
Heparin Precursors with Reduced Anticoagulant Properties Retain Antiviral and Protective Effects That Potentiate the Efficacy of Sofosbuvir against Zika Virus Infection in Human Neural Progenitor Cells
 , , , , and
, , , , and
Abstract
:1. Introduction
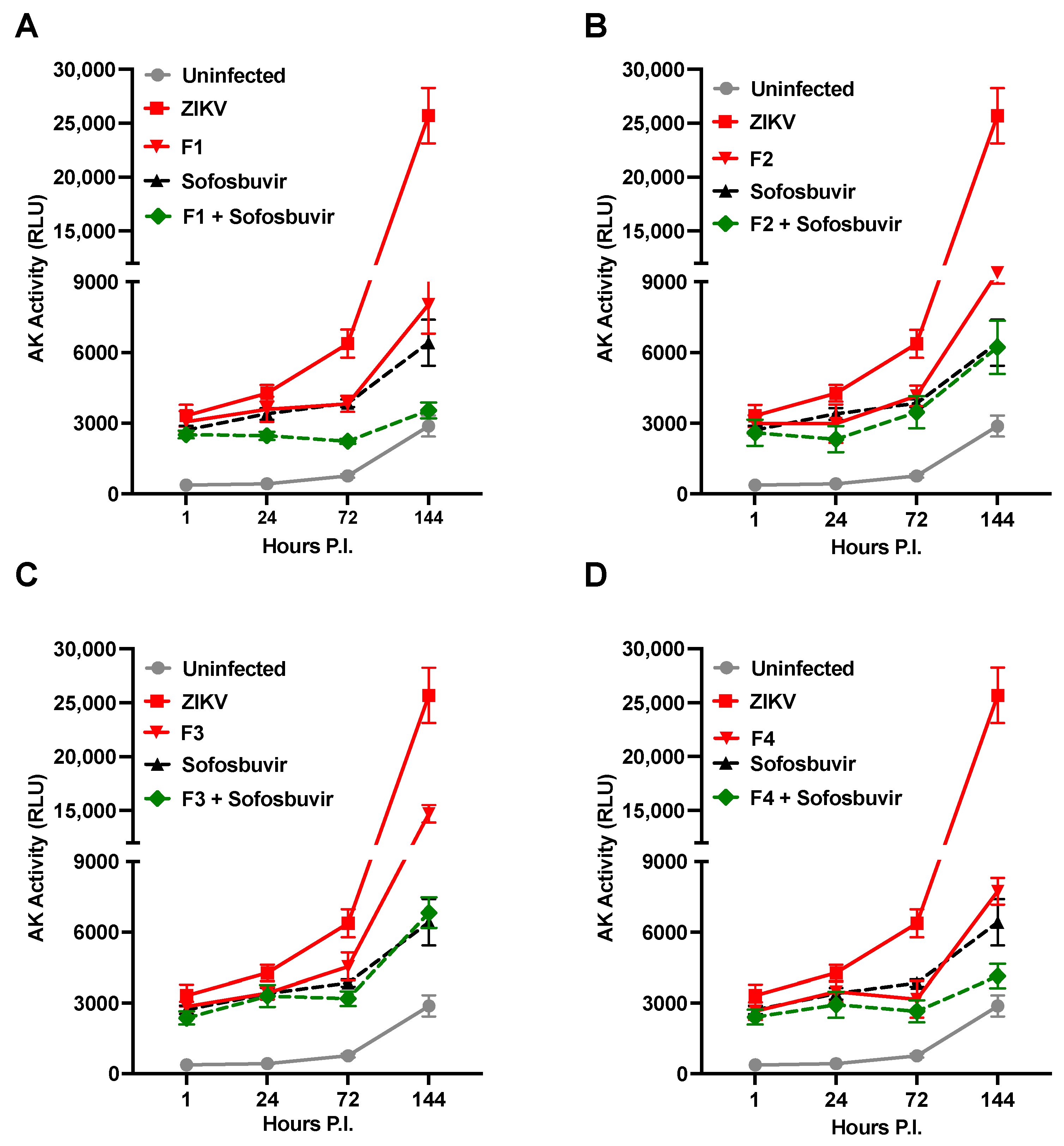
2. Results
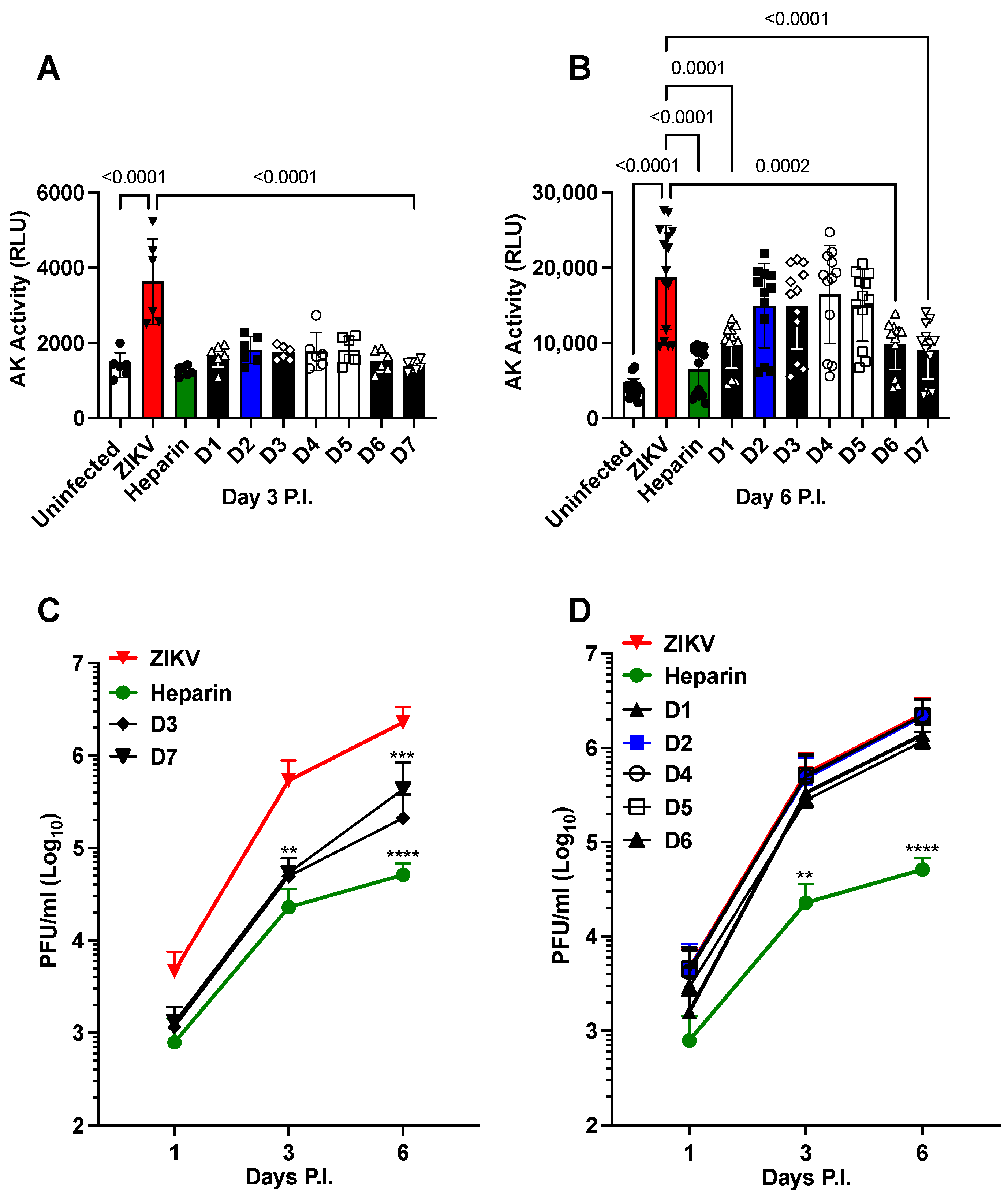
- (i)
- Chemically modified heparin derivatives (D1 to D7) prevent cell death independently of anticoagulant activity.
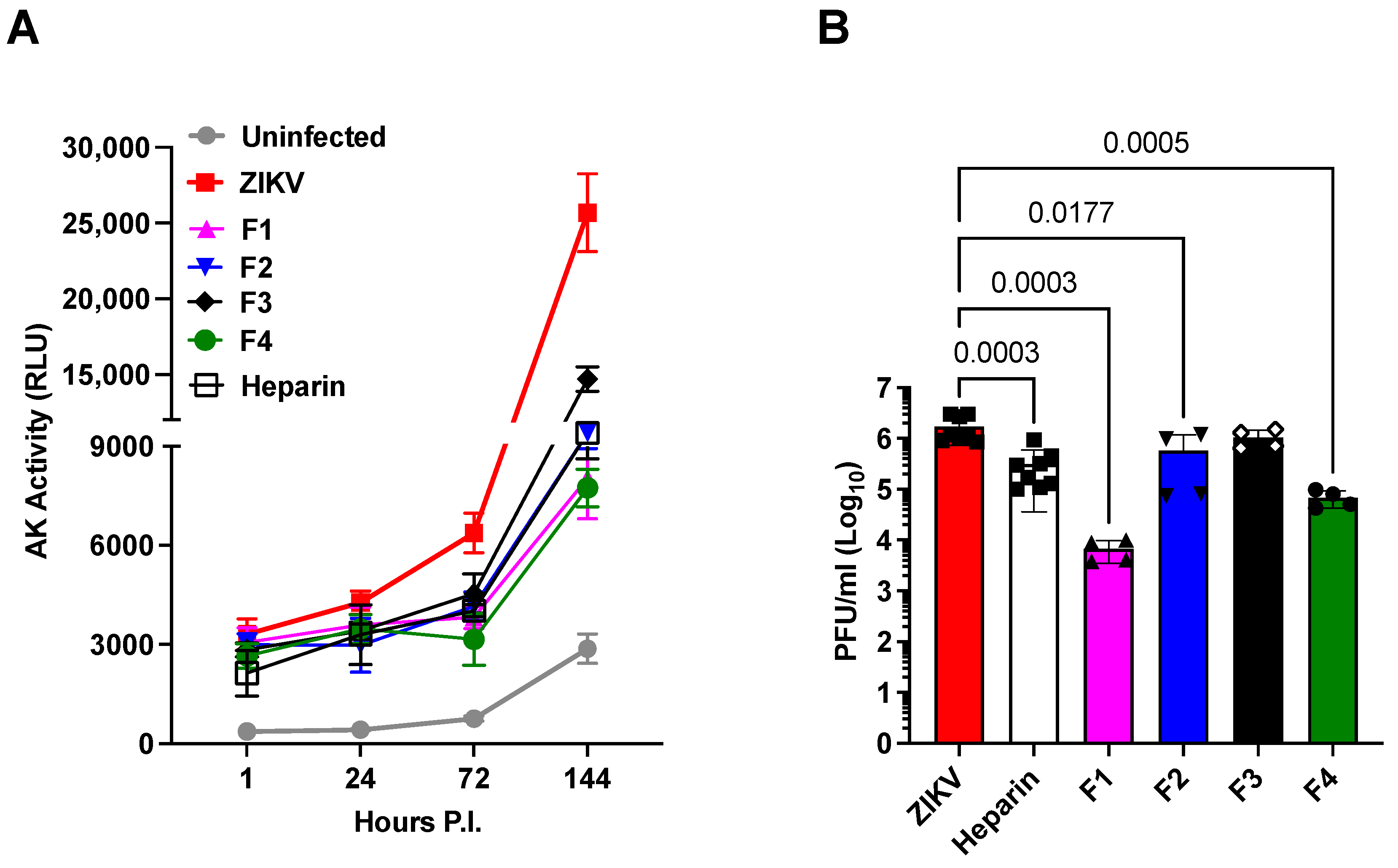
- (ii)
- Heparin precursor fractions (F1 to F4) possess antiviral activities independently of their reduced anticoagulant effects.
- (iii)
- The overall charge of the heparin precursor material fractions F1 to F4 does not correlate with their antiviral activities, and no single substitution predominates in the most active fractions, F1 and F4.
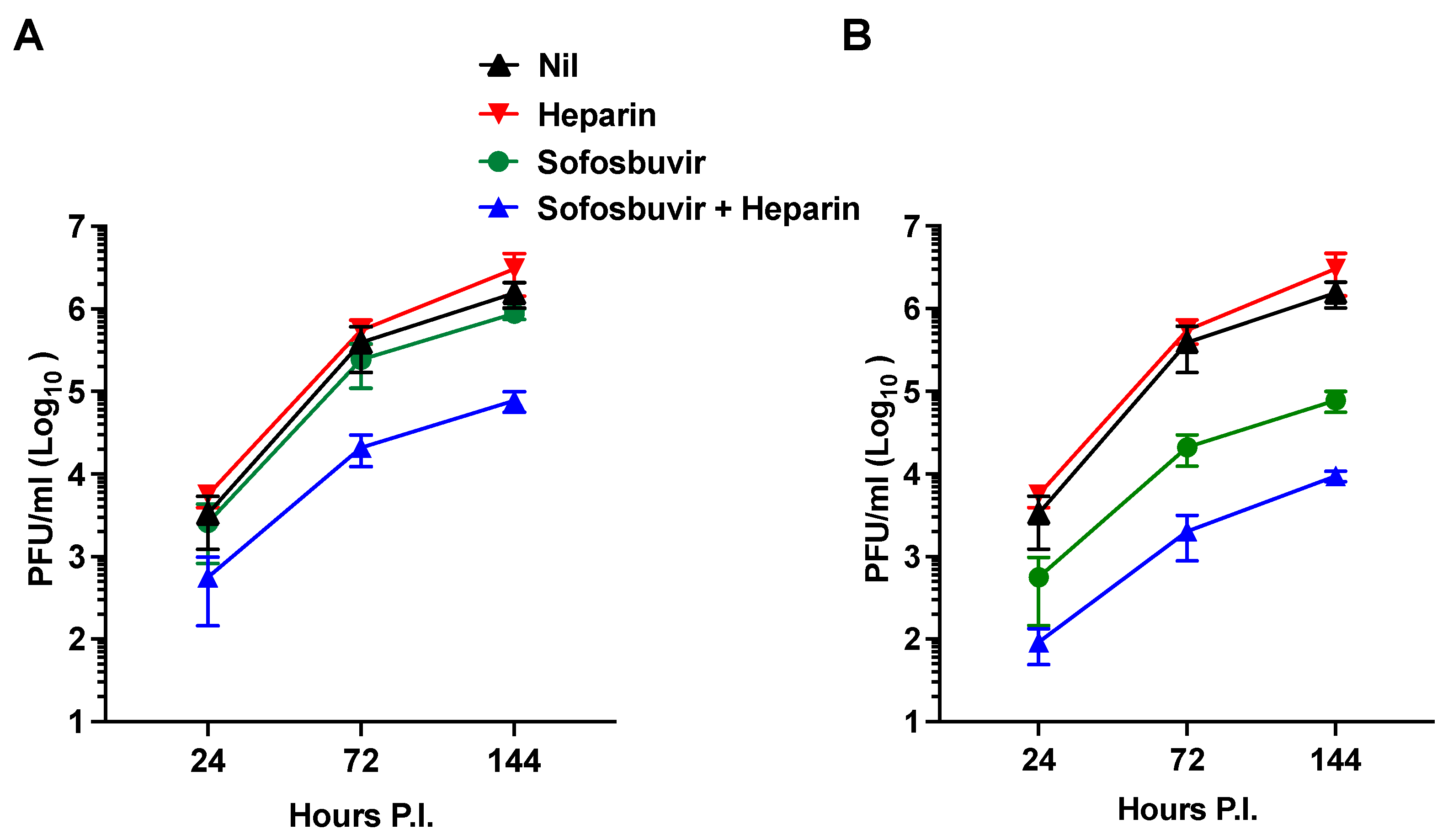
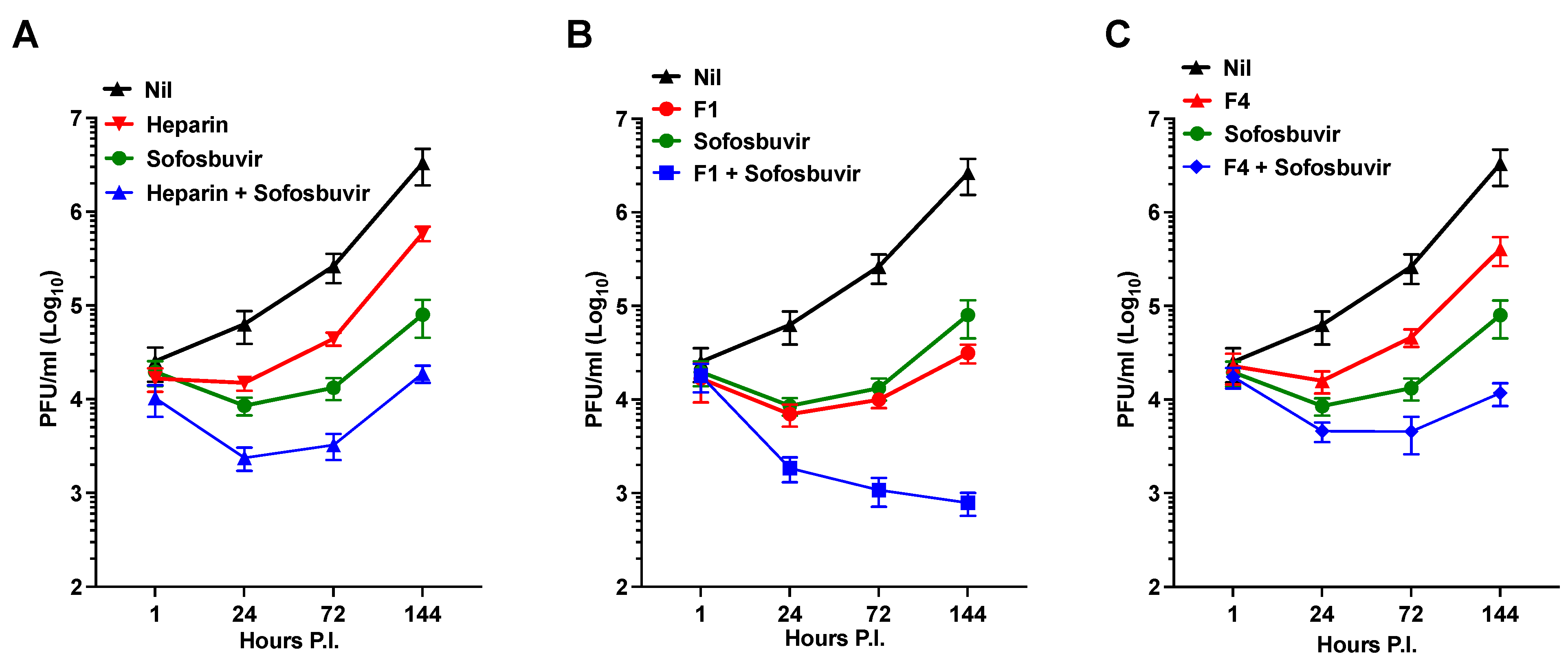
- (iv)
- Synergistic antiviral effects were observed for the dual use of Sofosbuvir, an established antiviral agent, and heparin.
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Viruses
5.2. Human iPSC-Derived NPCs and Neurospheres (NS)
5.3. Infections
5.4. Plaque Forming Assay (PFA)
5.5. Cell Death Detection Assay
5.6. Heparin and Chemically Modified Heparin Derivatives (D1 to D7) and Heparin Precursor Fractions (F1 to F4)
5.7. Anticoagulation Assays
5.8. Weight Average, Molecular Weight, and Dispersity
5.9. Degree of Sulfation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torri, G.; Naggi, A. Heparin centenary—An ever-young life-saving drug. Int. J. Cardiol. 2016, 212 (Suppl. S1), S1–S4. [Google Scholar] [CrossRef] [PubMed]
- Blann, A.D.; Landray, M.J.; Lip, G.Y. ABC of antithrombotic therapy: An overview of antithrombotic therapy. BMJ 2002, 325, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Hirsh, J.; Anand, S.S.; Halperin, J.L.; Fuster, V.; American Heart, A. Guide to anticoagulant therapy: Heparin: A statement for healthcare professionals from the American Heart Association. Circulation 2001, 103, 2994–3018. [Google Scholar] [CrossRef] [PubMed]
- Bernfield, M.; Gotte, M.; Park, P.W.; Reizes, O.; Fitzgerald, M.L.; Lincecum, J.; Zako, M. Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem. 1999, 68, 729–777. [Google Scholar] [CrossRef] [PubMed]
- Casu, B.; Naggi, A.; Torri, G. Heparin-derived heparan sulfate mimics to modulate heparan sulfate-protein interaction in inflammation and cancer. Matrix Biol. 2010, 29, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan sulfate proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3, a004952. [Google Scholar] [CrossRef]
- Shukla, D.; Liu, J.; Blaiklock, P.; Shworak, N.W.; Bai, X.; Esko, J.D.; Cohen, G.H.; Eisenberg, R.J.; Rosenberg, R.D.; Spear, P.G. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 1999, 99, 13–22. [Google Scholar] [CrossRef]
- Tamhankar, M.; Gerhardt, D.M.; Bennett, R.S.; Murphy, N.; Jahrling, P.B.; Patterson, J.L. Heparan sulfate is an important mediator of Ebola virus infection in polarized epithelial cells. Virol. J. 2018, 15, 135. [Google Scholar] [CrossRef]
- Clausen, T.M.; Sandoval, D.R.; Spliid, C.B.; Pihl, J.; Perrett, H.R.; Painter, C.D.; Narayanan, A.; Majowicz, S.A.; Kwong, E.M.; McVicar, R.N.; et al. SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell 2020, 183, 1043–1057.e15. [Google Scholar] [CrossRef]
- Mycroft-West, C.J.; Su, D.; Pagani, I.; Rudd, T.R.; Elli, S.; Gandhi, N.S.; Guimond, S.E.; Miller, G.J.; Meneghetti, M.C.Z.; Nader, H.B.; et al. Heparin Inhibits Cellular Invasion by SARS-CoV-2: Structural Dependence of the Interaction of the Spike S1 Receptor-Binding Domain with Heparin. Thromb. Haemost. 2020, 120, 1700–1715. [Google Scholar] [CrossRef]
- Cagno, V.; Tseligka, E.D.; Jones, S.T.; Tapparel, C. Heparan Sulfate Proteoglycans and Viral Attachment: True Receptors or Adaptation Bias? Viruses 2019, 11, 596. [Google Scholar] [CrossRef]
- Kim, S.Y.; Li, B.; Linhardt, R.J. Pathogenesis and Inhibition of Flaviviruses from a Carbohydrate Perspective. Pharmaceuticals 2017, 10, 44. [Google Scholar] [CrossRef] [PubMed]
- Ling, J.; Li, J.; Khan, A.; Lundkvist, A.; Li, J.P. Is heparan sulfate a target for inhibition of RNA virus infection? Am. J. Physiol. Cell Physiol. 2022, 322, C605–C613. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Wright, P.J.; Davidson, A.; Lobigs, M. Virulence attenuation of Dengue virus due to augmented glycosaminoglycan-binding affinity and restriction in extraneural dissemination. J. Gen. Virol. 2006, 87, 2791–2801. [Google Scholar] [CrossRef] [PubMed]
- Savidis, G.; McDougall, W.M.; Meraner, P.; Perreira, J.M.; Portmann, J.M.; Trincucci, G.; John, S.P.; Aker, A.M.; Renzette, N.; Robbins, D.R.; et al. Identification of Zika Virus and Dengue Virus Dependency Factors using Functional Genomics. Cell Rep. 2016, 16, 232–246. [Google Scholar] [CrossRef]
- Dick, G.W. Zika virus. II. Pathogenicity and physical properties. Trans. R. Soc. Trop. Med. Hyg. 1952, 46, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Dick, G.W. Epidemiological notes on some viruses isolated in Uganda; Yellow fever, Rift Valley fever, Bwamba fever, West Nile, Mengo, Semliki forest, Bunyamwera, Ntaya, Uganda S and Zika viruses. Trans. R. Soc. Trop. Med. Hyg. 1953, 47, 13–48. [Google Scholar] [CrossRef]
- Ramaiah, A.; Dai, L.; Contreras, D.; Sinha, S.; Sun, R.; Arumugaswami, V. Comparative analysis of protein evolution in the genome of pre-epidemic and epidemic Zika virus. Infect. Genet. Evol. 2017, 51, 74–85. [Google Scholar] [CrossRef]
- Cao-Lormeau, V.M.; Blake, A.; Mons, S.; Lastere, S.; Roche, C.; Vanhomwegen, J.; Dub, T.; Baudouin, L.; Teissier, A.; Larre, P.; et al. Guillain-Barre Syndrome outbreak associated with Zika virus infection in French Polynesia: A case-control study. Lancet 2016, 387, 1531–1539. [Google Scholar] [CrossRef]
- Parra, B.; Lizarazo, J.; Jiménez-Arango, J.A.; Zea-Vera, A.F.; González-Manrique, G.; Vargas, J.; Angarita, J.A.; Zuñiga, G.; Lopez-Gonzalez, R.; Beltran, C.L.; et al. Guillain-Barré Syndrome Associated with Zika Virus Infection in Colombia. N. Engl. J. Med. 2016, 375, 1513–1523. [Google Scholar] [CrossRef]
- Mlakar, J.; Korva, M.; Tul, N.; Popovic, M.; Poljsak-Prijatelj, M.; Mraz, J.; Kolenc, M.; Resman Rus, K.; Vesnaver Vipotnik, T.; Fabjan Vodusek, V.; et al. Zika Virus Associated with Microcephaly. N. Engl. J. Med. 2016, 374, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Microcephaly Epidemic Research, G. Microcephaly in Infants, Pernambuco State, Brazil, 2015. Emerg. Infect. Dis. 2016, 22, 1090–1093. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.C.; Diamond, M.S. The emergence of Zika virus and its new clinical syndromes. Nature 2018, 560, 573–581. [Google Scholar] [CrossRef]
- Freitas, D.A.; Souza-Santos, R.; Carvalho, L.M.A.; Barros, W.B.; Neves, L.M.; Brasil, P.; Wakimoto, M.D. Congenital Zika syndrome: A systematic review. PLoS ONE 2020, 15, e0242367. [Google Scholar] [CrossRef] [PubMed]
- Leao, V.H.P.; Aragao, M.M.; Pinho, R.S.; Hazin, A.N.; Paciorkowski, A.R.; Penalva de Oliveira, A.C.; Masruha, M.R. Congenital Zika Virus Infection: A Review with Emphasis on the Spectrum of Brain Abnormalities. Curr. Neurol. Neurosci. Rep. 2020, 20, 49. [Google Scholar] [CrossRef]
- Kraemer, M.U.G.; Reiner, R.C.; Brady, O.J.; Messina, J.P.; Gilbert, M.; Pigott, D.M.; Yi, D.; Johnson, K.; Earl, L.; Marczak, L.B.; et al. Past and future spread of the arbovirus vectors Aedes aegypti and Aedes albopictus. Nat. Microbiol. 2019, 4, 854–863. [Google Scholar] [CrossRef]
- Foy, B.D.; Kobylinski, K.C.; Chilson Foy, J.L.; Blitvich, B.J.; Travassos da Rosa, A.; Haddow, A.D.; Lanciotti, R.S.; Tesh, R.B. Probable non-vector-borne transmission of Zika virus, Colorado, USA. Emerg. Infect. Dis. 2011, 17, 880–882. [Google Scholar] [CrossRef]
- D’Ortenzio, E.; Matheron, S.; Yazdanpanah, Y.; de Lamballerie, X.; Hubert, B.; Piorkowski, G.; Maquart, M.; Descamps, D.; Damond, F.; Leparc-Goffart, I. Evidence of Sexual Transmission of Zika Virus. N. Engl. J. Med. 2016, 374, 2195–2198. [Google Scholar] [CrossRef]
- Musso, D.; Gubler, D.J. Zika Virus. Clin. Microbiol. Rev. 2016, 29, 487–524. [Google Scholar] [CrossRef]
- Motta, I.J.; Spencer, B.R.; Cordeiro da Silva, S.G.; Arruda, M.B.; Dobbin, J.A.; Gonzaga, Y.B.; Arcuri, I.P.; Tavares, R.C.; Atta, E.H.; Fernandes, R.F.; et al. Evidence for Transmission of Zika Virus by Platelet Transfusion. N. Engl. J. Med. 2016, 375, 1101–1103. [Google Scholar] [CrossRef]
- Albulescu, I.C.; Kovacikova, K.; Tas, A.; Snijder, E.J.; van Hemert, M.J. Suramin inhibits Zika virus replication by interfering with virus attachment and release of infectious particles. Antivir. Res. 2017, 143, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Pagani, I.; Ottoboni, L.; Podini, P.; Ghezzi, S.; Brambilla, E.; Bezukladova, S.; Corti, D.; Bianchi, M.E.; Capobianchi, M.R.; Poli, G.; et al. Heparin Protects Human Neural Progenitor Cells from Zika Virus-Induced Cell Death While Preserving Their Differentiation into Mature Neuroglial Cells. J. Virol. 2022, 96, e0112222. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Zhao, J.; Liu, X.; Fraser, K.; Lin, L.; Zhang, X.; Zhang, F.; Dordick, J.S.; Linhardt, R.J. Interaction of Zika Virus Envelope Protein with Glycosaminoglycans. Biochemistry 2017, 56, 1151–1162. [Google Scholar] [CrossRef]
- Hamel, R.; Dejarnac, O.; Wichit, S.; Ekchariyawat, P.; Neyret, A.; Luplertlop, N.; Perera-Lecoin, M.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; et al. Biology of Zika Virus Infection in Human Skin Cells. J. Virol. 2015, 89, 8880–8896. [Google Scholar] [CrossRef]
- Nowakowski, T.J.; Pollen, A.A.; Di Lullo, E.; Sandoval-Espinosa, C.; Bershteyn, M.; Kriegstein, A.R. Expression Analysis Highlights AXL as a Candidate Zika Virus Entry Receptor in Neural Stem Cells. Cell Stem Cell 2016, 18, 591–596. [Google Scholar] [CrossRef]
- Giraldo, M.I.; Xia, H.; Aguilera-Aguirre, L.; Hage, A.; van Tol, S.; Shan, C.; Xie, X.; Sturdevant, G.L.; Robertson, S.J.; McNally, K.L.; et al. Envelope protein ubiquitination drives entry and pathogenesis of Zika virus. Nature 2020, 585, 414–419. [Google Scholar] [CrossRef]
- Gerold, G.; Bruening, J.; Weigel, B.; Pietschmann, T. Protein Interactions during the Flavivirus and Hepacivirus Life Cycle. Mol. Cell Proteom. 2017, 16, S75–S91. [Google Scholar] [CrossRef]
- Tang, H.; Hammack, C.; Ogden, S.C.; Wen, Z.; Qian, X.; Li, Y.; Yao, B.; Shin, J.; Zhang, F.; Lee, E.M.; et al. Zika Virus Infects Human Cortical Neural Progenitors and Attenuates Their Growth. Cell Stem Cell 2016, 18, 587–590. [Google Scholar] [CrossRef]
- Dang, J.; Tiwari, S.K.; Lichinchi, G.; Qin, Y.; Patil, V.S.; Eroshkin, A.M.; Rana, T.M. Zika Virus Depletes Neural Progenitors in Human Cerebral Organoids through Activation of the Innate Immune Receptor TLR3. Cell Stem Cell 2016, 19, 258–265. [Google Scholar] [CrossRef]
- Onorati, M.; Li, Z.; Liu, F.; Sousa, A.M.M.; Nakagawa, N.; Li, M.; Dell’Anno, M.T.; Gulden, F.O.; Pochareddy, S.; Tebbenkamp, A.T.N.; et al. Zika Virus Disrupts Phospho-TBK1 Localization and Mitosis in Human Neuroepithelial Stem Cells and Radial Glia. Cell Rep. 2016, 16, 2576–2592. [Google Scholar] [CrossRef]
- Wen, Z.; Song, H.; Ming, G.L. How does Zika virus cause microcephaly? Genes. Dev. 2017, 31, 849–861. [Google Scholar] [CrossRef]
- Miner, J.J.; Cao, B.; Govero, J.; Smith, A.M.; Fernandez, E.; Cabrera, O.H.; Garber, C.; Noll, M.; Klein, R.S.; Noguchi, K.K.; et al. Zika Virus Infection during Pregnancy in Mice Causes Placental Damage and Fetal Demise. Cell 2016, 165, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Adams Waldorf, K.M.; Stencel-Baerenwald, J.E.; Kapur, R.P.; Studholme, C.; Boldenow, E.; Vornhagen, J.; Baldessari, A.; Dighe, M.K.; Thiel, J.; Merillat, S.; et al. Fetal brain lesions after subcutaneous inoculation of Zika virus in a pregnant nonhuman primate. Nat. Med. 2016, 22, 1256–1259. [Google Scholar] [CrossRef] [PubMed]
- Bell, T.M.; Field, E.J.; Narang, H.K. Zika virus infection of the central nervous system of mice. Arch. Gesamte Virusforsch. 1971, 35, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.; Zhang, F.; Wang, Y.; Lee, E.M.; Choi, I.Y.; Lim, H.; Mirakhori, F.; Li, R.; Huang, L.; Xu, T.; et al. Zika virus directly infects peripheral neurons and induces cell death. Nat. Neurosci. 2017, 20, 1209–1212. [Google Scholar] [CrossRef]
- Volpi, V.G.; Pagani, I.; Ghezzi, S.; Iannacone, M.; D’Antonio, M.; Vicenzi, E. Zika Virus Replication in Dorsal Root Ganglia Explants from Interferon Receptor1 Knockout Mice Causes Myelin Degeneration. Sci. Rep. 2018, 8, 10166. [Google Scholar] [CrossRef]
- de Paula Freitas, B.; Ventura, C.V.; Maia, M.; Belfort, R., Jr. Zika virus and the eye. Curr. Opin. Ophthalmol. 2017, 28, 595–599. [Google Scholar] [CrossRef]
- Miner, J.J.; Sene, A.; Richner, J.M.; Smith, A.M.; Santeford, A.; Ban, N.; Weger-Lucarelli, J.; Manzella, F.; Ruckert, C.; Govero, J.; et al. Zika Virus Infection in Mice Causes Panuveitis with Shedding of Virus in Tears. Cell Rep. 2016, 16, 3208–3218. [Google Scholar] [CrossRef]
- Almeida, R.D.N.; Braz-de-Melo, H.A.; Santos, I.O.; Correa, R.; Kobinger, G.P.; Magalhaes, K.G. The Cellular Impact of the ZIKA Virus on Male Reproductive Tract Immunology and Physiology. Cells 2020, 9, 1006. [Google Scholar] [CrossRef]
- Morelli, F.; Souza, R.P.; Cruz, T.E.D.; Damke, G.; Damke, E.; Suehiro, T.T.; Silva, V.; Consolaro, M.E.L. Zika virus infection in the genital tract of non-pregnant females: A systematic review. Rev. Inst. Med. Trop. Sao Paulo 2020, 62, e16. [Google Scholar] [CrossRef]
- Pagani, I.; Ghezzi, S.; Ulisse, A.; Rubio, A.; Turrini, F.; Garavaglia, E.; Candiani, M.; Castilletti, C.; Ippolito, G.; Poli, G.; et al. Human Endometrial Stromal Cells are Highly Permissive to Productive Infection by Zika Virus. Sci. Rep. 2017, 7, 44286. [Google Scholar] [CrossRef] [PubMed]
- Miner, J.J.; Diamond, M.S. Zika Virus Pathogenesis and Tissue Tropism. Cell Host Microbe 2017, 21, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Nicastri, E.; Castilletti, C.; Liuzzi, G.; Iannetta, M.; Capobianchi, M.R.; Ippolito, G. Persistent detection of Zika virus RNA in semen for six months after symptom onset in a traveller returning from Haiti to Italy, February 2016. Euro Surveill. 2016, 21, 30314. [Google Scholar] [CrossRef]
- Skidmore, M.A.; Kajaste-Rudnitski, A.; Wells, N.M.; Guimond, S.E.; Rudd, T.R.; Yates, E.A.; Vicenzi, E. Inhibition of influenza H5N1 invasion by modified heparin derivatives. MedChemComm 2015, 6, 640–646. [Google Scholar] [CrossRef]
- Zuberi, R.I.; Ge, X.N.; Jiang, S.; Bahaie, N.S.; Kang, B.N.; Hosseinkhani, R.M.; Frenzel, E.M.; Fuster, M.M.; Esko, J.D.; Rao, S.P.; et al. Deficiency of endothelial heparan sulfates attenuates allergic airway inflammation. J. Immunol. 2009, 183, 3971–3979. [Google Scholar] [CrossRef]
- Ghezzi, S.; Cooper, L.; Rubio, A.; Pagani, I.; Capobianchi, M.R.; Ippolito, G.; Pelletier, J.; Meneghetti, M.C.Z.; Lima, M.A.; Skidmore, M.A.; et al. Heparin prevents Zika virus induced-cytopathic effects in human neural progenitor cells. Antivir. Res. 2017, 140, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.L.; Hogwood, J.; Guo, W.; Yates, E.A.; Turnbull, J.E. By-Products of Heparin Production Provide a Diverse Source of Heparin-like and Heparan Sulfate Glycosaminoglycans. Sci. Rep. 2019, 9, 2679. [Google Scholar] [CrossRef]
- Patey, S.J.; Edwards, E.A.; Yates, E.A.; Turnbull, J.E. Heparin derivatives as inhibitors of BACE-1, the Alzheimer’s beta-secretase, with reduced activity against factor Xa and other proteases. J. Med. Chem. 2006, 49, 6129–6132. [Google Scholar] [CrossRef]
- Childs-Kean, L.M.; Hand, E.O. Simeprevir and sofosbuvir for treatment of chronic hepatitis C infection. Clin. Ther. 2015, 37, 243–267. [Google Scholar] [CrossRef]
- Murakami, E.; Niu, C.; Bao, H.; Micolochick Steuer, H.M.; Whitaker, T.; Nachman, T.; Sofia, M.A.; Wang, P.; Otto, M.J.; Furman, P.A. The mechanism of action of beta-D-2′-deoxy-2′-fluoro-2′-C-methylcytidine involves a second metabolic pathway leading to beta-D-2′-deoxy-2′-fluoro-2′-C-methyluridine 5′-triphosphate, a potent inhibitor of the hepatitis C virus RNA-dependent RNA polymerase. Antimicrob. Agents Chemother. 2008, 52, 458–464. [Google Scholar] [CrossRef]
- Bullard-Feibelman, K.M.; Govero, J.; Zhu, Z.; Salazar, V.; Veselinovic, M.; Diamond, M.S.; Geiss, B.J. The FDA-approved drug sofosbuvir inhibits Zika virus infection. Antiviral Res. 2017, 137, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.C.; Zaverucha-do-Valle, C.; Reis, P.A.; Barbosa-Lima, G.; Vieira, Y.R.; Mattos, M.; Silva, P.P.; Sacramento, C.; de Castro Faria Neto, H.C.; Campanati, L.; et al. Sofosbuvir protects Zika virus-infected mice from mortality, preventing short- and long-term sequelae. Sci. Rep. 2017, 7, 9409. [Google Scholar] [CrossRef]
- Mosier, P.D.; Krishnasamy, C.; Kellogg, G.E.; Desai, U.R. On the specificity of heparin/heparan sulfate binding to proteins. Anion-binding sites on antithrombin and thrombin are fundamentally different. PLoS ONE 2012, 7, e48632. [Google Scholar] [CrossRef] [PubMed]
- Board, N.L.; Moskovljevic, M.; Wu, F.; Siliciano, R.F.; Siliciano, J.D. Engaging innate immunity in HIV-1 cure strategies. Nat. Rev. Immunol. 2022, 22, 499–512. [Google Scholar] [CrossRef] [PubMed]
- Choay, J.; Lormeau, J.C.; Petitou, M.; Sinay, P.; Fareed, J. Structural studies on a biologically active hexasaccharide obtained from heparin. Ann. N. Y. Acad. Sci. 1981, 370, 644–649. [Google Scholar] [CrossRef]
- Thunberg, L.; Backstrom, G.; Lindahl, U. Further characterization of the antithrombin-binding sequence in heparin. Carbohydr. Res. 1982, 100, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Choay, J.; Petitou, M.; Lormeau, J.C.; Sinay, P.; Casu, B.; Gatti, G. Structure-activity relationship in heparin: A synthetic pentasaccharide with high affinity for antithrombin III and eliciting high anti-factor Xa activity. Biochem. Biophys. Res. Commun. 1983, 116, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Meneghetti, M.C.; Hughes, A.J.; Rudd, T.R.; Nader, H.B.; Powell, A.K.; Yates, E.A.; Lima, M.A. Heparan sulfate and heparin interactions with proteins. J. R. Soc. Interface 2015, 12, 0589. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Saraf, S.; Gangavarapu, K.; Watts, A.; Tan, A.L.; Oidtman, R.J.; Ladner, J.T.; Oliveira, G.; Matteson, N.L.; Kraemer, M.U.G.; et al. Travel Surveillance and Genomics Uncover a Hidden Zika Outbreak during the Waning Epidemic. Cell 2019, 178, 1057–1071. [Google Scholar] [CrossRef]
- McLaughlin, K.; Baczyk, D.; Potts, A.; Hladunewich, M.; Parker, J.D.; Kingdom, J.C. Low Molecular Weight Heparin Improves Endothelial Function in Pregnant Women at High Risk of Preeclampsia. Hypertension 2017, 69, 180–188. [Google Scholar] [CrossRef]
- Wat, J.M.; Hawrylyshyn, K.; Baczyk, D.; Greig, I.R.; Kingdom, J.C. Effects of glycol-split low molecular weight heparin on placental, endothelial, and anti-inflammatory pathways relevant to preeclampsia. Biol. Reprod. 2018, 99, 1082–1090. [Google Scholar] [CrossRef] [PubMed]
- Fung, A.; Jin, Z.; Dyatkina, N.; Wang, G.; Beigelman, L.; Deval, J. Efficiency of incorporation and chain termination determines the inhibition potency of 2′-modified nucleotide analogs against hepatitis C virus polymerase. Antimicrob. Agents Chemother. 2014, 58, 3636–3645. [Google Scholar] [CrossRef] [PubMed]
- Ban, H.; Nishishita, N.; Fusaki, N.; Tabata, T.; Saeki, K.; Shikamura, M.; Takada, N.; Inoue, M.; Hasegawa, M.; Kawamata, S.; et al. Efficient generation of transgene-free human induced pluripotent stem cells (iPSCs) by temperature-sensitive Sendai virus vectors. Proc. Natl. Acad. Sci. USA 2011, 108, 14234–14239. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, P.; Glatza, M.; Hemmer, K.; Tsytsyura, Y.; Thiel, C.S.; Hoing, S.; Moritz, S.; Parga, J.A.; Wagner, L.; Bruder, J.M.; et al. Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling. PLoS ONE 2013, 8, e59252. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AT Anti-Xa (%Hep) a | DS b | |
|---|---|---|
| Hep | 100 | 2.50 |
| D1 | 0.03 | 1.68 |
| D2 | 0.40 | 1.92 |
| D3 | 0.50 | 1.26 |
| D4 | 0.03 | 1.05 |
| D5 | 0.03 | 0.46 |
| D6 | 0.03 | 0.85 |
| D7 | 0.03 | 0.20 |
| APTT a | AT Anti-IIa b | AT Anti-Xa c | HCII-Anti-Xa d | Mw e | D f | DS g | |
|---|---|---|---|---|---|---|---|
| F1 | 11 | 6 | 11 | 6 | 30.6 | 2.2 | 1.8 |
| F2 | 34 | 27 | 28 | 23 | 18.7 | 2.0 | 2.1 |
| F3 | 11 | 9 | 11 | 7 | 33.5 | 2.1 | 1.6 |
| F4 | 37 | 35 | 37 | 3 | 26.3 | 1.7 | 2.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pagani, I.; Ottoboni, L.; Panina-Bordignon, P.; Martino, G.; Poli, G.; Taylor, S.; Turnbull, J.E.; Yates, E.; Vicenzi, E. Heparin Precursors with Reduced Anticoagulant Properties Retain Antiviral and Protective Effects That Potentiate the Efficacy of Sofosbuvir against Zika Virus Infection in Human Neural Progenitor Cells. Pharmaceuticals 2023, 16, 1385. https://doi.org/10.3390/ph16101385
Pagani I, Ottoboni L, Panina-Bordignon P, Martino G, Poli G, Taylor S, Turnbull JE, Yates E, Vicenzi E. Heparin Precursors with Reduced Anticoagulant Properties Retain Antiviral and Protective Effects That Potentiate the Efficacy of Sofosbuvir against Zika Virus Infection in Human Neural Progenitor Cells. Pharmaceuticals. 2023; 16(10):1385. https://doi.org/10.3390/ph16101385
Chicago/Turabian StylePagani, Isabel, Linda Ottoboni, Paola Panina-Bordignon, Gianvito Martino, Guido Poli, Sarah Taylor, Jeremy E. Turnbull, Edwin Yates, and Elisa Vicenzi. 2023. "Heparin Precursors with Reduced Anticoagulant Properties Retain Antiviral and Protective Effects That Potentiate the Efficacy of Sofosbuvir against Zika Virus Infection in Human Neural Progenitor Cells" Pharmaceuticals 16, no. 10: 1385. https://doi.org/10.3390/ph16101385
APA StylePagani, I., Ottoboni, L., Panina-Bordignon, P., Martino, G., Poli, G., Taylor, S., Turnbull, J. E., Yates, E., & Vicenzi, E. (2023). Heparin Precursors with Reduced Anticoagulant Properties Retain Antiviral and Protective Effects That Potentiate the Efficacy of Sofosbuvir against Zika Virus Infection in Human Neural Progenitor Cells. Pharmaceuticals, 16(10), 1385. https://doi.org/10.3390/ph16101385