

Review on Bortezomib Resistance in Multiple Myeloma and Potential Role of Emerging Technologies


Abstract
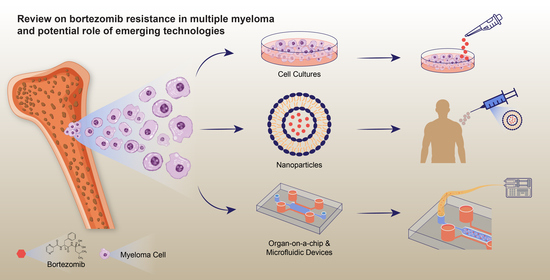
1. Introduction
2. Mechanisms of Bortezomib Resistance in MM Cell
2.1. Abnormal Drug Transport
2.2. Activation of Detoxification Systems
2.3. Changes in Drug Targets
2.4. Domination of Cell Cycle or Apoptosis
2.5. Distortion of Signalling Pathways
3. Emerging Technologies
3.1. Nanoparticles
3.2. 2D/3D Culture Systems
3.3. Microfluidic Systems
3.4. Organ on a Chip
4. Future Research Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Méndez-Ferrer, S.; Bonnet, D.; Steensma, D.P.; Hasserjian, R.P.; Ghobrial, I.M.; Gribben, J.G.; Andreeff, M.; Krause, D.S. Bone marrow niches in haematological malignancies. Nat. Rev. Cancer 2020, 20, 285–298. [Google Scholar] [CrossRef]
- Mateos, M.V.; Ludwig, H.; Bazarbachi, A.; Beksac, M.; Bladé, J.; Boccadoro, M.; Cavo, M.; Delforge, M.; Dimopoulos, M.A.; Facon, T.; et al. Insights on Multiple Myeloma Treatment Strategies. HemaSphere 2019, 3, e163. [Google Scholar] [CrossRef]
- Institute, N.C. Surveillance, Epidemiology, and End Results Program. Available online: https://seer.cancer.gov/ (accessed on 1 November 2022).
- Ito, S. Proteasome Inhibitors for the Treatment of Multiple Myeloma. Cancers 2020, 12, 265. [Google Scholar] [CrossRef]
- Lipchick, B.C.; Fink, E.E.; Nikiforov, M.A. Oxidative stress and proteasome inhibitors in multiple myeloma. Pharmacol. Res. 2016, 105, 210–215. [Google Scholar] [CrossRef]
- Auner, H.W.; Cenci, S. Recent advances and future directions in targeting the secretory apparatus in multiple myeloma. Br. J. Haematol. 2015, 168, 14–25. [Google Scholar] [CrossRef]
- Siegel, D.S.; Martin, T.; Wang, M.; Vij, R.; Jakubowiak, A.J.; Lonial, S.; Trudel, S.; Kukreti, V.; Bahlis, N.; Alsina, M.; et al. A phase 2 study of single-agent carfilzomib (PX-171-003-A1) in patients with relapsed and refractory multiple myeloma. Blood 2012, 120, 2817–2825. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Schjesvold, F.; Doronin, V.; Vinogradova, O.; Quach, H.; Leleu, X.; Montes, Y.G.; Ramasamy, K.; Pompa, A.; Levin, M.-D.; et al. Oral ixazomib-dexamethasone vs. oral pomalidomide-dexamethasone for lenalidomide-refractory, proteasome inhibitor-exposed multiple myeloma: A randomized Phase 2 trial. Blood Cancer J. 2022, 12, 9. [Google Scholar] [CrossRef]
- Adams, J.; Palombella, V.J.; Sausville, E.A.; Johnson, J.; Destree, A.; Lazarus, D.D.; Maas, J.; Pien, C.S.; Prakash, S.; Elliott, P.J. Proteasome inhibitors: A novel class of potent and effective antitumor agents. Cancer Res. 1999, 59, 2615–2622. [Google Scholar]
- Boccadoro, M.; Morgan, G.; Cavenagh, J. Preclinical evaluation of the proteasome inhibitor bortezomib in cancer therapy. Cancer Cell Int. 2005, 5, 18. [Google Scholar] [CrossRef]
- Ri, M.; Iida, S.; Ishida, T.; Ito, A.; Yano, H.; Inagaki, A.; Ding, J.; Kusumoto, S.; Komatsu, H.; Utsunomiya, A.; et al. Bortezomib-induced apoptosis in mature T-cell lymphoma cells partially depends on upregulation of Noxa and functional repression of Mcl-1. Cancer Sci. 2009, 100, 341–348. [Google Scholar] [CrossRef]
- Yerlikaya, A.; Okur, E.; Ulukaya, E. The p53-independent induction of apoptosis in breast cancer cells in response to proteasome inhibitor bortezomib. Tumor Biol. 2012, 33, 1385–1392. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bai, C.; Lu, D.; Wu, X.; Gao, L.; Zhang, W. Endoplasmic reticulum stress and autophagy participate in apoptosis induced by bortezomib in cervical cancer cells. Biotechnol. Lett. 2016, 38, 357–365. [Google Scholar] [CrossRef]
- Voutsadakis, I.A.; Patrikidou, A.; Tsapakidis, K.; Karagiannaki, A.; Hatzidaki, E.; Stathakis, N.E.; Papandreou, C.N. Additive inhibition of colorectal cancer cell lines by aspirin and bortezomib. Int. J. Colorectal. Dis. 2010, 25, 795–804. [Google Scholar] [CrossRef]
- Seki, N.; Toh, U.; Sayers, T.J.; Fujii, T.; Miyagi, M.; Akagi, Y.; Kusukawa, J.; Kage, M.; Shirouzu, K.; Yamana, H. Bortezomib sensitizes human esophageal squamous cell carcinoma cells to TRAIL-mediated apoptosis via activation of both extrinsic and intrinsic apoptosis pathways. Mol. Cancer Ther. 2010, 9, 1842–1851. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Ricker, J.L.; Malhotra, P.S.; Nottingham, L.; Bagain, L.; Lee, T.L.; Yeh, N.T.; Van Waes, C. Differential bortezomib sensitivity in head and neck cancer lines corresponds to proteasome, nuclear factor-κB and activator protein-1 related mechanisms. Mol. Cancer Ther. 2008, 7, 1949–1960. [Google Scholar] [CrossRef]
- Selimovic, D.; Porzig, B.B.O.W.; El-Khattouti, A.; Badura, H.E.; Ahmad, M.; Ghanjati, F.; Santourlidis, S.; Haikel, Y.; Hassan, M. Bortezomib/proteasome inhibitor triggers both apoptosis and autophagy-dependent pathways in melanoma cells. Cell. Signal. 2013, 25, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.L.; Flockhart, R.; Veal, G.J.; Lovat, P.E.; Redfern, C.P. Regulation of endoplasmic reticulum stress-induced cell death by ATF4 in neuroectodermal tumor cells. J. Biol. Chem. 2010, 285, 6091–6100. [Google Scholar] [CrossRef]
- Yang, Y.; Ikezoe, T.; Saito, T.; Kobayashi, M.; Koeffler, H.P.; Taguchi, H. Proteasome inhibitor PS-341 induces growth arrest and apoptosis of non-small cell lung cancer cells via the JNK/c-Jun/AP-1 signaling. Cancer Sci. 2004, 95, 176–180. [Google Scholar] [CrossRef]
- Awasthi, N.; Schwarz, M.A.; Schwarz, R.E. Combination effects of bortezomib with gemcitabine and EMAP II in experimental pancreatic cancer. Cancer Biol. Ther. 2010, 10, 99–107. [Google Scholar] [CrossRef]
- Befani, C.D.; Vlachostergios, P.J.; Hatzidaki, E.; Patrikidou, A.; Bonanou, S.; Simos, G.; Papandreou, C.N.; Liakos, P. Bortezomib represses HIF-1α protein expression and nuclear accumulation by inhibiting both PI3K/Akt/TOR and MAPK pathways in prostate cancer cells. J. Mol. Med. 2012, 90, 45–54. [Google Scholar] [CrossRef]
- Brooks, A.D.; Jacobsen, K.M.; Li, W.; Shanker, A.; Sayers, T.J. Bortezomib sensitizes human renal cell carcinomas to TRAIL apoptosis through increased activation of caspase-8 in the death-inducing signaling complex. Mol. Cancer Res. 2010, 8, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Ferro, E.G.; Xu, H.; Kramer, D.B.; Patell, R.; Kazi, D.S. Underperformance of Contemporary Phase III Oncology Trials and Strategies for Improvement. J. Natl. Compr. Cancer Netw. 2021, 19, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Zuchowska, A.; Skorupska, S. Multi-organ-on-chip approach in cancer research. Organs-on-a-Chip 2022, 4, 100014. [Google Scholar] [CrossRef]
- Schmitt, S.M.; Deshmukh, R.R.; Dou, Q.P. Proteasome Inhibitors and Lessons Learned from Their Mechanisms of Action and Resistance in Human Cancer. In Resistance to Proteasome Inhibitors in Cancer: Molecular Mechanisms and Strategies to Overcome Resistance; Dou, Q.P., Ed.; Springer International Publishing: Cham, Switzerland, 2014; pp. 1–46. [Google Scholar] [CrossRef]
- Maiso, P.; Huynh, D.; Moschetta, M.; Sacco, A.; Aljawai, Y.; Mishima, Y.; Asara, J.M.; Roccaro, A.M.; Kimmelman, A.C.; Ghobrial, I.M. Metabolic signature identifies novel targets for drug resistance in multiple myeloma. Cancer Res. 2015, 75, 2071–2082. [Google Scholar] [CrossRef]
- Wu, Q.; Yang, Z.; Nie, Y.; Shi, Y.; Fan, D. Multi-drug resistance in cancer chemotherapeutics: Mechanisms and lab approaches. Cancer Lett. 2014, 347, 159–166. [Google Scholar] [CrossRef]
- Grogan, T.M.; Spier, C.M.; Salmon, S.E.; Matzner, M.; Rybski, J.; Weinstein, R.S.; Scheper, R.J.; Dalton, W.S. P-glycoprotein expression in human plasma cell myeloma: Correlation with prior chemotherapy. Blood 1993, 81, 490–495. [Google Scholar] [CrossRef]
- Chernykh, Y.; Golenkov, A.; Vysotskaya, L.; Shushanov, S.; Rybalkina, E. Effect of Expression of Multidrug Resistance Genes in Newly Diagnosed Multiple Myeloma on the Clinical Course of the Disease. Blood 2016, 128, 5144. [Google Scholar] [CrossRef]
- Zhou, W.; Yang, Y.; Xia, J.; Wang, H.; Salama, M.E.; Xiong, W.; Xu, H.; Shetty, S.; Chen, T.; Zeng, Z.; et al. NEK2 induces drug resistance mainly through activation of efflux drug pumps and is associated with poor prognosis in myeloma and other cancers. Cancer Cell 2013, 23, 48–62. [Google Scholar] [CrossRef]
- Ishikawa, T.; Müller, M.; Klünemann, C.; Schaub, T.; Keppler, D. ATP-dependent primary active transport of cysteinyl leukotrienes across liver canalicular membrane. Role of the ATP-dependent transport system for glutathione S-conjugates. J. Biol. Chem. 1990, 265, 19279–19286. [Google Scholar] [CrossRef]
- Lagas, J.S.; Sparidans, R.W.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. Hepatic Clearance of Reactive Glucuronide Metabolites of Diclofenac in the Mouse Is Dependent on Multiple ATP-Binding Cassette Efflux Transporters. Mol. Pharmacol. 2010, 77, 687–694. [Google Scholar] [CrossRef]
- Sassi, Y.; Lipskaia, L.; Vandecasteele, G.; Nikolaev, V.O.; Hatem, S.N.; Cohen Aubart, F.; Russel, F.G.; Mougenot, N.; Vrignaud, C.; Lechat, P.; et al. Multidrug resistance-associated protein 4 regulates cAMP-dependent signaling pathways and controls human and rat SMC proliferation. J. Clin. Investig. 2008, 118, 2747–2757. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; van der Linden, M.; de Haas, M.; Baas, F.; Borst, P. Expression of human MRP6, a homologue of the multidrug resistance protein gene MRP1, in tissues and cancer cells. Cancer Res. 1999, 59, 175–182. [Google Scholar] [PubMed]
- Kozalak, G.; Oksuzoglu, E. Efficacy of Multi-Drug Resistance Transporters and Glutathione S-Transferase P-1 at Developing Bortezomib Resistance in Multiple Myeloma Cell Lines. Lat. Am. J. Pharm. 2021, 40, 2709–2716. [Google Scholar]
- Guo, Y.; Köck, K.; Ritter, C.A.; Chen, Z.-S.; Grube, M.; Jedlitschky, G.; Illmer, T.; Ayres, M.; Beck, J.F.; Siegmund, W.; et al. Expression of ABCC-Type Nucleotide Exporters in Blasts of Adult Acute Myeloid Leukemia: Relation to Long-term Survival. Clin. Cancer Res. 2009, 15, 1762–1769. [Google Scholar] [CrossRef]
- Morrow, C.S.; Smitherman, P.K.; Townsend, A.J. Combined expression of multidrug resistance protein (MRP) and glutathione S-transferase P1-1 (GSTP1-1) in MCF7 cells and high level resistance to the cytotoxicities of ethacrynic acid but not oxazaphosphorines or cisplatin. Biochem. Pharmacol. 1998, 56, 1013–1021. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, M.; He, P.; Chen, Y.; Wang, X.; Zhang, M. Identification of glutathione S-transferase π 1 as a prognostic proteomic biomarker for multiple myeloma using proteomic profiling. Oncol. Lett. 2020, 19, 2153–2162. [Google Scholar] [CrossRef]
- Soriano, G.P.; Besse, L.; Li, N.; Kraus, M.; Besse, A.; Meeuwenoord, N.; Bader, J.; Everts, B.; den Dulk, H.; Overkleeft, H.S.; et al. Proteasome inhibitor-adapted myeloma cells are largely independent from proteasome activity and show complex proteomic changes, in particular in redox and energy metabolism. Leukemia 2016, 30, 2198–2207. [Google Scholar] [CrossRef]
- Manik, C.; Mindaugas, A.; Thorsten, S.; Elisabeth, M.; Claudia, H.; Torsten, S.; Tanja, H.; Heike, S.; Stefanie, K.; Hermann, E.; et al. The PI3K/Akt signaling pathway regulates the expression of Hsp70, which critically contributes to Hsp90-chaperone function and tumor cell survival in multiple myeloma. Haematologica 2013, 98, 1132–1141. [Google Scholar] [CrossRef]
- Franke, N.E.; Niewerth, D.; Assaraf, Y.G.; van Meerloo, J.; Vojtekova, K.; van Zantwijk, C.H.; Zweegman, S.; Chan, E.T.; Kirk, C.J.; Geerke, D.P.; et al. Impaired bortezomib binding to mutant β5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia 2012, 26, 757–768. [Google Scholar] [CrossRef]
- Suzuki, E.; Demo, S.; Deu, E.; Keats, J.; Arastu-Kapur, S.; Bergsagel, P.L.; Bennett, M.K.; Kirk, C.J. Molecular mechanisms of bortezomib resistant adenocarcinoma cells. PLoS ONE 2011, 6, e27996. [Google Scholar] [CrossRef]
- Abdel-Wahab, A.F.; Mahmoud, W.; Al-Harizy, R.M. Targeting glucose metabolism to suppress cancer progression: Prospective of anti-glycolytic cancer therapy. Pharmacol. Res. 2019, 150, 104511. [Google Scholar] [CrossRef]
- Sekiguchi, N.; Ootsubo, K.; Wagatsuma, M.; Midorikawa, K.; Nagata, A.; Noto, S.; Yamada, K.; Takezako, N. The impact of C-Myc gene-related aberrations in newly diagnosed myeloma with bortezomib/dexamethasone therapy. Int. J. Hematol. 2014, 99, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Robak, P.; Drozdz, I.; Szemraj, J.; Robak, T. Drug resistance in multiple myeloma. Cancer Treat. Rev. 2018, 70, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Öksüzoğlu, E.; Kozalak, G. Inhibition of apoptosis may lead to the development of bortezomib resistance in multiple myeloma cancer cells. Turk. J. Biochem. 2021, 46, 65–71. [Google Scholar] [CrossRef]
- Spets, H.; Strömberg, T.; Georgii-Hemming, P.; Siljason, J.; Nilsson, K.; Jernberg-Wiklund, H. Expression of the bcl-2 family of pro- and anti-apoptotic genes in multiple myeloma and normal plasma cells: Regulation during interleukin-6(IL-6)-induced growth and survival. Eur. J. Haematol. 2002, 69, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Neri, P.; Gratton, K.; Ren, L.; Mansoor, A.; Duggan, P.; Stewart, D.A.; Bahlis, N.J. miRNA Expression in Multiple Myeloma as Predictive Model of Response to Bortezomib. Blood 2009, 114, 4918. [Google Scholar] [CrossRef]
- Chauhan, D.; Hideshima, T.; Anderson, K.C. Proteasome inhibition in multiple myeloma: Therapeutic implication. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 465–476. [Google Scholar] [CrossRef]
- Cusack, J.C. Rationale for the treatment of solid tumors with the proteasome inhibitor bortezomib. Cancer Treat. Rev. 2003, 29 (Suppl. S1), 21–31. [Google Scholar] [CrossRef]
- Schmidmaier, R.; Mörsdorf, K.; Baumann, P.; Emmerich, B.; Meinhardt, G. Evidence for cell adhesion-mediated drug resistance of multiple myeloma cells in vivo. Int. J. Biol. Markers 2006, 21, 218–222. [Google Scholar] [CrossRef]
- Cencini, E.; Fabbri, A.; Sicuranza, A.; Gozzetti, A.; Bocchia, M. The Role of Tumor-Associated Macrophages in Hematologic Malignancies. Cancers 2021, 13, 3597. [Google Scholar] [CrossRef]
- Ahmed, S.; Khan, H.; Aschner, M.; Mirzae, H.; Küpeli Akkol, E.; Capasso, R. Anticancer Potential of Furanocoumarins: Mechanistic and Therapeutic Aspects. Int. J. Mol. Sci. 2020, 21, 5622. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Waghray, D.; Zhang, Q. Inhibit or Evade Multidrug Resistance P-Glycoprotein in Cancer Treatment. J. Med. Chem. 2018, 61, 5108–5121. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L.; Ling, V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys Acta 1976, 455, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Christie, E.L.; Pattnaik, S.; Beach, J.; Copeland, A.; Rashoo, N.; Fereday, S.; Hendley, J.; Alsop, K.; Brady, S.L.; Lamb, G.; et al. Multiple ABCB1 transcriptional fusions in drug resistant high-grade serous ovarian and breast cancer. Nat. Commun. 2019, 10, 1295. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.I.; Williams, R.T.; Henderson, M.J.; Norris, M.D.; Haber, M. ABC transporters as mediators of drug resistance and contributors to cancer cell biology. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2016, 26, 1–9. [Google Scholar] [CrossRef]
- Verbrugge, S.E.; Assaraf, Y.G.; Dijkmans, B.A.; Scheffer, G.L.; Al, M.; den Uyl, D.; Oerlemans, R.; Chan, E.T.; Kirk, C.J.; Peters, G.J.; et al. Inactivating PSMB5 Mutations and P-Glycoprotein (Multidrug Resistance-Associated Protein/ATP-Binding Cassette B1) Mediate Resistance to Proteasome Inhibitors: Ex Vivo Efficacy of (Immuno)Proteasome Inhibitors in Mononuclear Blood Cells from Patients with Rheumatoid Arthritis. J. Pharmacol. Exp. Ther. 2012, 341, 174–182. [Google Scholar] [CrossRef]
- O’Connor, R.; Ooi, M.G.; Meiller, J.; Jakubikova, J.; Klippel, S.; Delmore, J.; Richardson, P.; Anderson, K.; Clynes, M.; Mitsiades, C.S.; et al. The interaction of bortezomib with multidrug transporters: Implications for therapeutic applications in advanced multiple myeloma and other neoplasias. Cancer Chemother. Pharmacol. 2013, 71, 1357–1368. [Google Scholar] [CrossRef]
- Doyle, L.A.; Yang, W.; Abruzzo, L.V.; Krogmann, T.; Gao, Y.; Rishi, A.K.; Ross, D.D. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. USA 1998, 95, 15665–15670. [Google Scholar] [CrossRef]
- Maliepaard, M.; Scheffer, G.L.; Faneyte, I.F.; van Gastelen, M.A.; Pijnenborg, A.C.; Schinkel, A.H.; van De Vijver, M.J.; Scheper, R.J.; Schellens, J.H. Subcellular localization and distribution of the breast cancer resistance protein transporter in normal human tissues. Cancer Res. 2001, 61, 3458–3464. [Google Scholar]
- Mo, W.; Zhang, J.T. Human ABCG2: Structure, function, and its role in multidrug resistance. Int. J. Biochem. Mol. Biol. 2012, 3, 1–27. [Google Scholar] [PubMed]
- Turner, J.G.; Gump, J.L.; Zhang, C.; Cook, J.M.; Marchion, D.; Hazlehurst, L.; Munster, P.; Schell, M.J.; Dalton, W.S.; Sullivan, D.M. ABCG2 expression, function, and promoter methylation in human multiple myeloma. Blood 2006, 108, 3881–3889. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, G.L.; Wijngaard, P.L.J.; Flens, M.J.; Izquierdo, M.A.; Slovak, M.L.; Pinedo, H.M.; Meijer, C.J.L.M.; Clevers, H.C.; Scheper, R.J. The drug resistance-related protein LRP is the human major vault protein. Nat. Med. 1995, 1, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.R.; Jaiswal, R.; Brown, R.D.; Luk, F.; Bebawy, M. Multiple myeloma and persistence of drug resistance in the age of novel drugs (Review). Int. J. Oncol. 2016, 49, 33–50. [Google Scholar] [CrossRef]
- Kulsoom, B.; Shamsi, T.S.; Afsar, N.A. Lung resistance-related protein (LRP) predicts favorable therapeutic outcome in Acute Myeloid Leukemia. Sci. Rep. 2019, 9, 378. [Google Scholar] [CrossRef]
- Huh, H.J.; Park, C.-J.; Jang, S.; Seo, E.-J.; Chi, H.-S.; Lee, J.-H.; Lee, K.-H.; Seo, J.J.; Moon, H.N.; Ghim, T. Prognostic Significance of Multidrug Resistance Gene 1 (MDR1), Multidrug Resistance-related Protein (MRP) and Lung Resistance Protein (LRP) mRNA Expression in Acute Leukemia. J. Korean Med. Sci. 2006, 21, 253–258. [Google Scholar] [CrossRef]
- Mándoky, L.; Géczi, L.; Doleschall, Z.; Bodrogi, I.; Csuka, O.; Kásler, M.; Bak, M. Expression and prognostic value of the lung resistance-related protein (LRP) in germ cell testicular tumors. Anticancer Res. 2004, 24, 1097–1104. [Google Scholar]
- Burger, H.; Foekens, J.A.; Look, M.P.; Meijer-van Gelder, M.E.; Klijn, J.G.; Wiemer, E.A.; Stoter, G.; Nooter, K. RNA expression of breast cancer resistance protein, lung resistance-related protein, multidrug resistance-associated proteins 1 and 2, and multidrug resistance gene 1 in breast cancer: Correlation with chemotherapeutic response. Clin. Cancer Res. 2003, 9, 827–836. [Google Scholar]
- Raaijmakers, H.G.; Izquierdo, M.A.; Lokhorst, H.M.; de Leeuw, C.; Belien, J.A.; Bloem, A.C.; Dekker, A.W.; Scheper, R.J.; Sonneveld, P. Lung-resistance-related protein expression is a negative predictive factor for response to conventional low but not to intensified dose alkylating chemotherapy in multiple myeloma. Blood 1998, 91, 1029–1036. [Google Scholar] [CrossRef]
- Cole, S.P.C.; Bhardwaj, G.; Gerlach, J.H.; Mackie, J.E.; Grant, C.E.; Almquist, K.C.; Stewart, A.J.; Kurz, E.U.; Duncan, A.M.V.; Deeley, R.G. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science 1992, 258, 1650–1654. [Google Scholar] [CrossRef]
- Johnson, Z.L.; Chen, J. Structural Basis of Substrate Recognition by the Multidrug Resistance Protein MRP1. Cell 2017, 168, 1075–1085.e1079. [Google Scholar] [CrossRef]
- Kumar, A.; Jaitak, V. Natural products as multidrug resistance modulators in cancer. Eur. J. Med. Chem. 2019, 176, 268–291. [Google Scholar] [CrossRef] [PubMed]
- Loe, D.W.; Deeley, R.G.; Cole, S.P.C. Characterization of Vincristine Transport by the Mr 190,000 Multidrug Resistance Protein (MRP): Evidence for Cotransport with Reduced Glutathione1. Cancer Res. 1998, 58, 5130–5136. [Google Scholar] [PubMed]
- Renes, J.; de Vries, E.G.; Nienhuis, E.F.; Jansen, P.L.; Müller, M. ATP- and glutathione-dependent transport of chemotherapeutic drugs by the multidrug resistance protein MRP1. Br. J. Pharmacol. 1999, 126, 681–688. [Google Scholar] [CrossRef]
- Nasr, R.; Lorendeau, D.; Khonkarn, R.; Dury, L.; Pérès, B.; Boumendjel, A.; Cortay, J.C.; Falson, P.; Chaptal, V.; Baubichon-Cortay, H. Molecular analysis of the massive GSH transport mechanism mediated by the human Multidrug Resistant Protein 1/ABCC1. Sci. Rep. 2020, 10, 7616. [Google Scholar] [CrossRef] [PubMed]
- Hopper-Borge, E.; Xu, X.; Shen, T.; Shi, Z.; Chen, Z.-S.; Kruh, G.D. Human Multidrug Resistance Protein 7 (ABCC10) Is a Resistance Factor for Nucleoside Analogues and Epothilone B. Cancer Res. 2008, 69, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Sarkadi, B.; Homolya, L.; Szakács, G.; Váradi, A. Human multidrug resistance ABCB and ABCG transporters: Participation in a chemoimmunity defense system. Physiol. Rev. 2006, 86, 1179–1236. [Google Scholar] [CrossRef]
- Petrini, M.; Di Simone, D.; Favati, A.; Mattii, L.; Valentini, P.; Grassi, B. GST-pi and P-170 co-expression in multiple myeloma. Br. J. Haematol. 1995, 90, 393–397. [Google Scholar] [CrossRef]
- Ishikawa, T. The ATP-dependent glutathione S-conjugate export pump. Trends Biochem. Sci. 1992, 17, 463–468. [Google Scholar] [CrossRef]
- Zaman, G.J.; Lankelma, J.; Tellingen, O.V.; Beijnen, J.; Dekker, H.; Paulusma, C.; Elferink, R.P.O.; Baas, F.; Borst, P. Role of glutathione in the export of compounds from cells by the multidrug-resistance-associated protein. Proc. Natl. Acad. Sci. USA 1995, 92, 7690–7694. [Google Scholar] [CrossRef]
- Keppler, D.; König, J. Expression and localization of the conjugate export pump encoded by the MRP2 (cMRP/cMOAJ) gene in liver. FASEB J. 1997, 11, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, R.N. Enzyme-catalyzed detoxication reactions: Mechanisms and stereochemistry. CRC Crit. Rev. Biochem. 1987, 22, 39–88. [Google Scholar] [CrossRef] [PubMed]
- Sau, A.; Pellizzari Tregno, F.; Valentino, F.; Federici, G.; Caccuri, A.M. Glutathione transferases and development of new principles to overcome drug resistance. Arch. Biochem. Biophys. 2010, 500, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Tew, K.D.; Dutta, S.; Schultz, M. Inhibitors of glutathione S-transferases as therapeutic agents. Adv. Drug Deliv. Rev. 1997, 26, 91–104. [Google Scholar] [CrossRef]
- Holkova, B.; Grant, S. Proteasome inhibitors in mantle cell lymphoma. Best Pract. Res. Clin. Haematol. 2012, 25, 133–141. [Google Scholar] [CrossRef]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J., Jr.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef]
- Egan, P.; Drain, S.; Conway, C.; Bjourson, A.J.; Alexander, H.D. Towards Stratified Medicine in Plasma Cell Myeloma. Int. J. Mol. Sci. 2016, 17, 1760. [Google Scholar] [CrossRef]
- Nikesitch, N.; Ling, S.C. Molecular mechanisms in multiple myeloma drug resistance. J. Clin. Pathol. 2016, 69, 97–101. [Google Scholar] [CrossRef]
- Yun, Z.; Zhichao, J.; Hao, Y.; Ou, J.; Ran, Y.; Wen, D.; Qun, S. Targeting autophagy in multiple myeloma. Leuk. Res. 2017, 59, 97–104. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Lippincott-Schwartz, J.; Yin, X.M.; Weiss, W.A.; Takebe, N.; Timmer, W.; DiPaola, R.S.; Lotze, M.T.; White, E. Principles and current strategies for targeting autophagy for cancer treatment. Clin. Cancer Res. 2011, 17, 654–666. [Google Scholar] [CrossRef]
- Milani, M.; Rzymski, T.; Mellor, H.R.; Pike, L.; Bottini, A.; Generali, D.; Harris, A.L. The Role of ATF4 Stabilization and Autophagy in Resistance of Breast Cancer Cells Treated with Bortezomib. Cancer Res. 2009, 69, 4415–4423. [Google Scholar] [CrossRef] [PubMed]
- Huber, E.M.; Heinemeyer, W.; Groll, M. Bortezomib-resistant mutant proteasomes: Structural and biochemical evaluation with carfilzomib and ONX 0914. Structure 2015, 23, 407–417. [Google Scholar] [CrossRef]
- Kubiczkova, L.; Pour, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. Proteasome inhibitors—Molecular basis and current perspectives in multiple myeloma. J. Cell. Mol. Med. 2014, 18, 947–961. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, A.; France, J.; Sy, M.S.; Harding, C.V. Down-regulation of the transporter for antigen presentation, proteasome subunits, and class I major histocompatibility complex in tumor cell lines. Cancer Res. 1998, 58, 3660–3667. [Google Scholar] [PubMed]
- Kitamura, A.; Maekawa, Y.; Uehara, H.; Izumi, K.; Kawachi, I.; Nishizawa, M.; Toyoshima, Y.; Takahashi, H.; Standley, D.M.; Tanaka, K.; et al. A mutation in the immunoproteasome subunit PSMB8 causes autoinflammation and lipodystrophy in humans. J. Clin. Investig. 2011, 121, 4150–4160. [Google Scholar] [CrossRef]
- Furukawa, Y.; Kikuchi, J. Molecular basis of clonal evolution in multiple myeloma. Int. J. Hematol. 2020, 111, 496–511. [Google Scholar] [CrossRef]
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348. [Google Scholar] [CrossRef]
- Solary, E.; Droin, N.; Bettaieb, A.; Corcos, L.; Dimanche-Boitrel, M.T.; Garrido, C. Positive and negative regulation of apoptotic pathways by cytotoxic agents in hematological malignancies. Leukemia 2000, 14, 1833–1849. [Google Scholar] [CrossRef]
- Abdi, J.; Chen, G.; Chang, H. Drug resistance in multiple myeloma: Latest findings and new concepts on molecular mechanisms. Oncotarget 2013, 4, 2186–2207. [Google Scholar] [CrossRef]
- Mitsiades, N.; Mitsiades, C.S.; Poulaki, V.; Chauhan, D.; Fanourakis, G.; Gu, X.; Bailey, C.; Joseph, M.; Libermann, T.A.; Treon, S.P.; et al. Molecular sequelae of proteasome inhibition in human multiple myeloma cells. Proc. Natl. Acad. Sci. USA 2002, 99, 14374–14379. [Google Scholar] [CrossRef]
- Balsas, P.; López-Royuela, N.; Galán-Malo, P.; Anel, A.; Marzo, I.; Naval, J. Cooperation between Apo2L/TRAIL and bortezomib in multiple myeloma apoptosis. Biochem. Pharmacol. 2009, 77, 804–812. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hamrouni, A.; Wolowiec, D.; Coiteux, V.; Kuliczkowski, K.; Hetuin, D.; Saudemont, A.; Quesnel, B. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-{gamma} and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood 2007, 110, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Geng, J.; Wang, Y.; Wang, L.; Huang, M.; Chen, J.; Zhang, K.; Xue, L.; Liu, X.; Mao, X.; et al. FOXM1 evokes 5-fluorouracil resistance in colorectal cancer depending on ABCC10. Oncotarget 2017, 8, 8574–8589. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef] [PubMed]
- Bi, C.; Chng, W.J. MicroRNA: Important Player in the Pathobiology of Multiple Myeloma. BioMed Res. Int. 2014, 2014, 521586. [Google Scholar] [CrossRef]
- Pichiorri, F.; Suh, S.S.; Ladetto, M.; Kuehl, M.; Palumbo, T.; Drandi, D.; Taccioli, C.; Zanesi, N.; Alder, H.; Hagan, J.P.; et al. MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 12885–12890. [Google Scholar] [CrossRef]
- Chen, L.; Li, C.; Zhang, R.; Gao, X.; Qu, X.; Zhao, M.; Qiao, C.; Xu, J.; Li, J. miR-17-92 cluster microRNAs confers tumorigenicity in multiple myeloma. Cancer Lett. 2011, 309, 62–70. [Google Scholar] [CrossRef]
- Zhang, Y.K.; Wang, H.; Leng, Y.; Li, Z.L.; Yang, Y.F.; Xiao, F.J.; Li, Q.F.; Chen, X.Q.; Wang, L.S. Overexpression of microRNA-29b induces apoptosis of multiple myeloma cells through down regulating Mcl-1. Biochem. Biophys. Res. Commun. 2011, 414, 233–239. [Google Scholar] [CrossRef]
- Löffler, D.; Brocke-Heidrich, K.; Pfeifer, G.; Stocsits, C.; Hackermüller, J.; Kretzschmar, A.K.; Burger, R.; Gramatzki, M.; Blumert, C.; Bauer, K.; et al. Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood 2007, 110, 1330–1333. [Google Scholar] [CrossRef]
- Gururajan, M.; Haga, C.L.; Das, S.; Leu, C.M.; Hodson, D.; Josson, S.; Turner, M.; Cooper, M.D. MicroRNA 125b inhibition of B cell differentiation in germinal centers. Int. Immunol. 2010, 22, 583–592. [Google Scholar] [CrossRef]
- Morelli, E.; Biamonte, L.; Federico, C.; Amodio, N.; Di Martino, M.T.; Gallo Cantafio, M.E.; Manzoni, M.; Scionti, F.; Samur, M.K.; Gullà, A.; et al. Therapeutic vulnerability of multiple myeloma to MIR17PTi, a first-in-class inhibitor of pri-miR-17-92. Blood 2018, 132, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.C.; Lin, S.F. Mechanisms of Drug Resistance in Relapse and Refractory Multiple Myeloma. BioMed Res. Int. 2015, 2015, 341430. [Google Scholar] [CrossRef] [PubMed]
- Fabre, C.; Mimura, N.; Bobb, K.; Kong, S.Y.; Gorgun, G.; Cirstea, D.; Hu, Y.; Minami, J.; Ohguchi, H.; Zhang, J.; et al. Dual inhibition of canonical and noncanonical NF-κB pathways demonstrates significant antitumor activities in multiple myeloma. Clin. Cancer Res. 2012, 18, 4669–4681. [Google Scholar] [CrossRef] [PubMed]
- Markovina, S.; Callander, N.S.; O’Connor, S.L.; Kim, J.; Werndli, J.E.; Raschko, M.; Leith, C.P.; Kahl, B.S.; Kim, K.; Miyamoto, S. Bortezomib-resistant nuclear factor-kappaB activity in multiple myeloma cells. Mol. Cancer Res. 2008, 6, 1356–1364. [Google Scholar] [CrossRef]
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665–674. [Google Scholar] [CrossRef]
- Markovina, S.; Callander, N.S.; O’Connor, S.L.; Xu, G.; Shi, Y.; Leith, C.P.; Kim, K.; Trivedi, P.; Kim, J.; Hematti, P.; et al. Bone marrow stromal cells from multiple myeloma patients uniquely induce bortezomib resistant NF-kappaB activity in myeloma cells. Mol. Cancer 2010, 9, 176. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, Y.; Saha, M.N.; Chen, J.; Evans, K.; Qiu, L.; Reece, D.; Chen, G.A.; Chang, H. Targeting phospho-MARCKS overcomes drug-resistance and induces antitumor activity in preclinical models of multiple myeloma. Leukemia 2015, 29, 715–726. [Google Scholar] [CrossRef]
- Wang, J.; Hendrix, A.; Hernot, S.; Lemaire, M.; De Bruyne, E.; Van Valckenborgh, E.; Lahoutte, T.; De Wever, O.; Vanderkerken, K.; Menu, E. Bone marrow stromal cell–derived exosomes as communicators in drug resistance in multiple myeloma cells. Blood 2014, 124, 555–566. [Google Scholar] [CrossRef]
- Yang, Y.; Shi, J.; Tolomelli, G.; Xu, H.; Xia, J.; Wang, H.; Zhou, W.; Zhou, Y.; Das, S.; Gu, Z.; et al. RARα2 expression confers myeloma stem cell features. Blood 2013, 122, 1437–1447. [Google Scholar] [CrossRef]
- Federico, C.; Alhallak, K.; Sun, J.; Duncan, K.; Azab, F.; Sudlow, G.P.; de la Puente, P.; Muz, B.; Kapoor, V.; Zhang, L.; et al. Tumor microenvironment-targeted nanoparticles loaded with bortezomib and ROCK inhibitor improve efficacy in multiple myeloma. Nat. Commun. 2020, 11, 6037. [Google Scholar] [CrossRef]
- Omstead, D.T.; Mejia, F.; Sjoerdsma, J.; Kim, B.; Shin, J.; Khan, S.; Wu, J.; Kiziltepe, T.; Littlepage, L.E.; Bilgicer, B. In vivo evaluation of CD38 and CD138 as targets for nanoparticle-based drug delivery in multiple myeloma. J. Hematol. Oncol. 2020, 13, 145. [Google Scholar] [CrossRef] [PubMed]
- de la Puente, P.; Luderer, M.J.; Federico, C.; Jin, A.; Gilson, R.C.; Egbulefu, C.; Alhallak, K.; Shah, S.; Muz, B.; Sun, J.; et al. Enhancing proteasome-inhibitory activity and specificity of bortezomib by CD38 targeted nanoparticles in multiple myeloma. J. Control. Release 2018, 270, 158–176. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Chu, B.; Wei, X.; Chen, Y.; Yang, Y.; Hu, D.; Huang, J.; Wang, F.; Chen, M.; Zheng, Y.; et al. Cancer-Cell-Biomimetic Nanoparticles for Targeted Therapy of Multiple Myeloma Based on Bone Marrow Homing. Adv. Mater. 2022, 34, 2107883. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Chai, Z.; Lu, L.; Ruan, H.; Wang, R.; Zhan, C.; Xie, C.; Pan, J.; Liu, M.; Wang, H.; et al. Bortezomib Dendrimer Prodrug-Based Nanoparticle System. Adv. Funct. Mater. 2019, 29, 1807941. [Google Scholar] [CrossRef]
- Gu, Z.; Wang, X.; Cheng, R.; Cheng, L.; Zhong, Z. Hyaluronic acid shell and disulfide-crosslinked core micelles for in vivo targeted delivery of bortezomib for the treatment of multiple myeloma. Acta Biomater. 2018, 80, 288–295. [Google Scholar] [CrossRef]
- Nigro, A.; Frattaruolo, L.; Fava, M.; De Napoli, I.; Greco, M.; Comandè, A.; De Santo, M.; Pellegrino, M.; Ricci, E.; Giordano, F.; et al. Bortezomib-Loaded Mesoporous Silica Nanoparticles Selectively Alter Metabolism and Induce Death in Multiple Myeloma Cells. Cancers 2020, 12, 2709. [Google Scholar] [CrossRef]
- Patra, C.R.; Verma, R.; Kumar, S.; Greipp, P.R.; Mukhopadhyay, D.; Mukherjee, P. Fabrication of Gold Nanoparticle for Potential Application in Multiple Myeloma. J. Biomed. Nanotechnol. 2008, 4, 499–507. [Google Scholar] [CrossRef]
- Che, F.; Chen, J.; Dai, J.; Liu, X. Inhibition of Multiple Myeloma Using 5-Aza-2′-Deoxycytidine and Bortezomib-Loaded Self-Assembling Nanoparticles. Cancer Manag. Res. 2020, 12, 6969–6976. [Google Scholar] [CrossRef]
- Waldschmidt, J.M.; Fruttiger, S.J.; Wider, D.; Jung, J.; Thomsen, A.R.; Hartmann, T.N.; Duyster, J.; Hug, M.J.; Azab, K.A.; Jung, M.; et al. Ex vivo propagation in a novel 3D high-throughput co-culture system for multiple myeloma. J. Cancer Res. Clin. Oncol. 2022, 148, 1045–1055. [Google Scholar] [CrossRef]
- Wu, D.; Wang, Z.; Li, J.; Song, Y.; Perez, M.E.M.; Wang, Z.; Cao, X.; Cao, C.; Maharjan, S.; Anderson, K.C.; et al. A 3D-Bioprinted Multiple Myeloma Model. Adv. Healthc. Mater. 2022, 11, 2100884. [Google Scholar] [CrossRef]
- Clara-Trujillo, S.; Tolosa, L.; Cordón, L.; Sempere, A.; Gallego Ferrer, G.; Gómez Ribelles, J.L. Novel microgel culture system as semi-solid three-dimensional in vitro model for the study of multiple myeloma proliferation and drug resistance. Biomater. Adv. 2022, 135, 212749. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-R.; Yu, Y.; Kramer, A.; Hon, R.; Wilson, M.; Brown, J.; Yang, L. An Ex Vivo 3D Tumor Microenvironment-Mimicry Culture to Study TAM Modulation of Cancer Immunotherapy. Cells 2022, 11, 1583. [Google Scholar] [CrossRef] [PubMed]
- Reagan, M.R.; Mishima, Y.; Glavey, S.V.; Zhang, Y.; Manier, S.; Lu, Z.N.; Memarzadeh, M.; Zhang, Y.; Sacco, A.; Aljawai, Y.; et al. Investigating osteogenic differentiation in multiple myeloma using a novel 3D bone marrow niche model. Blood 2014, 124, 3250–3259. [Google Scholar] [CrossRef] [PubMed]
- Freitas Misakyan, M.F.; Wijeratne, E.M.K.; Issa, M.E.; Xu, Y.-M.; Monteillier, A.; Gunatilaka, A.A.L.; Cuendet, M. Structure–Activity Relationships of Withanolides as Antiproliferative Agents for Multiple Myeloma: Comparison of Activity in 2D Models and a 3D Coculture Model. J. Nat. Prod. 2021, 84, 2321–2335. [Google Scholar] [CrossRef]
- Jakubikova, J.; Cholujova, D.; Hideshima, T.; Gronesova, P.; Soltysova, A.; Harada, T.; Joo, J.; Kong, S.Y.; Szalat, R.E.; Richardson, P.G.; et al. A novel 3D mesenchymal stem cell model of the multiple myeloma bone marrow niche: Biologic and clinical applications. Oncotarget 2016, 7, 77326–77341. [Google Scholar] [CrossRef]
- Sui, C.; Zilberberg, J.; Lee, W. Microfluidic device engineered to study the trafficking of multiple myeloma cancer cells through the sinusoidal niche of bone marrow. Sci. Rep. 2022, 12, 1439. [Google Scholar] [CrossRef]
- Ouyang, D.; Li, Y.; He, W.; Lin, W.; Hu, L.; Wang, C.; Xu, L.; Park, J.; You, L. Mechanical segregation and capturing of clonal circulating plasma cells in multiple myeloma using micropillar-integrated microfluidic device. Biomicrofluidics 2019, 13, 064114. [Google Scholar] [CrossRef]
- Carreras, P.; Gonzalez, I.; Gallardo, M.; Ortiz-Ruiz, A.; Martinez-Lopez, J. Droplet Microfluidics for the ex Vivo Expansion of Human Primary Multiple Myeloma Cells. Micromachines 2020, 11, 261. [Google Scholar] [CrossRef]
- Moore, T.A.; Brodersen, P.; Young, E.W.K. Multiple Myeloma Cell Drug Responses Differ in Thermoplastic vs. PDMS Microfluidic Devices. Anal. Chem. 2017, 89, 11391–11398. [Google Scholar] [CrossRef]
- Zeng, Y.; Gao, L.; Luo, X.; Chen, Y.; Kabeer, M.H.; Chen, X.; Stucky, A.; Loudon, W.G.; Li, S.C.; Zhang, X.; et al. Microfluidic enrichment of plasma cells improves treatment of multiple myeloma. Mol. Oncol. 2018, 12, 1004–1011. [Google Scholar] [CrossRef]
- Pak, C.; Callander, N.S.; Young, E.W.; Titz, B.; Kim, K.; Saha, S.; Chng, K.; Asimakopoulos, F.; Beebe, D.J.; Miyamoto, S. MicroC(3): An ex vivo microfluidic cis-coculture assay to test chemosensitivity and resistance of patient multiple myeloma cells. Integr. Biol. 2015, 7, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Glaser, D.E.; Curtis, M.B.; Sariano, P.A.; Rollins, Z.A.; Shergill, B.S.; Anand, A.; Deely, A.M.; Shirure, V.S.; Anderson, L.; Lowen, J.M.; et al. Organ-on-a-chip model of vascularized human bone marrow niches. Biomaterials 2022, 280, 121245. [Google Scholar] [CrossRef]
- Yetisgin, A.A.; Cetinel, S.; Zuvin, M.; Kosar, A.; Kutlu, O. Therapeutic Nanoparticles and Their Targeted Delivery Applications. Molecules 2020, 25, 2193. [Google Scholar] [CrossRef] [PubMed]
- Iannazzo, D.; Ettari, R.; Giofrè, S.; Eid, A.H.; Bitto, A. Recent Advances in Nanotherapeutics for Multiple Myeloma. Cancers 2020, 12, 3144. [Google Scholar] [CrossRef] [PubMed]
- Zheleznyak, A.; Shokeen, M.; Achilefu, S. Nanotherapeutics for multiple myeloma. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnology 2018, 10, e1526. [Google Scholar] [CrossRef]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: Importance and enhancement techniques. Int. Sch. Res. Not. Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef]
- Barenholz, Y.C. Doxil®—The first FDA-approved nano-drug: From an idea to a product. In Handbook of Harnessing Biomaterials in Nanomedicine; Jenny Stanford Publishing: Singapore, 2021; pp. 463–528. [Google Scholar]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; Rodriguez-Torres, M.D.P.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnology 2018, 16, 71. [Google Scholar] [CrossRef]
- Lee, K.H.; Kim, T.H. Recent Advances in Multicellular Tumor Spheroid Generation for Drug Screening. Biosensors 2021, 11, 445. [Google Scholar] [CrossRef]
- Regmi, S.; Poudel, C.; Adhikari, R.; Luo, K.Q. Applications of Microfluidics and Organ-on-a-Chip in Cancer Research. Biosensors 2022, 12, 459. [Google Scholar] [CrossRef]
- Mehta, P.; Rahman, Z.; Ten Dijke, P.; Boukany, P.E. Microfluidics meets 3D cancer cell migration. Trends Cancer 2022, 8, 683–697. [Google Scholar] [CrossRef]
- Gharib, G.; Bütün, İ.; Muganlı, Z.; Kozalak, G.; Namlı, İ.; Sarraf, S.S.; Ahmadi, V.E.; Toyran, E.; van Wijnen, A.J.; Koşar, A. Biomedical Applications of Microfluidic Devices: A Review. Biosensors 2022, 12, 1023. [Google Scholar] [CrossRef] [PubMed]
- Young, E.W.; Pak, C.; Kahl, B.S.; Yang, D.T.; Callander, N.S.; Miyamoto, S.; Beebe, D.J. Microscale functional cytomics for studying hematologic cancers. Blood 2012, 119, e76–e85. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudian, M.; Valizadeh, H.; Löbenberg, R.; Zakeri-Milani, P. Bortezomib-loaded lipidic-nano drug delivery systems; formulation, therapeutic efficacy, and pharmacokinetics. J. Microencapsul. 2021, 38, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Cetin, A.E.; Stevens, M.M.; Calistri, N.L.; Fulciniti, M.; Olcum, S.; Kimmerling, R.J.; Munshi, N.C.; Manalis, S.R. Determining therapeutic susceptibility in multiple myeloma by single-cell mass accumulation. Nat. Commun. 2017, 8, 1613. [Google Scholar] [CrossRef]
- Sung, H.W.; Choi, S.-E.; Chu, C.H.; Ouyang, M.; Kalyan, S.; Scott, N.; Hur, S.C. Sensitizing drug-resistant cancer cells from blood using microfluidic electroporator. PLoS ONE 2022, 17, e0264907. [Google Scholar] [CrossRef]
- Postek, W.; Garstecki, P. Droplet Microfluidics for High-Throughput Analysis of Antibiotic Susceptibility in Bacterial Cells and Populations. Acc. Chem. Res. 2022, 55, 605–615. [Google Scholar] [CrossRef]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef]
- Pires de Mello, C.P.; Carmona-Moran, C.; McAleer, C.W.; Perez, J.; Coln, E.A.; Long, C.J.; Oleaga, C.; Riu, A.; Note, R.; Teissier, S.; et al. Microphysiological heart-liver body-on-a-chip system with a skin mimic for evaluating topical drug delivery. Lab Chip 2020, 20, 749–759. [Google Scholar] [CrossRef]
- Maschmeyer, I.; Lorenz, A.K.; Schimek, K.; Hasenberg, T.; Ramme, A.P.; Hübner, J.; Lindner, M.; Drewell, C.; Bauer, S.; Thomas, A.; et al. A four-organ-chip for interconnected long-term co-culture of human intestine, liver, skin and kidney equivalents. Lab Chip 2015, 15, 2688–2699. [Google Scholar] [CrossRef]
- Sung, J.H.; Wang, Y.I.; Narasimhan Sriram, N.; Jackson, M.; Long, C.; Hickman, J.J.; Shuler, M.L. Recent Advances in Body-on-a-Chip Systems. Anal. Chem. 2019, 91, 330–351. [Google Scholar] [CrossRef]
- Ma, C.; Peng, Y.; Li, H.; Chen, W. Organ-on-a-Chip: A New Paradigm for Drug Development. Trends Pharmacol. Sci. 2021, 42, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Imura, Y.; Sato, K.; Yoshimura, E. Micro Total Bioassay System for Ingested Substances: Assessment of Intestinal Absorption, Hepatic Metabolism, and Bioactivity. Anal. Chem. 2010, 82, 9983–9988. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.R.; Ghoshal, D.; Mejías, J.C.; Rubio, D.F.; Keith, E.; Roy, K. A multi-niche microvascularized human bone marrow (hBM) on-a-chip elucidates key roles of the endosteal niche in hBM physiology. Biomaterials 2021, 270, 120683. [Google Scholar] [CrossRef]
- Galván-Chacón, V.P.; Zampouka, A.; Hesse, B.; Bohner, M.; Habibovic, P.; Barata, D. Bone-on-a-Chip: A Microscale 3D Biomimetic Model to Study Bone Regeneration. Adv. Eng. Mater. 2022, 24, 2101467. [Google Scholar] [CrossRef]
- Das, B.; Seesala, S.V.; Pal, P.; Roy, T.; Roy, P.G.; Dhara, S. A vascularized bone-on-a-chip model development via exploring mechanical stimulation for evaluation of fracture healing therapeutics. Vitr. Model. 2022, 1, 73–83. [Google Scholar] [CrossRef]
- Chou, D.B.; Frismantas, V.; Milton, Y.; David, R.; Pop-Damkov, P.; Ferguson, D.; MacDonald, A.; Vargel Bölükbaşı, Ö.; Joyce, C.E.; Moreira Teixeira, L.S.; et al. On-chip recapitulation of clinical bone marrow toxicities and patient-specific pathophysiology. Nat. Biomed. Eng. 2020, 4, 394–406. [Google Scholar] [CrossRef]
- Chen, W.; Yang, Y.; Chen, Y.; Du, F.; Zhan, H. Cost-effectiveness of bortezomib for multiple myeloma: A systematic review. Clin. Outcomes Res. 2016, 8, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xiao, L.; Tao, J.; Srinivasan, V.; Boyce, B.F.; Ebetino, F.H.; Oyajobi, B.O.; Boeckman, R.K.; Xing, L. Synthesis of a Bone-Targeted Bortezomib with In Vivo Anti-Myeloma Effects in Mice. Pharmaceutics 2018, 10, 154. [Google Scholar] [CrossRef] [PubMed]
- Lourenço, D.; Lopes, R.; Pestana, C.; Queirós, A.C.; João, C.; Carneiro, E.A. Patient-Derived Multiple Myeloma 3D Models for Personalized Medicine— Are We There Yet? Int. J. Mol. Sci. 2022, 23, 12888. [Google Scholar] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Effect | Ref. |
|---|---|---|
| Adult T-cell leukemia/Cutaneous T-cell lymphoma | Inactivation of nfκb pathway and up-regulation of NOXA | [11] |
| Breast cancer | Activation of caspase-3 in p53-null breast cancer cells | [12] |
| Cervical cancer | Increased expression of caspase-3, PARP, and increased the level of ER stress-associated and autophagy-related proteins | [13] |
| Colorectal carcinoma | Prevention of NF-κb signaling | [14] |
| Esophageal squamous cell carcinomas | TRAIL-induced apoptosis and increased Association of caspase-8 and the Fas-associated death domain | [15] |
| Head and neck squamous cell carcinomas | Inhibition of NF-κb and AP-1 activities | [16] |
| Melanoma | Activation of ER-stress and mitochondrial-dysregulation associated pathways | [17] |
| Neuroblastoma | Induction of eif2α signalling and ATF-4 dependent ER stress | [18] |
| Non-small lung cancer | Up-regulation of p21(waf1) and p53, and down-regulation of bcl-2 via the JNK/c-Jun/AP-1 signaling | [19] |
| Pancreatic cancer | Repression in Bcl-2 and an Increase in Bax and p53 | [20] |
| Prostate cancer | Inhibition of HIF-1α and suppression of PI3K/Akt/mtor and MAPK pathways | [21] |
| Renal cell carcinoma | Increase in caspase-8 activity | [22] |
| Resistance Mechanism | Main Factors | Contribution | Ref. |
|---|---|---|---|
| Abnormal drug transport | P-gp, BCRP, LRP, MRP1-9 | Increases Bortezomib excretion | [28,29,30,31,32,33,34,35,36] |
| Activation of detoxification systems | GSH/GST levels | Increases Bortezomib excretion | [37,38] |
| Changes in drug targets | Unfolded Protein Response, Autophagy, PSMB5 and PSMB8 mutations | Prevents Bortezomib binding to proteasome by interrupting the UPS | [39,40,41,42] |
| Domination of cell cycle or apoptosis | P-53, c-Myc, MAF, Bcl-2/Bax ratio, anti-apoptotic factors, miRNAs | Regulates cell survival and death | [43,44,45,46,47,48] |
| Distortion of signaling pathways | NF-kB, JAK/STAT3, PI3K/AKT, SFM-DR, CAM-DR | Maintains interaction with BM microenvironment | [49,50,51,52] |
| Emerging Technology | Method | Anticancer Agent | Purpose | Ref. |
|---|---|---|---|---|
| Nanoparticles | Liposome | Bortezomib | Drug delivery | [122,123] |
| Chitosan | Bortezomib | Drug delivery | [124] | |
| Poly(ε-caprolactone)-poly(ethylene glycol)-poly(ε-caprolactone) with the MM cell membrane | Bortezomib | Drug delivery | [125] | |
| PEGylated dendrimer | Bortezomib | Drug delivery | [126] | |
| Hyaluronic acid shell and disulfide-crosslinked core micelles | Bortezomib | Drug delivery | [127] | |
| Mesoporous silica | Bortezomib | Drug delivery | [128] | |
| Gold nanoparticle | Bortezomib | Drug delivery | [129] | |
| Polyethylene glycol and polycaprolactone | 5-Aza-2ʹ-deoxycytidine Bortezomib | Drug delivery | [130] | |
| 3D culture systems | Conical agarose microwell array | Bortezomib and Auranofin | 3D high-throughput co-culture system | [131] |
| Coaxial extrusion bioprinting | Bortezomib Tocilizumab | 3D-Bioprinted multiple myeloma model | [132] | |
| Microspheres Microgels | Bortezomib Dexamethasone | Dynamic 3D multiple myeloma culture | [133] | |
| TAM modulation of cancer immunotherapy | - | Ex vivo 3D TME-mimicry culture | [134] | |
| 3D myeloma coculture with bone cell/cancer cell | - | Investigation of MM cells osteogenesis, angiogenesis, tumor growth, and drug response | [135] | |
| 2D/3D coculture | Withaferin A | Cytotoxicity | [136] | |
| 2D/Hydrogel based 3D ex vivo co-culture system | Pomalidomide Lenalidomide Thalidomide Bortezomib Carfilzomib Doxorubicin Dexamethasone Melphalan | MM pathogenesis and drug resistance in the BM niche | [137] | |
| Microfluidic systems | Traffic and metastasis of MM cells | - | Mimic bone marrows’ stroma, sinusoidal endothelium and circulation | [138] |
| Capture clonal plasma cells | - | Micropillar-integrated microfluidic device | [139] | |
| Micromanipulation and encapsulation using a droplet-based microfluidic device | Bortezomib Lenalidomide | Ex vivo platform of primary multiple myeloma cells for drug screening | [140] | |
| Thermoplastic PDMS microfluidic devices | Bortezomib Carfilzomib | The importance of material selection in microfluidic device design, for drug cytotoxicity | [141] | |
| Enrichment of plasma cells by MF-CD45-TACs (microfluidic–CD45 depletion–tetrameric antibody complexes) | - | Detection of cytogenetic abnormalities in MM patients | [142] | |
| Propagation of primary CD138+ MM cells in microfluidic-cis-culture (MicroC3) to simulate patients’ own tumor microenvironment | Bortezomib | Chemosensitivity and resistance assay | [143] | |
| Organ-on-chip | Organ-on-a-chip model of vascularized human bone marrow niches | Doxorubicin | 3D in vitro model of human bone marrow function and drug response | [144] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kozalak, G.; Bütün, İ.; Toyran, E.; Koşar, A. Review on Bortezomib Resistance in Multiple Myeloma and Potential Role of Emerging Technologies. Pharmaceuticals 2023, 16, 111. https://doi.org/10.3390/ph16010111
Kozalak G, Bütün İ, Toyran E, Koşar A. Review on Bortezomib Resistance in Multiple Myeloma and Potential Role of Emerging Technologies. Pharmaceuticals. 2023; 16(1):111. https://doi.org/10.3390/ph16010111
Chicago/Turabian StyleKozalak, Gül, İsmail Bütün, Erçil Toyran, and Ali Koşar. 2023. "Review on Bortezomib Resistance in Multiple Myeloma and Potential Role of Emerging Technologies" Pharmaceuticals 16, no. 1: 111. https://doi.org/10.3390/ph16010111
APA StyleKozalak, G., Bütün, İ., Toyran, E., & Koşar, A. (2023). Review on Bortezomib Resistance in Multiple Myeloma and Potential Role of Emerging Technologies. Pharmaceuticals, 16(1), 111. https://doi.org/10.3390/ph16010111