M.; Rossetti, A.; Castelli, R.; Lodola, A.; Gravina, G.L.; Tognolini, M.; Giorgio, C. A Pharmacological Investigation of Eph-Ephrin Antagonism in Prostate Cancer: UniPR1331 Efficacy Evidence. Pharmaceuticals 2023, 16, 1452. https://doi.org/10.3390/ph16101452
Festuccia C, Corrado M, Rossetti A, Castelli R, Lodola A, Gravina GL, Tognolini M, Giorgio C. A Pharmacological Investigation of Eph-Ephrin Antagonism in Prostate Cancer: UniPR1331 Efficacy Evidence. Pharmaceuticals. 2023; 16(10):1452. https://doi.org/10.3390/ph16101452
Chicago/Turabian StyleFestuccia, Claudio, Miriam Corrado, Alessandra Rossetti, Riccardo Castelli, Alessio Lodola, Giovanni Luca Gravina, Massimiliano Tognolini, and Carmine Giorgio. 2023. "A Pharmacological Investigation of Eph-Ephrin Antagonism in Prostate Cancer: UniPR1331 Efficacy Evidence" Pharmaceuticals 16, no. 10: 1452. https://doi.org/10.3390/ph16101452
APA StyleFestuccia, C., Corrado, M., Rossetti, A., Castelli, R., Lodola, A., Gravina, G. L., Tognolini, M., & Giorgio, C. (2023). A Pharmacological Investigation of Eph-Ephrin Antagonism in Prostate Cancer: UniPR1331 Efficacy Evidence. Pharmaceuticals, 16(10), 1452. https://doi.org/10.3390/ph16101452