
The Implication of Topoisomerase II Inhibitors in Synthetic Lethality for Cancer Therapy



Abstract
1. Introduction
2. DNA Topoisomerases
3. DNA Topoisomerase Inhibitors
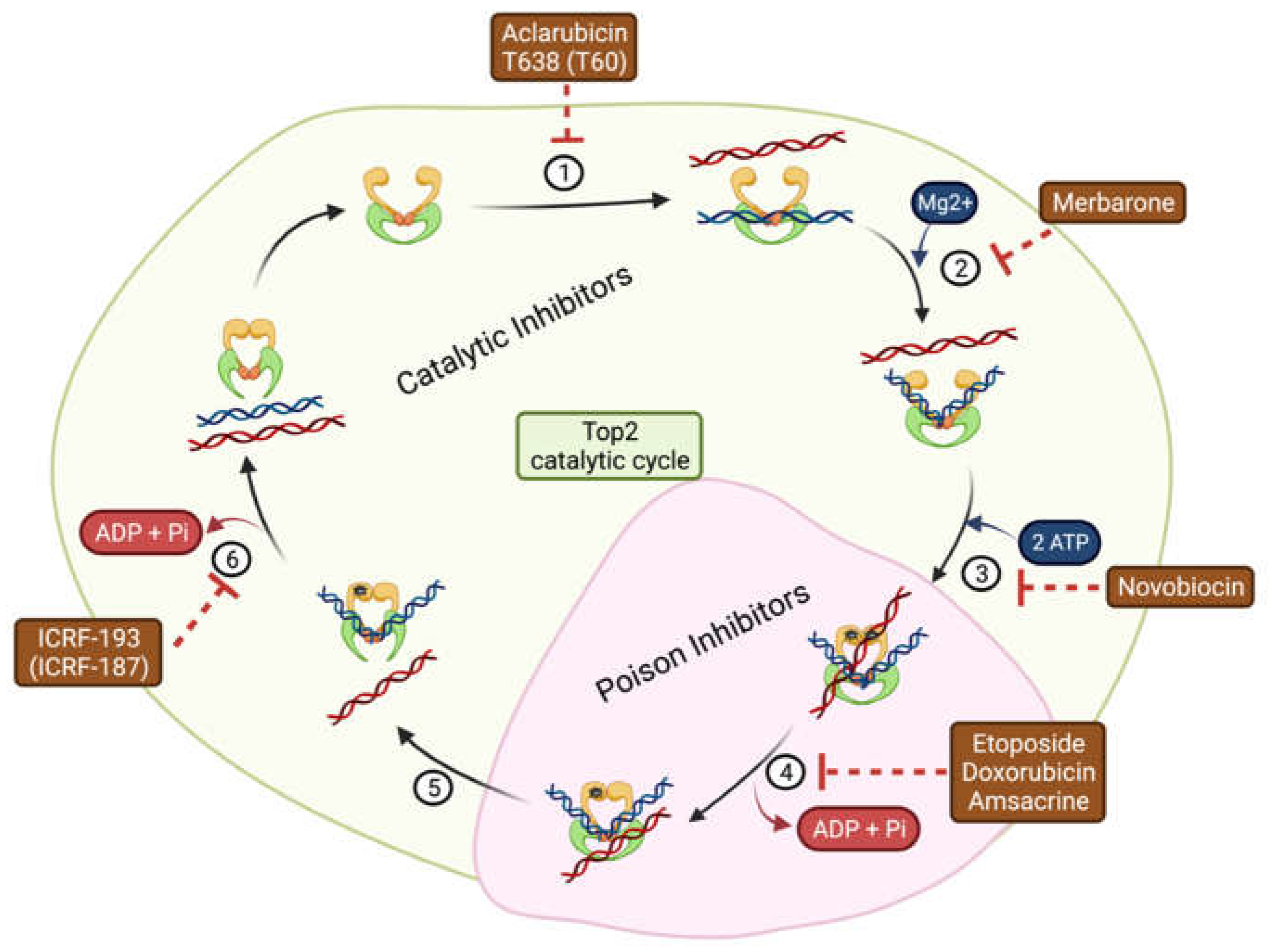
3.1. DNA Topoisomerase II Inhibitors
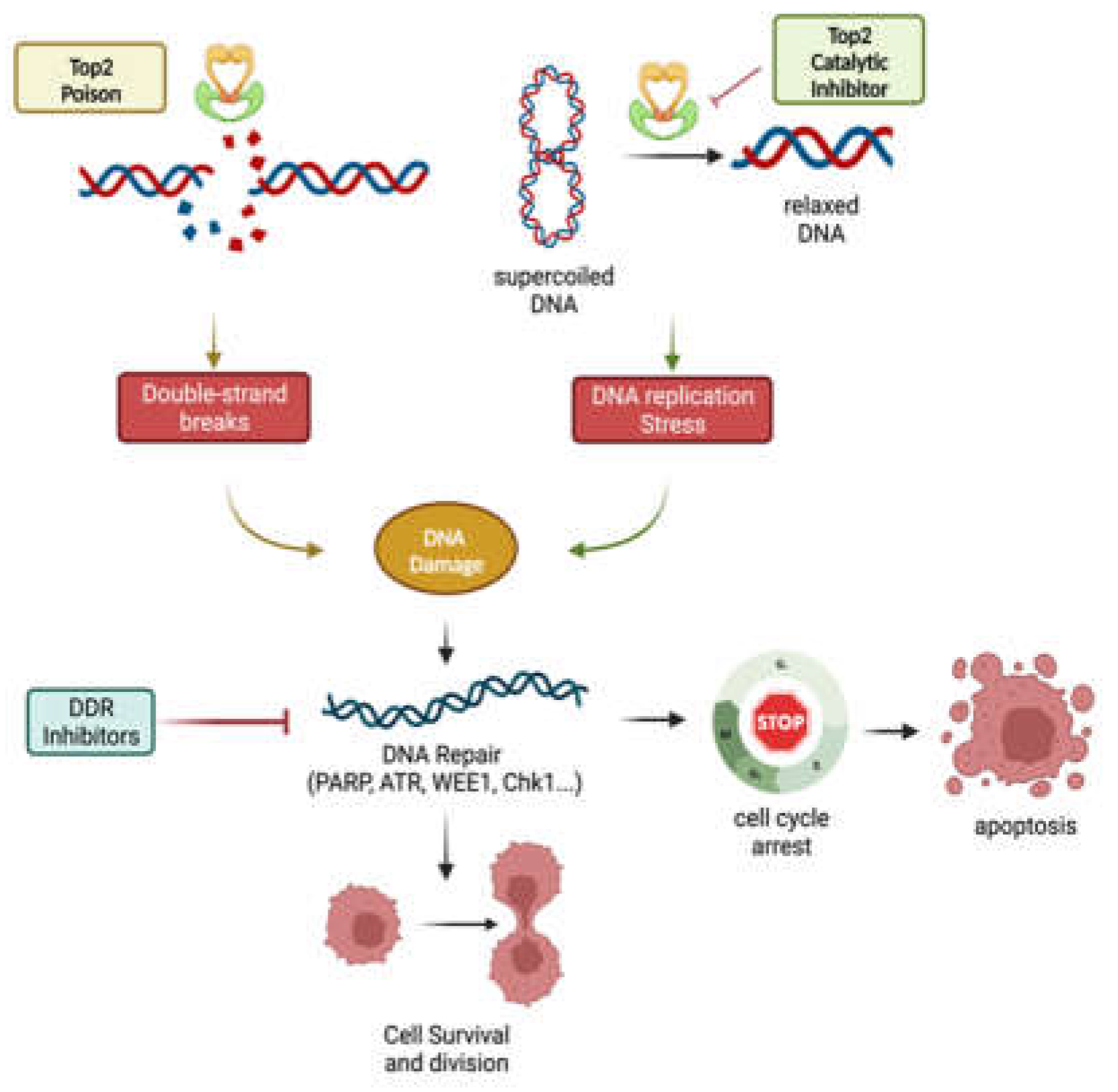
3.1.1. DNA Topoisomerase II Poisons
3.1.2. DNA Topoisomerase II Catalytic Inhibitors
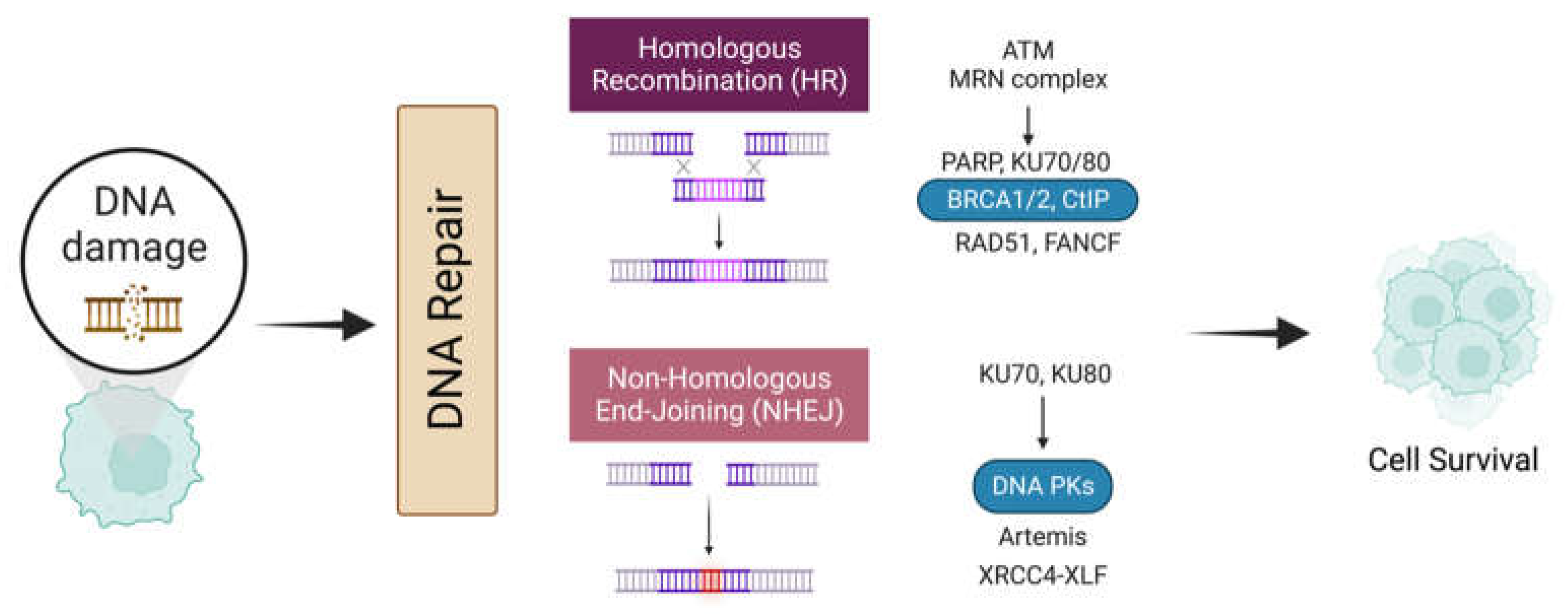
4. Double-Strand Break (DSB) Repair
4.1. DNA Repair Mechanism
4.1.1. Homologous Recombination
4.1.2. Non-Homologous End-Joining
5. Topoisomerase II inhibition Synergizes with DNA Repair Inhibitors
5.1. PARP Inhibitors
5.2. ATM/ATR Inhibitors
5.3. WEE1/CHK1 Inhibitors
5.4. DNA-PK Inhibitors
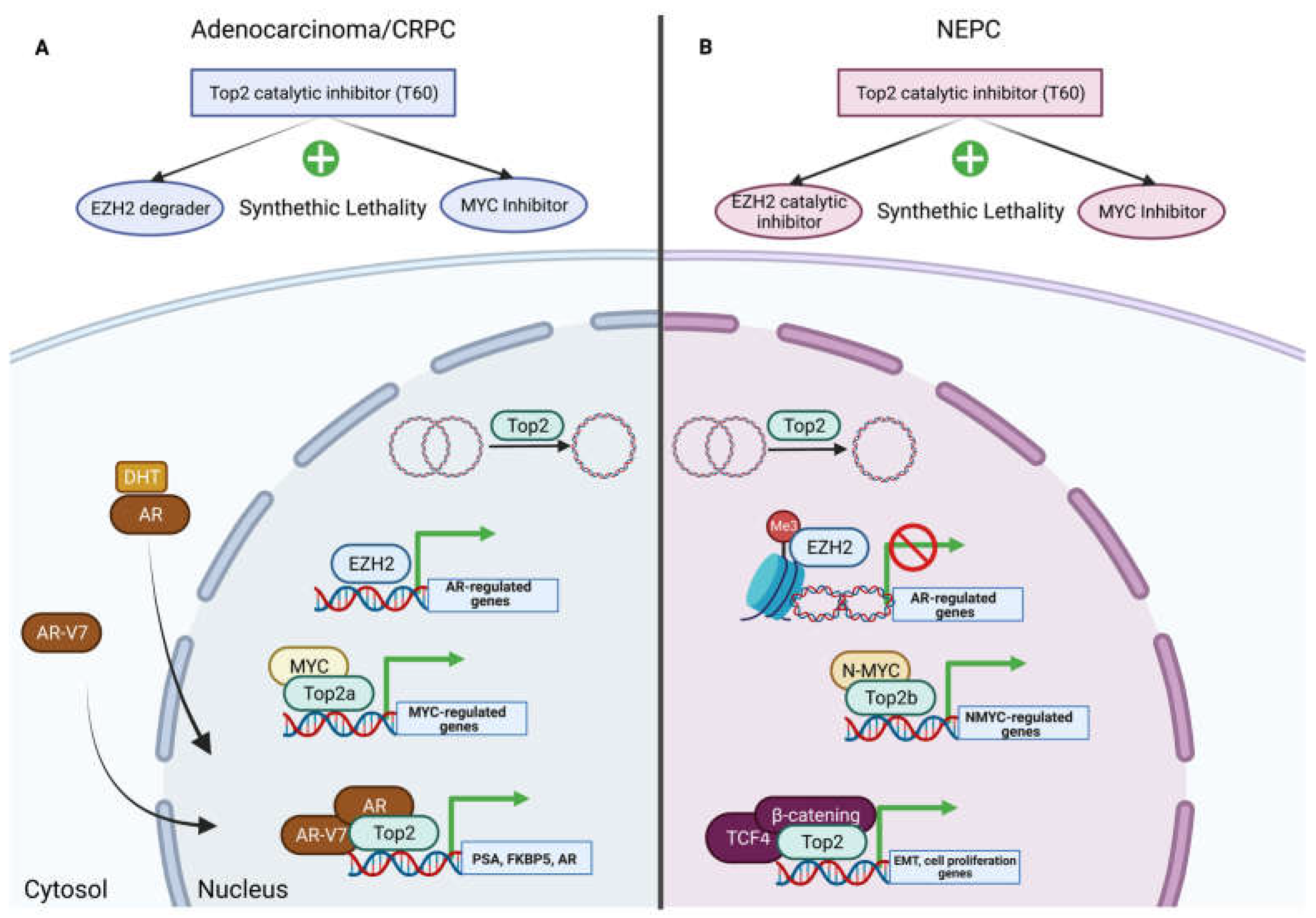
6. Chromatin Remodeling and Gene Transcription
6.1. EZH2 and Topoisomerase II
6.2. Myc and Topoisomerase II
7. Top2 Catalytic Inhibitors and Synthetic Lethality
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Delgado, J.L.; Hsieh, C.M.; Chan, N.L.; Hiasa, H. Topoisomerases as Anticancer Targets. Biochem. J. 2018, 475, 373–398. [Google Scholar] [CrossRef] [PubMed]
- Bjornsti, M.A.; Kaufmann, S.H. Topoisomerases and Cancer Chemotherapy: Recent Advances and Unanswered Questions. F1000Research 2019, 8, F1000 Faculty Rev-1704. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Garraway, L.A.; Ashworth, A.; Weber, B. Synthetic Lethality as an Engine for Cancer Drug Target Discovery. Nat. Rev. Drug Discov. 2020, 19, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Dziadkowiec, K.N.; Gasiorowska, E.; Nowak-Markwitz, E.; Jankowska, A. PARP Inhibitors: Review of Mechanisms of Action and BRCA1/2 Mutation Targeting. Prz. Menopauzalny 2016, 15, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Bayliss, R.; Burgess, S.G.; Leen, E.; Richards, M.W. A Moving Target: Structure and Disorder in Pursuit of Myc Inhibitors. Biochem. Soc. Trans. 2017, 45, 709–717. [Google Scholar] [CrossRef]
- Carabet, L.A.; Lallous, N.; Leblanc, E.; Ban, F.; Morin, H.; Lawn, S.; Ghaidi, F.; Lee, J.; Mills, I.G.; Gleave, M.E.; et al. Computer-Aided Drug Discovery of Myc-Max Inhibitors as Potential Therapeutics for Prostate Cancer. Eur. J. Med. Chem. 2018, 160, 108–119. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef]
- Das, S.K.; Kuzin, V.; Cameron, D.P.; Sanford, S.; Jha, R.K.; Nie, Z.; Rosello, M.T.; Holewinski, R.; Andresson, T.; Wisniewski, J.; et al. MYC Assembles and Stimulates Topoisomerases 1 and 2 in a “Topoisome”. Mol. Cell 2022, 82, 140–158.e12. [Google Scholar] [CrossRef]
- Adema, V.; Colla, S. EZH2 Inhibitors: The Unpacking Revolution. Cancer Res. 2022, 82, 359–361. [Google Scholar] [CrossRef]
- Champoux, J.J. DNA Topoisomerases: Structure, Function, and Mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef]
- Corbett, K.D.; Berger, J.M. Structure, Molecular Mechanisms, and Evolutionary Relationships in DNA Topoisomerases. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 95–118. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Sun, Y.; Huang, S.Y.N.; Nitiss, J.L. Roles of Eukaryotic Topoisomerases in Transcription, Replication and Genomic Stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef] [PubMed]
- Spakman, D.; Bakx, J.A.M.; Biebricher, A.S.; Peterman, E.J.G.; Wuite, G.J.L.; King, G.A. Unravelling the Mechanisms of Type 1A Topoisomerases Using Single-Molecule Approaches. Nucleic Acids Res. 2021, 49, 5470–5492. [Google Scholar] [CrossRef]
- Type I DNA Topoisomerases|Journal of Medicinal Chemistry. Available online: https://pubs.acs.org/doi/10.1021/acs.jmedchem.6b00966 (accessed on 27 December 2022).
- McKie, S.J.; Neuman, K.C.; Maxwell, A. DNA Topoisomerases: Advances in Understanding of Cellular Roles and Multi-Protein Complexes via Structure-Function Analysis. BioEssays 2021, 43, e2000286. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. DNA Topoisomerase II and Its Growing Repertoire of Biological Functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Dalvie, E.D.; Osheroff, N. DNA Recognition/Processing | DNA Topoisomerases: Type II☆. In Encyclopedia of Biological Chemistry III, 3rd ed.; Jez, J., Ed.; Elsevier: Oxford, UK, 2021; pp. 479–486. ISBN 978-0-12-822040-5. [Google Scholar]
- Lang, A.J.; Mirski, S.E.L.; Cummings, H.J.; Yu, Q.; Gerlach, J.H.; Cole, S.P.C. Structural Organization of the Human TOP2A and TOP2B Genes. Gene 1998, 221, 255–266. [Google Scholar] [CrossRef]
- Lee, J.H.; Berger, J.M. Cell Cycle-Dependent Control and Roles of DNA Topoisomerase II. Genes 2019, 10, 859. [Google Scholar] [CrossRef]
- Bredel, M.; Slavc, I.; Birner, P.; Czech, T.; Haberler, C.; Ströbel, T.; Wolfsberger, S.; Budka, H.; Hainfellner, J.A. DNA Topoisomerase IIalpha Expression in Optic Pathway Gliomas of Childhood. Eur. J. Cancer 2002, 38, 393–400. [Google Scholar] [CrossRef]
- Feng, Y.; Zhang, H.; Gao, W.; Wen, S.; Huangfu, H.; Sun, R.; Bai, W.; Wang, B. Expression of DNA Topoisomerase II-α: Clinical Significance in Laryngeal Carcinoma. Oncol. Lett. 2014, 8, 1575–1580. [Google Scholar] [CrossRef]
- Zhao, Q.; Li, H.; Zhu, L.; Hu, S.; Xi, X.; Liu, Y.; Liu, J.; Zhong, T. Bioinformatics Analysis Shows That TOP2A Functions as a Key Candidate Gene in the Progression of Cervical Cancer. Biomed. Rep. 2020, 13, 21. [Google Scholar] [CrossRef]
- Wang, X.; Wang, J.; Lyu, L.; Gao, X.; Cai, Y.; Tang, B. Oncogenic Role and Potential Regulatory Mechanism of Topoisomerase IIα in a Pan-Cancer Analysis. Sci. Rep. 2022, 12, 1–16. [Google Scholar] [CrossRef]
- Haffner, M.C.; Aryee, M.J.; Toubaji, A.; Esopi, D.M.; Albadine, R.; Gurel, B.; Isaacs, W.B.; Bova, G.S.; Liu, W.; Xu, J.; et al. Androgen-Induced TOP2B-Mediated Double-Strand Breaks and Prostate Cancer Gene Rearrangements. Nat. Genet. 2010, 42, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Manville, C.M.; Smith, K.; Sondka, Z.; Rance, H.; Cockell, S.; Cowell, I.G.; Lee, K.C.; Morris, N.J.; Padget, K.; Jackson, G.H.; et al. Genome-Wide ChIP-Seq Analysis of Human TOP2B Occupancy in MCF7 Breast Cancer Epithelial Cells. Biol. Open 2015, 4, 1436–1447. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Wu, Q.; Luan, S.; Yin, Z.; He, C.; Yin, L.; Zou, Y.; Yuan, Z.; Li, L.; Song, X.; et al. A Comprehensive Review of Topoisomerase Inhibitors as Anticancer Agents in the Past Decade. Eur. J. Med. Chem. 2019, 171, 129–168. [Google Scholar] [CrossRef]
- Pommier, Y. Drugging Topoisomerases: Lessons and Challenges. ACS Chem. Biol. 2013, 8, 82–95. [Google Scholar] [CrossRef]
- Berger, J.M.; Gamblin, S.J.; Harrison, S.C.; Wang, J.C. Structure and Mechanism of DNA Topoisomerase II. Nature 1996, 379, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Burden, D.A.; Osheroff, N. Mechanism of Action of Eukaryotic Topoisomerase II and Drugs Targeted to the Enzyme. Biochim. Biophys. Acta (BBA)-Gene Struct. Expr. 1998, 1400, 139–154. [Google Scholar] [CrossRef]
- Bromberg, K.D.; Osheroff, N. Mechanism of Action of Topoisomerase II-Targeted Anticancer Drugs. In DNA Topoisomerases in Cancer Therapy: Present and Future; Andoh, T., Ed.; Springer: Boston, MA, USA, 2003; pp. 53–78. ISBN 978-1-4615-0141-1. [Google Scholar]
- Wang, L.; Luo, J.; Chen, G.; Fang, M.; Wei, X.; Li, Y.; Liu, Z.; Zhang, Y.; Gao, S.; Shen, J.; et al. Chidamide, Decitabine, Cytarabine, Aclarubicin, and Granulocyte Colony-Stimulating Factor (CDCAG) in Patients with Relapsed/Refractory Acute Myeloid Leukemia: A Single-Arm, Phase 1/2 Study. Clin. Epigenetics 2020, 12, 132. [Google Scholar] [CrossRef] [PubMed]
- Matias-Barrios, V.M.; Radaeva, M.; Song, Y.; Alperstein, Z.; Lee, A.R.; Schmitt, V.; Lee, J.; Ban, F.; Xie, N.; Qi, J.; et al. Discovery of New Catalytic Topoisomerase II Inhibitors for Anticancer Therapeutics. Front. Oncol. 2021, 10, 633142. [Google Scholar] [CrossRef]
- Matias-Barrios, V.M.; Radaeva, M.; Ho, C.-H.; Lee, J.; Adomat, H.; Lallous, N.; Cherkasov, A.; Dong, X. Optimization of New Catalytic Topoisomerase II Inhibitors as an Anti-Cancer Therapy. Cancers 2021, 13, 3675. [Google Scholar] [CrossRef]
- Barrios, M.; Manuel, V. New Catalytic Topoisomerase II Inhibitors Discovered for Anticancer Therapeutics, University of British Columbia. Front. Oncol. 2021, 10, 633142. [Google Scholar] [CrossRef] [PubMed]
- Patterson-Fortin, J.; Jadhav, H.; Phan, T.; D’Andrea, A.; Shapiro, G.I. Abstract 1133: Novobiocin-Mediated Polymerase Theta Inhibition Induces CGAS/STING Pathway Activation and T-Cell Infiltration in BRCA-Associated Cancers. Cancer Res. 2022, 82, 1133. [Google Scholar] [CrossRef]
- Kraut, E.H.; Bendetti, J.; Balcerzak, S.P.; Doroshow, J.H. Phase II Trial of Merbarone in Soft Tissue Sarcoma. A Southwest Oncology Group Study. Invest New Drugs 1992, 10, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.L.; Kerrigan, J.E.; Lin, C.P.; Azarova, A.M.; Tsai, Y.C.; Ban, Y.; Liu, L.F. Topoisomerase IIbeta Mediated DNA Double-Strand Breaks: Implications in Doxorubicin Cardiotoxicity and Prevention by Dexrazoxane. Cancer Res. 2007, 67, 8839–8846. [Google Scholar] [CrossRef]
- Reyhanoglu, G.; Tadi, P. Etoposide; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Johnson-Arbor, K.; Dubey, R. Doxorubicin. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Kessler, T.; Mohr, M.; Müller-Tidow, C.; Krug, U.; Brunnberg, U.; Mohr, B.; Schliemann, C.; Sauerland, C.; Serve, H.; Büchner, T.; et al. Amsacrine Containing Induction Therapy in Elderly AML Patients: Comparison to Standard Induction Regimens in a Matched-Pair Analysis. Leuk Res. 2008, 32, 491–494. [Google Scholar] [CrossRef]
- Liu, L.F. DNA Topoisomerase Poisons as Antitumor Drugs. Annu. Rev. Biochem. 1989, 58, 351–375. [Google Scholar] [CrossRef]
- Meresse, P.; Dechaux, E.; Monneret, C.; Bertounesque, E. Etoposide: Discovery and Medicinal Chemistry. Curr. Med. Chem. 2012, 11, 2443–2466. [Google Scholar] [CrossRef]
- Montgomery, B.; Lin, D.W. Chapter 10-Toxicities of Chemotherapy for Genitourinary Malignancies. In Complications of Urologic Surgery, 4th ed.; Taneja, S.S., Ed.; W.B. Saunders: Philadelphia, PA, USA, 2010; pp. 117–123. ISBN 978-1-4160-4572-4. [Google Scholar]
- Maréchal, A.; Zou, L. DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harb. Perspect Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Wijdeven, R.H.; Pang, B.; van der Zanden, S.Y.; Qiao, X.; Blomen, V.; Hoogstraat, M.; Lips, E.H.; Janssen, L.; Wessels, L.; Brummelkamp, T.R.; et al. Genome-Wide Identification and Characterization of Novel Factors Conferring Resistance to Topoisomerase II Poisons in Cancer. Cancer Res. 2015, 75, 4176–4187. [Google Scholar] [CrossRef]
- Larsen, A.K.; Escargueil, A.E.; Skladanowski, A. Catalytic Topoisomerase II Inhibitors in Cancer Therapy. Pharmacol. Ther. 2003, 99, 167–181. [Google Scholar] [CrossRef]
- Portugal, J.; Mansilla, S.; Bataller, M. Mechanisms of Drug-Induced Mitotic Catastrophe in Cancer Cells. Curr. Pharm. Des. 2010, 16, 69–78. [Google Scholar] [CrossRef]
- Weiss, G.; Loyevsky, M.; Gordeuk, V.R. Dexrazoxane (ICRF-187). Gen. Pharmacol. 1999, 32, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xie, N.; Gleave, M.E.; Dong, X.; Li, H.; Xie, N.; Gleave, M.E.; Dong, X. Catalytic Inhibitors of DNA Topoisomerase II Suppress the Androgen Receptor Signaling and Prostate Cancer Progression. Oncotarget 2015, 6, 20474–20484. [Google Scholar] [CrossRef] [PubMed]
- Kawatani, M.; Takayama, H.; Muroi, M.; Kimura, S.; Maekawa, T.; Osada, H. Identification of a Small-Molecule Inhibitor of DNA Topoisomerase II by Proteomic Profiling. Chem. Biol. 2011, 18, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Wu, Y.; Sun, Y.; Liu, N.; Wu, S.; Zhang, W.; Sheng, C. Identification of Potent Catalytic Inhibitors of Human DNA Topoisomerase II by Structure-Based Virtual Screening. Med. Chem. Commun. 2018, 9, 1142–1146. [Google Scholar] [CrossRef]
- Yang, G.; Liu, C.; Chen, S.H.; Kassab, M.A.; Hoff, J.D.; Walter, N.G.; Yu, X. Super-Resolution Imaging Identifies PARP1 and the Ku Complex Acting as DNA Double-Strand Break Sensors. Nucleic Acids Res. 2018, 46, 3446–3457. [Google Scholar] [CrossRef]
- Jazayeri, A.; Balestrini, A.; Garner, E.; Haber, J.E.; Costanzo, V. Mre11–Rad50–Nbs1-Dependent Processing of DNA Breaks Generates Oligonucleotides That Stimulate ATM Activity. EMBO J. 2008, 27, 1953–1962. [Google Scholar] [CrossRef]
- Bartocci, C.; Denchi, E.L. Put a RING on It: Regulation and Inhibition of RNF8 and RNF168 RING Finger E3 Ligases at DNA Damage Sites. Front Genet 2013, 4, 128. [Google Scholar] [CrossRef]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA Double Strand Break Repair Pathway Choice in Somatic Mammalian Cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Chapman, J.R.; Taylor, M.R.G.; Boulton, S.J. Playing the End Game: DNA Double-Strand Break Repair Pathway Choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef]
- Branzei, D.; Foiani, M. Regulation of DNA Repair throughout the Cell Cycle. Nat. Rev. Mol. Cell Biol. 2008, 9, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Torgovnick, A.; Schumacher, B. DNA Repair Mechanisms in Cancer Development and Therapy. Front Genet 2015, 6, 157. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.; Yamane, K. DNA Mismatch Repair: Molecular Mechanism, Cancer, and Ageing. Mech. Ageing Dev. 2008, 129, 391–407. [Google Scholar] [CrossRef]
- Li, W.; Xie, C.; Yang, Z.; Chen, J.; Lu, N.-H. Abnormal DNA-PKcs and Ku 70/80 Expression May Promote Malignant Pathological Processes in Gastric Carcinoma. World J. Gastroenterol. 2013, 19, 6894–6901. [Google Scholar] [CrossRef] [PubMed]
- Alshareeda, A.T.; Negm, O.H.; Albarakati, N.; Green, A.R.; Nolan, C.; Sultana, R.; Madhusudan, S.; Benhasouna, A.; Tighe, P.; Ellis, I.O.; et al. Clinicopathological Significance of KU70/KU80, a Key DNA Damage Repair Protein in Breast Cancer. Breast Cancer Res. Treat 2013, 139, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.F.; Knudsen, K.E. Beyond DNA Repair: DNA-PK Function in Cancer. Cancer Discov. 2014, 4, 1126–1139. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, E.; Bertucci, F.; Sabatier, R.; Gonçalves, A. Targeting BRCA Deficiency in Breast Cancer: What Are the Clinical Evidences and the Next Perspectives? Cancers 2018, 10, 506. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Weng, J. A Pan-Cancer Bioinformatic Analysis of RAD51 Regarding the Values for Diagnosis, Prognosis, and Therapeutic Prediction. Front. Oncol. 2022, 12m, 858756. [Google Scholar] [CrossRef]
- Reginato, G.; Cejka, P. The MRE11 Complex: A Versatile Toolkit for the Repair of Broken DNA. DNA Repair 2020, 91–92, 102869. [Google Scholar] [CrossRef]
- Chen, R.; Wold, M.S. Replication Protein A: Single-Stranded DNA’s First Responder: Dynamic DNA-Interactions Allow Replication Protein A to Direct Single-Strand DNA Intermediates into Different Pathways for Synthesis or Repair. Bioessays 2014, 36, 1156–1161. [Google Scholar] [CrossRef]
- Ma, C.J.; Kwon, Y.; Sung, P.; Greene, E.C. Human RAD52 Interactions with Replication Protein A and the RAD51 Presynaptic Complex. J. Biol. Chem. 2017, 292, 11702–11713. [Google Scholar] [CrossRef] [PubMed]
- Ranjha, L.; Howard, S.M.; Cejka, P. Main Steps in DNA Double-Strand Break Repair: An Introduction to Homologous Recombination and Related Processes. Chromosoma 2018, 127, 187–214. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Kantake, N.; Sugiyama, T.; Kowalczykowski, S.C. Rad51 Protein Controls Rad52-Mediated DNA Annealing. J. Biol. Chem. 2008, 283, 14883–14892. [Google Scholar] [CrossRef]
- Kwon, Y.; Sung, P. Biochemical Analysis of D-Loop Extension and DNA Strand Displacement Synthesis. Methods Mol. Biol. 2021, 2153, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Wright, W.D.; Shah, S.S.; Heyer, W.D. Homologous Recombination and the Repair of DNA Double-Strand Breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef] [PubMed]
- Multiple Pathways of Recombination Induced by Double-Strand Breaks in Saccharomyces Cerevisiae-PMC. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC98970/ (accessed on 27 December 2022).
- Malkova, A.; Ira, G. Break-Induced Replication: Functions and Molecular Mechanism. Curr. Opin. Genet Dev. 2013, 23, 271–279. [Google Scholar] [CrossRef]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254. [Google Scholar] [CrossRef]
- Wu, Y.; Zhao, H. CTBP1 Strengthens the Cisplatin Resistance of Gastric Cancer Cells by Upregulating RAD51 Expression. Oncol Lett 2021, 22, 810. [Google Scholar] [CrossRef]
- Xu, Y.; Chen, K.; Cai, Y.; Cheng, C.; Zhang, Z.; Xu, G. Overexpression of Rad51 Predicts Poor Prognosis and Silencing of Rad51 Increases Chemo-Sensitivity to Doxorubicin in Neuroblastoma. Am. J. Transl. Res. 2019, 11, 5788–5799. [Google Scholar]
- García-Sanz, P.; Triviño, J.C.; Mota, A.; Pérez López, M.; Colás, E.; Rojo-Sebastián, A.; García, Á.; Gatius, S.; Ruiz, M.; Prat, J.; et al. Chromatin Remodelling and DNA Repair Genes Are Frequently Mutated in Endometrioid Endometrial Carcinoma. Int. J. Cancer 2017, 140, 1551–1563. [Google Scholar] [CrossRef]
- Cybulski, C.; Górski, B.; Debniak, T.; Gliniewicz, B.; Mierzejewski, M.; Masojć, B.; Jakubowska, A.; Matyjasik, J.; Złowocka, E.; Sikorski, A.; et al. NBS1 Is a Prostate Cancer Susceptibility Gene. Cancer Res. 2004, 64, 1215–1219. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.J.; Chen, D.J. DNA Double Strand Break Repair via Non-Homologous End-Joining. Transl. Cancer Res. 2013, 2, 130–143. [Google Scholar] [CrossRef]
- Valikhani, M.; Rahimian, E.; Ahmadi, S.E.; Chegeni, R.; Safa, M. Involvement of Classic and Alternative Non-Homologous End Joining Pathways in Hematologic Malignancies: Targeting Strategies for Treatment. Exp. Hematol. Oncol. 2021, 10, 51. [Google Scholar] [CrossRef]
- Blasiak, J. Single-Strand Annealing in Cancer. Int. J. Mol. Sci. 2021, 22, 2167. [Google Scholar] [CrossRef]
- McVey, M.; Lee, S.E. MMEJ Repair of Double-Strand Breaks (Director’s Cut): Deleted Sequences and Alternative Endings. Trends Genet. 2008, 24, 529–538. [Google Scholar] [CrossRef]
- Lu, G.; Duan, J.; Shu, S.; Wang, X.; Gao, L.; Guo, J.; Zhang, Y. Ligase I and Ligase III Mediate the DNA Double-Strand Break Ligation in Alternative End-Joining. Proc. Natl. Acad. Sci. USA 2016, 113, 1256–1260. [Google Scholar] [CrossRef]
- Shamanna, R.A.; Lu, H.; De Freitas, J.K.; Tian, J.; Croteau, D.L.; Bohr, V.A. WRN Regulates Pathway Choice between Classical and Alternative Non-Homologous End Joining. Nat. Commun. 2016, 7, 13785. [Google Scholar] [CrossRef]
- Yao, J.; Huang, A.; Zheng, X.; Liu, T.; Lin, Z.; Zhang, S.; Yang, Q.; Zhang, T.; Ma, H. 53BP1 Loss Induces Chemoresistance of Colorectal Cancer Cells to 5-Fluorouracil by Inhibiting the ATM–CHK2–P53 Pathway. J. Cancer Res. Clin. Oncol. 2017, 143, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Expression of DNA Damage Checkpoint 53BP1 Is Correlated with Prognosis, Cell Proliferation and Apoptosis in Colorectal Cancer-PMC. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4525819/ (accessed on 27 December 2022).
- Matsuda, K.; Kawasaki, T.; Akazawa, Y.; Hasegawa, Y.; Kondo, H.; Suzuki, K.; Iseki, M.; Nakashima, M. Expression Pattern of P53-Binding Protein 1 as a New Molecular Indicator of Genomic Instability in Bladder Urothelial Carcinoma. Sci. Rep. 2018, 8, 15477. [Google Scholar] [CrossRef] [PubMed]
- Sishc, B.J.; Davis, A.J. The Role of the Core Non-Homologous End Joining Factors in Carcinogenesis and Cancer. Cancers 2017, 9, 81. [Google Scholar] [CrossRef]
- CtIP Contributes to Non-Homologous End Joining Formation through Interacting with Ligase IV and Promotion of TMZ Resistance in Glioma Cells-PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/30915754/ (accessed on 27 December 2022).
- Reczek, C.R.; Shakya, R.; Miteva, Y.; Szabolcs, M.; Ludwig, T.; Baer, R. The DNA Resection Protein CtIP Promotes Mammary Tumorigenesis. Oncotarget 2016, 7, 32172–32183. [Google Scholar] [CrossRef] [PubMed]
- Expression of POLQ in Cancer-Summary-The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000051341-POLQ/pathology (accessed on 27 December 2022).
- To Assess the Efficacy and Safety of Olaparib Maintenance Monotherapy in the Treatment of Ovarian Cancer-Full Text online. Available online: https://clinicaltrials.gov/ct2/show/NCT02476968 (accessed on 27 December 2022).
- AstraZeneca. An Open Label, Single Arm, Multicentre Study to Assess the Clinical Effectiveness and Safety of Lynparza (Olaparib) Capsules Maintenance Monotherapy in Platinum Sensitive Relapsed Somatic or Germline BRCA Mutated Ovarian Cancer Patients Who Are in Complete or Partial Response Following Platinum Based Chemotherapy (ORZORA). clinicaltrials.gov. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT03286842 (accessed on 27 December 2022).
- AstraZeneca. A Phase I, Open-Label Study to Assess the Safety, Tolerability, Pharmacokinetics and Preliminary Efficacy of Ascending Doses of AZD0156 Monotherapy or in Combination With Either Cytotoxic Chemotherapies or Novel Anti-Cancer Agents in Patients With Advanced Malignancies; clinicaltrials.gov. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT02588105 (accessed on 27 December 2022).
- A Study to Assess the Safety and Tolerability of AZD1390 Given With Radiation Therapy in Patients With Brain Cancer-Full Text View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03423628 (accessed on 27 December 2022).
- Walls, G.M.; Oughton, J.B.; Chalmers, A.J.; Brown, S.; Collinson, F.; Forster, M.D.; Franks, K.N.; Gilbert, A.; Hanna, G.G.; Hannaway, N.; et al. CONCORDE: A Phase I Platform Study of Novel Agents in Combination with Conventional Radiotherapy in Non-Small-Cell Lung Cancer. Clin. Transl. Radiat. Oncol. 2020, 25, 61–66. Available online: https://clinicaltrials.gov/ct2/show/NCT04550104 (accessed on 27 December 2022). [CrossRef]
- Testing the Safety of M6620 (VX-970) When Given With Standard Whole Brain Radiation Therapy for the Treatment of Brain Metastases From Non-Small Cell Lung Cancer, Small Cell Lung Cancer, or Neuroendocrine Tumors-Full Text View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02589522 (accessed on 27 December 2022).
- Pal, S.K.; Frankel, P.H.; Mortazavi, A.; Milowsky, M.; Vaishampayan, U.; Parikh, M.; Lyou, Y.; Weng, P.; Parikh, R.; Teply, B.; et al. Effect of Cisplatin and Gemcitabine with or without Berzosertib in Patients with Advanced Urothelial Carcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT02567409 (accessed on 27 December 2022). [CrossRef]
- Safety and Tolerability Study of AZD7762 in Combination with Gemcitabine-Full Text View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00937664 (accessed on 27 December 2022).
- Adavosertib and Irinotecan Hydrochloride in Treating Younger Patients With Relapsed or Refractory Solid Tumors-Full Text View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02095132 (accessed on 27 December 2022).
- Adavosertib, Radiation Therapy, and Temozolomide in Treating Patients With Newly Diagnosed or Recurrent Glioblastoma-Full Text View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01849146 (accessed on 27 December 2022).
- First-in-Human Study of the Safety, Tolerability, and Pharmacokinetic/Pharmacodynamic Profile of VX-984 in Combination With Chemotherapy-Full Text View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02644278 (accessed on 27 December 2022).
- Study of Peposertib in Combination With Capecitabine and RT in Rectal Cancer-Full Text View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03770689 (accessed on 27 December 2022).
- Gelmon, K.A.; Fasching, P.A.; Couch, F.J.; Balmaña, J.; Delaloge, S.; Labidi-Galy, I.; Bennett, J.; McCutcheon, S.; Walker, G.; O’Shaughnessy, J.; et al. Clinical Effectiveness of Olaparib Monotherapy in Germline BRCA-Mutated, HER2-Negative Metastatic Breast Cancer in a Real-World Setting: Phase IIIb LUCY Interim Analysis. Eur. J. Cancer 2021, 152, 68–77. [Google Scholar] [CrossRef]
- Bochum, S.; Berger, S.; Martens, U.M. Olaparib. Recent Results Cancer Res. 2018, 211, 217–233. [Google Scholar] [CrossRef]
- Gupta, N.; Huang, T.T.; Horibata, S.; Lee, J.M. Cell Cycle Checkpoints and beyond: Exploiting the ATR/CHK1/WEE1 Pathway for the Treatment of PARP Inhibitor–Resistant Cancer-ScienceDirect. Pharmacol. Res. 2022, 178, 106162. [Google Scholar] [CrossRef]
- Cal, M.; Szakonyi, Z.; Flörkemeier, I.; Hillmann, J.S.; Weimer, J.P.; Hildebrandt, J.; Hedemann, N.; Rogmans, C.; Dempfle, A.; Arnold, N.; et al. Combined PARP and Dual Topoisomerase Inhibition Potentiates Genome Instability and Cell Death in Ovarian Cancer. Int. J. Mol. Sci. 2022, 23, 10503. [Google Scholar] [CrossRef]
- Boerner, J.L.; Nechiporchik, N.; Mueller, K.L.; Polin, L.; Heilbrun, L.; Boerner, S.A.; Zoratti, G.L.; Stark, K.; Lorusso, P.M.; Burger, A. Protein Expression of DNA Damage Repair Proteins Dictates Response to Topoisomerase and PARP Inhibitors in Triple-Negative Breast Cancer. PLoS ONE 2015, 10, e0119614. [Google Scholar] [CrossRef]
- Sasanuma, H.; Tsuda, M.; Morimoto, S.; Saha, L.K.; Rahman, M.M.; Kiyooka, Y.; Fujiike, H.; Cherniack, A.D.; Itou, J.; Moreu, E.C.; et al. BRCA1 Ensures Genome Integrity by Eliminating Estrogen-Induced Pathological Topoisomerase II-DNA Complexes. Proc. Natl. Acad. Sci. USA 2018, 115, E10642–E10651. [Google Scholar] [CrossRef]
- Treszezamsky, A.D.; Kachnic, L.A.; Feng, Z.; Zhang, J.; Tokadjian, C.; Powell, S.N. BRCA1- and BRCA2-Deficient Cells Are Sensitive to Etoposide-Induced DNA Double-Strand Breaks via Topoisomerase II. Cancer Res. 2007, 67, 7078–7081. [Google Scholar] [CrossRef]
- ATM Orchestrates the DNA-Damage Response to Counter Toxic Non-Homologous End-Joining at Broken Replication Forks-PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/30622252/ (accessed on 27 December 2022).
- Wagner, J.M.; Kaufmann, S.H. Prospects for the Use of ATR Inhibitors to Treat Cancer. Pharmaceuticals 2010, 3, 1311–1334. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.D.; Cable, P.L.; Garrus, J.E.; Sullivan, F.X.; von Carlowitz, I.; Huerou, Y.L.; Wallace, E.; Woessner, R.D.; Gross, S. Chk1 Inhibition and Wee1 Inhibition Combine Synergistically to Impede Cellular Proliferation. Cancer Biol. Ther. 2011, 12, 788–796. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, C.S.; Syljuåsen, R.G. Safeguarding Genome Integrity: The Checkpoint Kinases ATR, CHK1 and WEE1 Restrain CDK Activity during Normal DNA Replication. Nucleic Acids Res. 2012, 40, 477–486. [Google Scholar] [CrossRef]
- Mir, S.E.; De Witt Hamer, P.C.; Krawczyk, P.M.; Balaj, L.; Claes, A.; Niers, J.M.; Van Tilborg, A.A.G.; Zwinderman, A.H.; Geerts, D.; Kaspers, G.J.L.; et al. In Silico Analysis of Kinase Expression Identifies WEE1 as a Gatekeeper against Mitotic Catastrophe in Glioblastoma. Cancer Cell 2010, 18, 244–257. [Google Scholar] [CrossRef]
- Regulation of G2/M Transition by Inhibition of WEE1 and PKMYT1 Kinases-PMC. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6149964/ (accessed on 27 December 2022).
- ATM, ATR, CHK1, CHK2 and WEE1 Inhibitors in Cancer and Cancer Stem Cells-PMC. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6072143/ (accessed on 27 December 2022).
- Arroyo, M.; Cañuelo, A.; Calahorra, J.; Hastert, F.D.; Sánchez, A.; Clarke, D.J.; Marchal, J.A. Mitotic Entry upon Topo II Catalytic Inhibition Is Controlled by Chk1 and Plk1. FEBS J. 2020, 287, 4933–4951. [Google Scholar] [CrossRef] [PubMed]
- WEE1 Inhibition Enhances the Antitumor Immune Response to PD-L1 Blockade by the Concomitant Activation of STING and STAT1 Pathways in SCLC-ScienceDirect. Available online: https://www.sciencedirect.com/science/article/pii/S221112472200585X (accessed on 27 December 2022).
- Ghelli Luserna Di Rorà, A.; Beeharry, N.; Imbrogno, E.; Ferrari, A.; Robustelli, V.; Righi, S.; Sabattini, E.; Verga Falzacappa, M.V.; Ronchini, C.; Testoni, N.; et al. Targeting WEE1 to Enhance Conventional Therapies for Acute Lymphoblastic Leukemia. J Hematol. Oncol. 2018, 11, 99. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- Hu, S.; Hui, Z.; Lirussi, F.; Garrido, C.; Ye, X.Y.; Xie, T. Small Molecule DNA-PK Inhibitors as Potential Cancer Therapy: A Patent Review (2010–Present). Expert Opin. Ther. Pat. 2021, 31, 435–452. [Google Scholar] [CrossRef]
- Damia, G. Targeting DNA-PK in Cancer. Mutat. Res.-Fundam. Mol. Mech. Mutagen. 2020, 821, 111692. [Google Scholar] [CrossRef]
- Timme, C.R.; Rath, B.H.; O’Neill, J.W.; Camphausen, K.; Tofilon, P.J. The DNA-PK Inhibitor VX-984 Enhances the Radiosensitivity of Glioblastoma Cells Grown in Vitro and as Orthotopic Xenografts. Mol. Cancer Ther. 2018, 17, 1207–1216. [Google Scholar] [CrossRef]
- Zenke, F.T.; Zimmermann, A.; Sirrenberg, C.; Dahmen, H.; Kirkin, V.; Pehl, U.; Grombacher, T.; Wilm, C.; Fuchss, T.; Amendt, C.; et al. Pharmacologic Inhibitor of DNA-PK, M3814, Potentiates Radiotherapy and Regresses Human Tumors in Mouse Models. Mol. Cancer Ther. 2020, 19, 1091–1101. [Google Scholar] [CrossRef]
- A Study Combining the Peposertib (M3814) Pill With Standard Chemotherapy in Patients With Ovarian Cancer With an Expansion in High Grade Serous Ovarian Cancer and Low Grade Serous Ovarian Cancer-Full Text View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04092270 (accessed on 27 December 2022).
- Lal, S.; Sun, B.-C.; Chen, Y.; Huang, T.; Bhola, N.; Morton, V.; Chen, K.; Xia, S.; Zhang, H.; Ye, Q.; et al. Abstract 2594: Discovery and Characterization of ZL-2201, a Potent, Highly-Selective, and Orally Bioavailable Small-Molecule DNA-PK Inhibitor. Cancer Res. 2022, 82, 2594. [Google Scholar] [CrossRef]
- Lockwood, N.; Martini, S.; Lopez-Pardo, A.; Deiss, K.; Segeren, H.A.; Semple, R.K.; Collins, I.; Repana, D.; Cobbaut, M.; Soliman, T.; et al. Genome-Protective Topoisomerase 2a-Dependent G2 Arrest Requires P53 in HTERT-Positive Cancer Cells. Cancer Res. 2022, 82, 1762–1773. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Shen, R.; Liu, X.; Yang, X.; Xie, K.; Guo, Z.; Wang, D. Histone Post-Translational Modification and the DNA Damage Response. Genes Dis. 2022, 128, 28–36. [Google Scholar] [CrossRef]
- Lu, Y.; Chan, Y.-T.; Tan, H.-Y.; Li, S.; Wang, N.; Feng, Y. Epigenetic Regulation in Human Cancer: The Potential Role of Epi-Drug in Cancer Therapy. Mol. Cancer 2020, 19, 79. [Google Scholar] [CrossRef]
- Lu, L.-Y.; Kuang, H.; Korakavi, G.; Yu, X. Topoisomerase II Regulates the Maintenance of DNA Methylation. J. Biol. Chem. 2015, 290, 851–860. [Google Scholar] [CrossRef]
- Li, L.; Wang, Y. Cross-Talk between the H3K36me3 and H4K16ac Histone Epigenetic Marks in DNA Double-Strand Break Repair. J. Biol. Chem. 2017, 292, 11951–11959. [Google Scholar] [CrossRef] [PubMed]
- Top2a Promotes the Development of Social Behavior via PRC2 and H3K27me3. Science Advances. Available online: https://www.science.org/doi/10.1126/sciadv.abm7069 (accessed on 27 December 2022).
- Bunch, H.; Lawney, B.P.; Lin, Y.F.; Asaithamby, A.; Murshid, A.; Wang, Y.E.; Chen, B.P.C.; Calderwood, S.K. Transcriptional Elongation Requires DNA Break-Induced Signalling. Nat. Commun. 2015, 6, 10191. [Google Scholar] [CrossRef] [PubMed]
- Ju, B.G.; Lunyak, V.V.; Perissi, V.; Garcia-Bassets, I.; Rose, D.W.; Glass, C.K.; Rosenfeld, M.G. A Topoisomerase IIβ-Mediated DsDNA Break Required for Regulated Transcription. Science 2006, 312, 1798–1802. [Google Scholar] [CrossRef]
- Thakurela, S.; Garding, A.; Jung, J.; Schübeler, D.; Burger, L.; Tiwari, V.K. Gene Regulation and Priming by Topoisomerase IIα in Embryonic Stem Cells. Nat. Commun. 2013 4:1 2013, 4, 1–13. [Google Scholar] [CrossRef]
- Eich, M.L.; Athar, M.; Ferguson, J.E.; Varambally, S. EZH2-Targeted Therapies in Cancer: Hype or a Reality. Cancer Res. 2020, 80, 5449–5458. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Nishikori, M.; Otsuka, Y.; Arima, H.; Kitawaki, T.; Takaori-Kondo, A. The EZH2 Inhibitor Tazemetostat Upregulates the Expression of CCL17/TARC in B-Cell Lymphoma and Enhances T-Cell Recruitment. Cancer Sci. 2021, 112, 4604–4616. [Google Scholar] [CrossRef] [PubMed]
- Morschhauser, F.; Tilly, H.; Chaidos, A.; McKay, P.; Phillips, T.; Assouline, S.; Batlevi, C.L.; Campbell, P.; Ribrag, V.; Damaj, G.L.; et al. Tazemetostat for Patients with Relapsed or Refractory Follicular Lymphoma: An Open-Label, Single-Arm, Multicentre, Phase 2 Trial. Lancet Oncol. 2020, 21, 1433–1442. [Google Scholar] [CrossRef]
- Bryant, R.J.; Cross, N.A.; Eaton, C.L.; Hamdy, F.C.; Cunliffe, V.T. EZH2 Promotes Proliferation and Invasiveness of Prostate Cancer Cells. Prostate 2007, 67, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Bryant, R.J.; Winder, S.J.; Cross, S.S.; Hamdy, F.C.; Cunliffe, V.T. The Polycomb Group Protein EZH2 Regulates Actin Polymerization in Human Prostate Cancer Cells. Prostate 2008, 68, 255–263. [Google Scholar] [CrossRef]
- Dardenne, E.; Beltran, H.; Benelli, M.; Gayvert, K.; Berger, A.; Puca, L.; Cyrta, J.; Sboner, A.; Noorzad, Z.; MacDonald, T.; et al. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell 2016, 30, 563–577. [Google Scholar] [CrossRef]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.A.B.; Otte, A.P.; et al. The Polycomb Group Protein EZH2 Is Involved in Progression of Prostate Cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, D.; Zhou, T.; Song, H.; Hulsurkar, M.; Su, N.; Liu, Y.; Wang, Z.; Shao, L.; Ittmann, M.; et al. Androgen Deprivation Promotes Neuroendocrine Differentiation and Angiogenesis through CREB-EZH2-TSP1 Pathway in Prostate Cancers. Nat. Commun. 2018, 9, 4080. [Google Scholar] [CrossRef]
- Park, S.H.; Fong, K.-W.; Mong, E.; Martin, M.C.; Schiltz, G.E.; Yu, J. Going beyond Polycomb: EZH2 Functions in Prostate Cancer. Oncogene 2021, 40, 5788–5798. [Google Scholar] [CrossRef]
- Discovery of a First-in-Class EZH2 Selective Degrader|Nature Chemical Biology. Available online: https://www.nature.com/articles/s41589-019-0421-4 (accessed on 27 December 2022).
- Targeting EZH2 in Cancer-PMC. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4918227/ (accessed on 27 December 2022).
- Porazzi, P.; Petruk, S.; Pagliaroli, L.; De Dominici, M.; Deming, D.; Puccetti, M.V.; Kushinsky, S.; Kumar, G.; Minieri, V.; Barbieri, E.; et al. Targeting Chemotherapy to De-Condensed H3K27me3-Marked Chromatin of AML Cells Enhances Leukemia Suppression. Cancer Res. 2022, 82, 458–471. [Google Scholar] [CrossRef]
- Labbé, D.P.; Sweeney, C.J.; Brown, M.; Galbo, P.; Rosario, S.; Wadosky, K.M.; Ku, S.-Y.; Sjöström, M.; Alshalalfa, M.; Erho, N.; et al. TOP2A and EZH2 Provide Early Detection of an Aggressive Prostate Cancer Subgroup. Clin. Cancer Res. 2017, 23, 7072–7083. [Google Scholar] [CrossRef] [PubMed]
- Fillmore, C.M.; Xu, C.; Desai, P.T.; Berry, J.M.; Rowbotham, S.P.; Lin, Y.J.; Zhang, H.; Marquez, V.E.; Hammerman, P.S.; Wong, K.K.; et al. EZH2 Inhibition Sensitizes BRG1 and EGFR Mutant Lung Tumours to TopoII Inhibitors. Nature 2015, 520, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. C-Myc Target Genes Involved in Cell Growth, Apoptosis, and Metabolism. Mol. Cell Biol. 1999, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Xu, L.; Chang, Y.; Zeng, T.; Chen, X.; Wang, A.; Groth, J.; Foo, W.C.; Liang, C.; Hu, H.; et al. N-Myc Promotes Therapeutic Resistance Development of Neuroendocrine Prostate Cancer by Differentially Regulating MiR-421/ATM Pathway. Mol. Cancer 2019, 18. [Google Scholar] [CrossRef]
- Xie, Y.; Ning, S.; Hu, J. Jianpeng Molecular Mechanisms of Neuroendocrine Differentiation in Prostate Cancer Progression. J. Cancer Res. Clin. Oncol. 2022, 148, 1813–1823. [Google Scholar] [CrossRef]
- Yamada, Y.; Beltran, H. Clinical and Biological Features of Neuroendocrine Prostate Cancer. Curr. Oncol. Rep. 2021, 23. [Google Scholar] [CrossRef]
- Zeng, W.; Sun, H.; Meng, F.; Liu, Z.; Xiong, J.; Zhou, S.; Li, F.; Hu, J.; Hu, Z.; Liu, Z. Nuclear C-MYC Expression Level Is Associated with Disease Progression and Potentially Predictive of Two Year Overall Survival in Prostate Cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 1878–1888. [Google Scholar]
- C-Myc Antagonises the Transcriptional Activity of the Androgen Receptor in Prostate Cancer Affecting Key Gene Networks-PMC. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5405195/ (accessed on 27 December 2022).
- Kallio, H.M.L.; Hieta, R.; Latonen, L.; Brofeldt, A.; Annala, M.; Kivinummi, K.; Tammela, T.L.; Nykter, M.; Isaacs, W.B.; Lilja, H.G.; et al. Constitutively Active Androgen Receptor Splice Variants AR-V3, AR-V7 and AR-V9 Are Co-Expressed in Castration-Resistant Prostate Cancer Metastases. Br. J. Cancer 2018, 119, 347–356. [Google Scholar] [CrossRef]
- Lee, J.K.; Phillips, J.W.; Smith, B.A.; Park, J.W.; Stoyanova, T.; McCaffrey, E.F.; Baertsch, R.; Sokolov, A.; Meyerowitz, J.G.; Mathis, C.; et al. N-Myc Drives Neuroendocrine Prostate Cancer Initiated from Human Prostate Epithelial Cells. Cancer Cell 2016, 29, 536–547. [Google Scholar] [CrossRef]
- Choi, S.H.; Wright, J.B.; Gerber, S.A.; Cole, M.D. Myc Protein Is Stabilized by Suppression of a Novel E3 Ligase Complex in Cancer Cells. Genes Dev. 2010, 24, 1236–1241. [Google Scholar] [CrossRef]
- Fletcher, S.; Prochownik, E.V. Small-Molecule Inhibitors of the Myc Oncoprotein. Biochim. Biophys. Acta-Gene Regul. Mech. 2015, 1849, 525–543. [Google Scholar] [CrossRef] [PubMed]
- TOP2A Induces Malignant Character of Pancreatic Cancer through Activating β-Catenin Signaling Pathway-PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/29045811/ (accessed on 27 December 2022).
- Liu, Y.; Ma, J.; Song, J.S.; Zhou, H.Y.; Li, J.H.; Luo, C.; Geng, X.; Zhao, H.X. DNA Topoisomerase II Alpha Promotes the Metastatic Characteristics of Glioma Cells by Transcriptionally Activating β-Catenin. Bioengineered 2022, 13, 2207–2216. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Shitashige, M.; Satow, R.; Honda, K.; Ono, M.; Yun, J.; Tomida, A.; Tsuruo, T.; Hirohashi, S.; Yamada, T. Functional Interaction of DNA Topoisomerase IIα With the β-Catenin and T-Cell Factor-4 Complex. Gastroenterology 2007, 133, 1569–1578. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Subfamily | Mechanism | Human Proteins | Function | Co-Factor | Substrate |
|---|---|---|---|---|---|---|
| I | IA | Strand passage | Top3a | Relaxed DNA Sc, decatenation activity | Mg2+ | DNA Sc, hemicatenanes, double Holliday junctions, and D loops. |
| Top3b | Relaxed DNA Sc, RNA helicase activity | DNA HSc, RNA knots, and R loops. | ||||
| IB | Controlled rotation | Top1 | Relaxed negative and positive DNA Sc | None | DNA positive and negative Sc. | |
| Top1mt | ||||||
| IC | Not found in humans | |||||
| II | IIA | Strand passage | Top2a | Relaxed negative and positive DNA Sc, potent decatenation activity | Mg2+, ATP | DNA positive and negative Sc, DNA knots, and DNA catenanes. |
| Top2b | ||||||
| IIB | Not found in humans | |||||
| Name | Mechanism of Action | Mode of Inhibition | Application | Refs. |
|---|---|---|---|---|
| Aclarubicin | Prevents binding of Top2 to DNA | Catalytic | Acute myeloid leukemia | [31] |
| T638 (T60) | Inhibition compared to etoposide with less cytotoxicity in K562 cancer cells and xenografts | [32,33,34] | ||
| Novobiocin | Binds to ATP binding site | BRCA-deficient tumors with acquired PARP inhibitor resistance | [35] | |
| Merbarone | Blocks DNA Cleavage | Limitations due to nephrotoxicity | [36] | |
| ICRF-193 (ICRF-187) | Blocks ATP hydrolysis and traps enzymes in the closed clamp | Addresses cardiotoxicity caused by Top2 poison | [37] | |
| Etoposide | Stabilizes covalent cleavage complexes | Poison | Small cell lung cancer, lymphomas, refractory testicular tumors | [38] |
| Doxorubicin | Leukemia, ovarian and breast carcinomas | [39] | ||
| Amsacrine | Acute myeloid leukemia | [40] |
| DNA Repair Protein | Inhibitor | Application | Clinical Trial | Refs. |
|---|---|---|---|---|
| PARP | Olaparib | BRCA or HR+ ovarian cancer mutations | NCT02476968 NCT03286842 | [92,93] |
| ATM | AZD0156 | Advanced solid tumors | NCT02588105 | [94] |
| AZD1390 | Non-small cell lung cancer and brain cancer | NCT03423628 NCT04550104 | [95,96] | |
| ATR | M6620 | Metastatic urothelial cancer and solid tumors | NCT02589522 NCT02567409 | [97,98] |
| CHK1 | AZD7762 | Refractory solid tumors | NCT00937664 | [99] |
| WEE1 | AZD1775 | Glioblastoma and refractory solid tumors | NCT02095132 NCT01849146 | [100,101] |
| DNA-PK | VX-984 | Advanced solid tumors | NCT02644278 | [102] |
| M3814 | Advanced rectal cancer | NCT03770689 | [103] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matias-Barrios, V.M.; Dong, X. The Implication of Topoisomerase II Inhibitors in Synthetic Lethality for Cancer Therapy. Pharmaceuticals 2023, 16, 94. https://doi.org/10.3390/ph16010094
Matias-Barrios VM, Dong X. The Implication of Topoisomerase II Inhibitors in Synthetic Lethality for Cancer Therapy. Pharmaceuticals. 2023; 16(1):94. https://doi.org/10.3390/ph16010094
Chicago/Turabian StyleMatias-Barrios, Victor M., and Xuesen Dong. 2023. "The Implication of Topoisomerase II Inhibitors in Synthetic Lethality for Cancer Therapy" Pharmaceuticals 16, no. 1: 94. https://doi.org/10.3390/ph16010094
APA StyleMatias-Barrios, V. M., & Dong, X. (2023). The Implication of Topoisomerase II Inhibitors in Synthetic Lethality for Cancer Therapy. Pharmaceuticals, 16(1), 94. https://doi.org/10.3390/ph16010094