
P-Glycoprotein (MDR1/ABCB1) Restricts Brain Accumulation of the Novel EGFR Inhibitor EAI045 and Oral Elacridar Coadministration Enhances Its Brain Accumulation and Oral Exposure

 , and
, and
Abstract
:1. Introduction
2. Results
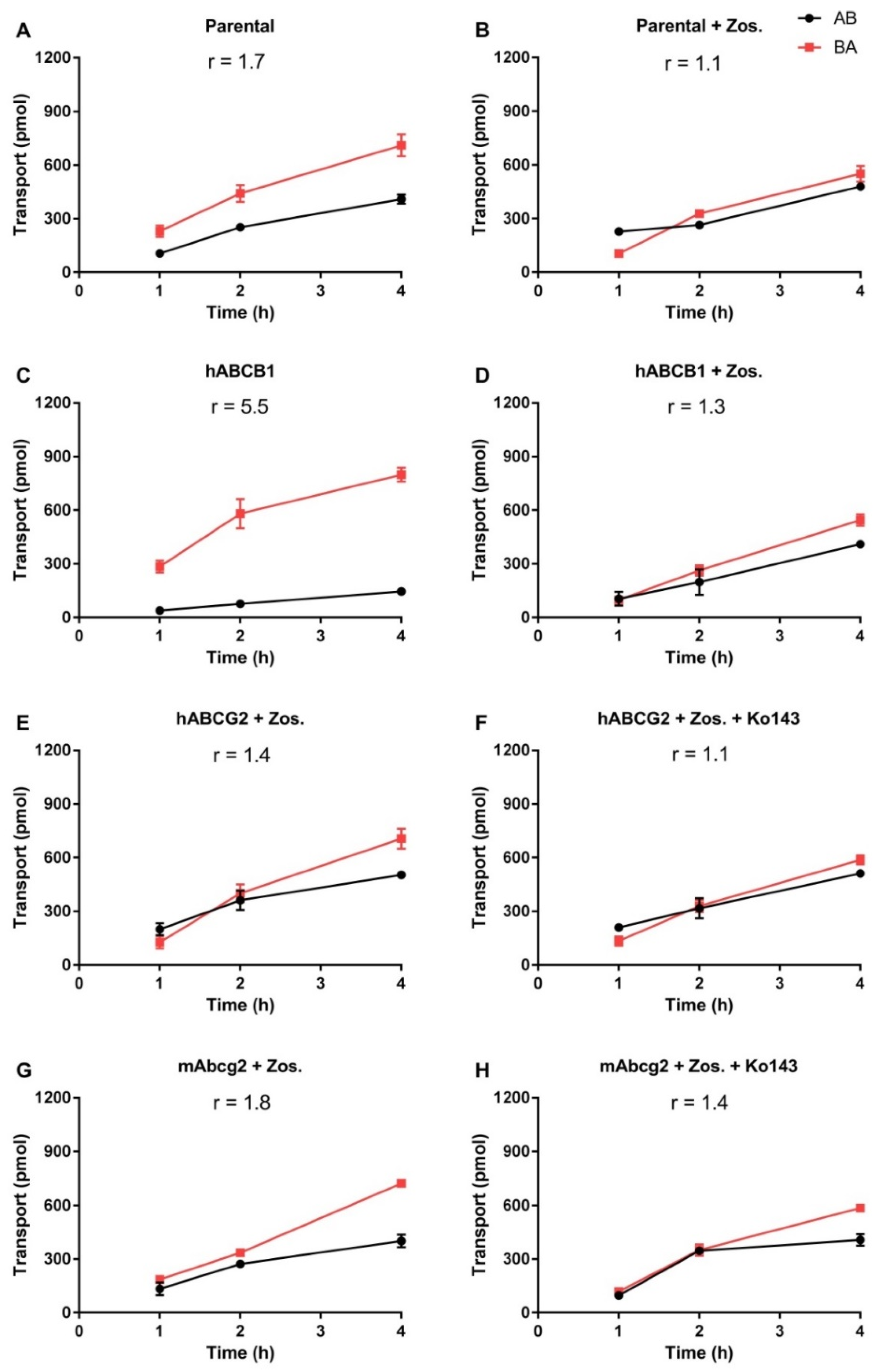
2.1. In Vitro Transport of EAI045
2.2. Impact of ABCB1 and ABCG2 on Oral EAI045 Plasma and Tissue Distribution over 4 h
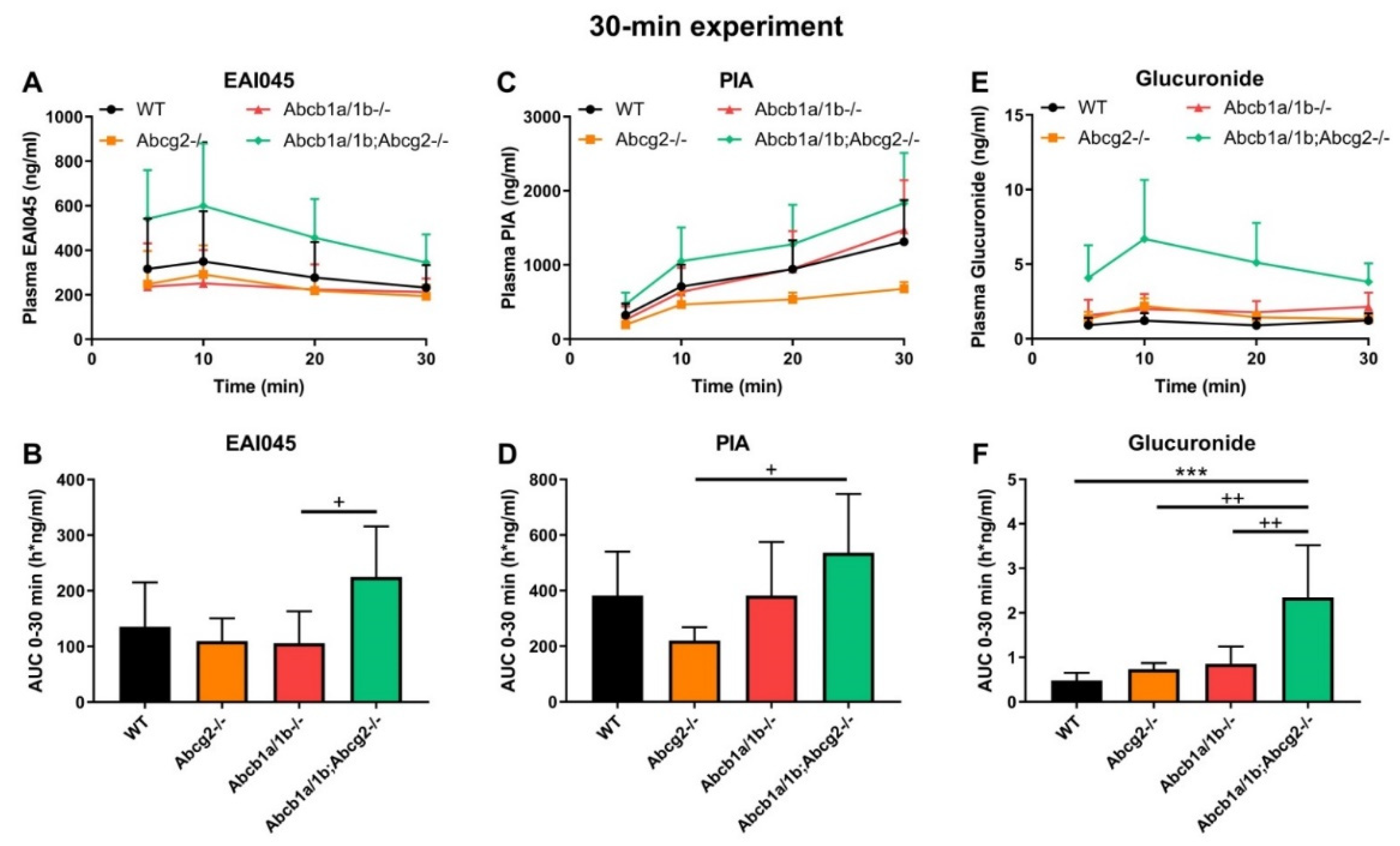
2.3. Effect of ABCB1 and ABCG2 on EAI045 Plasma and Tissue Distribution over 30 min
2.4. Effect of ABCB1 and ABCG2 on Plasma and Tissue Distribution of EAI045 Metabolites
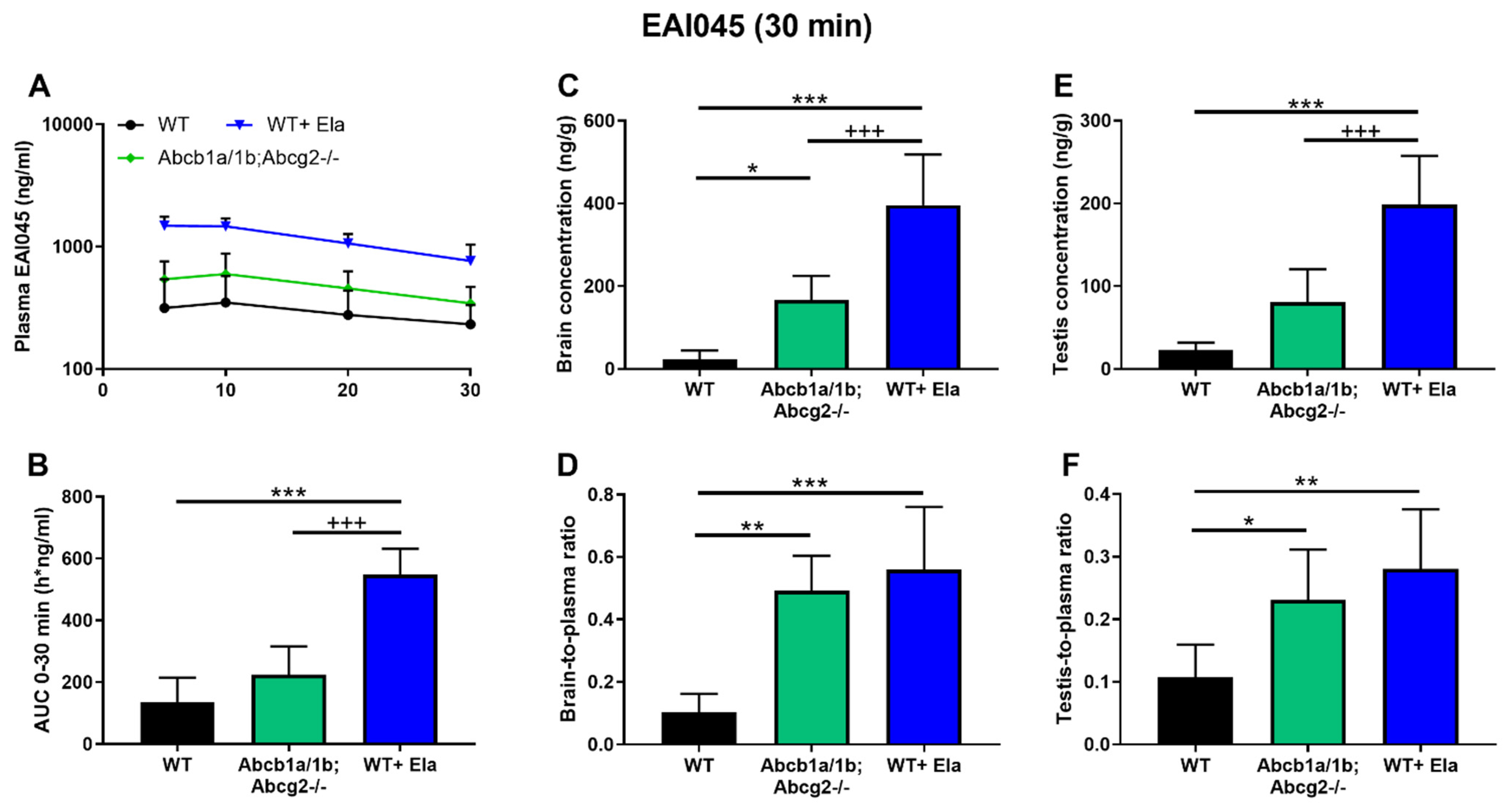
2.5. Co-Administration of Elacridar Boosts Brain and Testis Distribution of EAI045
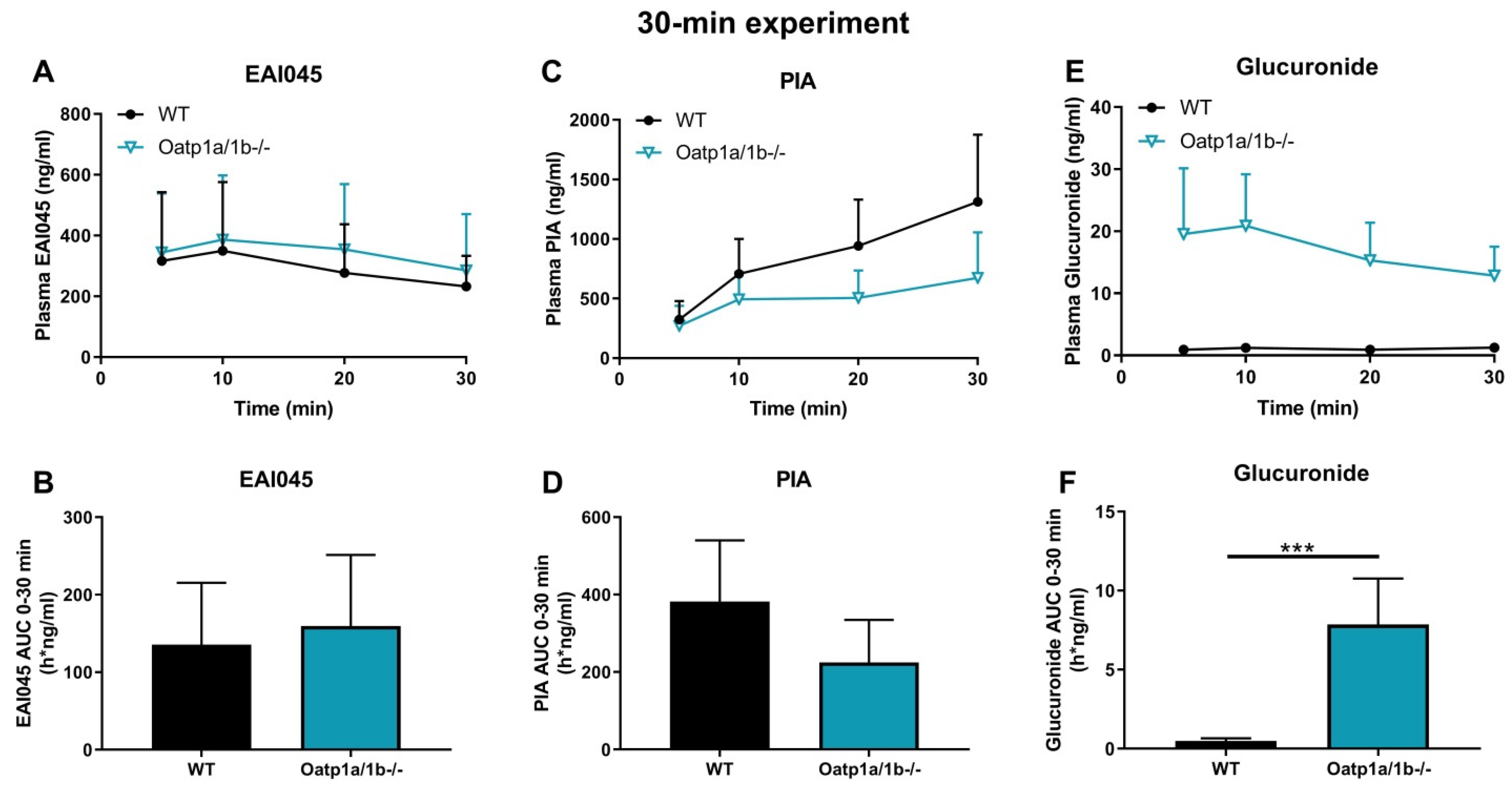
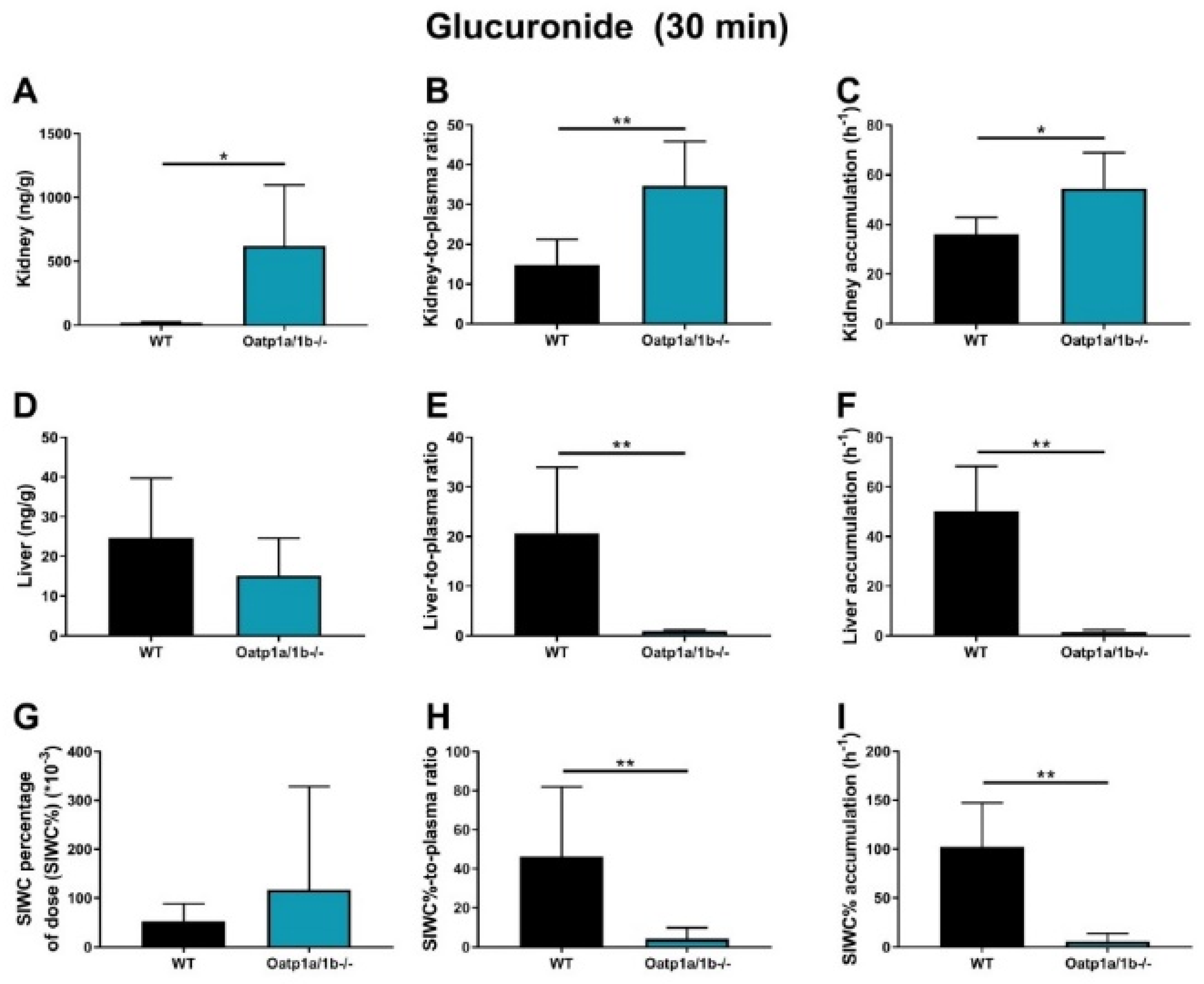
2.6. Impact of OATP1A/1B on Plasma and Tissue Distribution of EAI045 and Its Metabolites
2.7. Assessment of the In Vivo Role of CYP3A in EAI045 Pharmacokinetics
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Transport Assays
4.3. Animals
4.4. Working Solutions
4.5. Plasma and Tissue Pharmacokinetics of EAI045
4.6. LC-MS/MS Analysis
4.7. Pharmacokinetic and Statistical Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Zarogoulidis, K.; Zarogoulidis, P.; Darwiche, K.; Boutsikou, E.; Machairiotis, N.; Tsakiridis, K.; Katsikogiannis, N.; Kougioumtzi, I.; Karapantzos, I.; Huang, H. Treatment of non-small cell lung cancer (NSCLC). J. Thorac. Dis. 2013, 5, S389. [Google Scholar] [PubMed]
- Navada, S.; Lai, P.; Schwartz, A.; Kalemkerian, G. Temporal trends in small cell lung cancer: Analysis of the national Surveillance, Epidemiology, and End-Results (SEER) database. J. Clin. Oncol. 2006, 24, 7082. [Google Scholar] [CrossRef]
- Sher, T.; Dy, G.K.; Adjei, A.A. Small cell lung cancer. In Mayo Clinic Proceedings; Elsevier: Amsterdam, The Netherlands, 2008; pp. 355–367. [Google Scholar]
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. In Mayo Clinic Proceedings; Elsevier: Amsterdam, The Netherlands, 2008; pp. 584–594. [Google Scholar]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.; Kuang, Y. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non–small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. [Google Scholar] [CrossRef]
- Niederst, M.J.; Hu, H.; Mulvey, H.E.; Lockerman, E.L.; Garcia, A.R.; Piotrowska, Z.; Sequist, L.V.; Engelman, J.A. The allelic context of the C797S mutation acquired upon treatment with third-generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin. Cancer Res. 2015, 21, 3924–3933. [Google Scholar] [CrossRef]
- Helena, A.Y.; Tian, S.K.; Drilon, A.E.; Borsu, L.; Riely, G.J.; Arcila, M.E.; Ladanyi, M. Acquired resistance of EGFR-mutant lung cancer to a T790M-specific EGFR inhibitor: Emergence of a third mutation (C797S) in the EGFR tyrosine kinase domain. JAMA Oncol. 2015, 1, 982–984. [Google Scholar]
- Wang, S.; Tsui, S.T.; Liu, C.; Song, Y.; Liu, D. EGFR C797S mutation mediates resistance to third-generation inhibitors in T790M-positive non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 59. [Google Scholar] [CrossRef]
- Jia, Y.; Yun, C.-H.; Park, E.; Ercan, D.; Manuia, M.; Juarez, J.; Xu, C.; Rhee, K.; Chen, T.; Zhang, H. Overcoming EGFR (T790M) and EGFR (C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016, 534, 129–132. [Google Scholar] [CrossRef]
- Goldstein, N.I.; Prewett, M.; Zuklys, K.; Rockwell, P.; Mendelsohn, J. Biological efficacy of a chimeric antibody to the epidermal growth factor receptor in a human tumor xenograft model. Clin. Cancer Res. 1995, 1, 1311–1318. [Google Scholar]
- Li, S.; Schmitz, K.R.; Jeffrey, P.D.; Wiltzius, J.J.; Kussie, P.; Ferguson, K.M. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 2005, 7, 301–311. [Google Scholar] [CrossRef]
- Cho, J.; Chen, L.; Sangji, N.; Okabe, T.; Yonesaka, K.; Francis, J.M.; Flavin, R.J.; Johnson, W.; Kwon, J.; Yu, S. Cetuximab response of lung cancer–derived EGF receptor mutants is associated with asymmetric dimerization. Cancer Res. 2013, 73, 6770–6779. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Maher, J.M.; Chen, C.; Klaassen, C.D. Tissue distribution and ontogeny of mouse organic anion transporting polypeptides (Oatps). Drug Metab. Dispos. 2005, 33, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Klaassen, C.D. Tissue distribution, ontogeny, and hormonal regulation of xenobiotic transporters in mouse kidneys. Drug Metab. Dispos. 2009, 37, 2178–2185. [Google Scholar] [CrossRef] [PubMed]
- Endicott, J.A.; Ling, V. The biochemistry of P-glycoprotein-mediated multidrug resistance. Annu. Rev. Biochem. 1989, 58, 137–171. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Pastan, I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu. Rev. Biochem. 1993, 62, 385–427. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP–dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef]
- Thiebaut, F.; Tsuruo, T.; Hamada, H.; Gottesman, M.M.; Pastan, I.; Willingham, M.C. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc. Natl. Acad. Sci. USA 1987, 84, 7735–7738. [Google Scholar] [CrossRef]
- Sugawara, I.; Kataoka, I.; Morishita, Y.; Hamada, H.; Tsuruo, T.; Itoyama, S.; Mori, S. Tissue distribution of P-glycoprotein encoded by a multidrug-resistant gene as revealed by a monoclonal antibody, MRK 16. Cancer Res. 1988, 48, 1926–1929. [Google Scholar]
- Cordon-Cardo, C.; O’Brien, J.P.; Casals, D.; Rittman-Grauer, L.; Biedler, J.L.; Melamed, M.R.; Bertino, J.R. Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc. Natl. Acad. Sci. USA 1989, 86, 695–698. [Google Scholar] [CrossRef]
- Kennedy, B.G.; Mangini, N.J. P-glycoprotein expression in human retinal pigment epithelium. Mol. Vis. 2002, 8, 422–430. [Google Scholar]
- Constable, P.A.; Lawrenson, J.G.; Dolman, D.E.; Arden, G.B.; Abbott, N.J. P-Glycoprotein expression in human retinal pigment epithelium cell lines. Exp. Eye Res. 2006, 83, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.H.; Jonker, J.W. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: An overview. Adv. Drug Deliv. Rev. 2003, 55, 3–29. [Google Scholar] [CrossRef]
- Hagenbuch, B.; Meier, P.J. Organic anion transporting polypeptides of the OATP/SLC21 family: Phylogenetic classification as OATP/SLCO superfamily, new nomenclature and molecular/functional properties. Pflügers Arch. 2004, 447, 653–665. [Google Scholar] [CrossRef] [PubMed]
- Iusuf, D.; van Esch, A.; Hobbs, M.; Taylor, M.; Kenworthy, K.E.; van de Steeg, E.; Wagenaar, E.; Schinkel, A.H. Murine Oatp1a/1b uptake transporters control rosuvastatin systemic exposure without affecting its apparent liver exposure. Mol. Pharmacol. 2013, 83, 919–929. [Google Scholar] [CrossRef] [Green Version]
- Gartzke, D.; Delzer, J.; Laplanche, L.; Uchida, Y.; Hoshi, Y.; Tachikawa, M.; Terasaki, T.; Sydor, J.; Fricker, G. Genomic knockout of endogenous canine P-glycoprotein in wild-type, human P-glycoprotein and human BCRP transfected MDCKII cell lines by zinc finger nucleases. Pharm. Res. 2015, 32, 2060–2071. [Google Scholar] [CrossRef]
- Kamiie, J.; Ohtsuki, S.; Iwase, R.; Ohmine, K.; Katsukura, Y.; Yanai, K.; Sekine, Y.; Uchida, Y.; Ito, S.; Terasaki, T. Quantitative atlas of membrane transporter proteins: Development and application of a highly sensitive simultaneous LC/MS/MS method combined with novel in-silico peptide selection criteria. Pharm. Res. 2008, 25, 1469–1483. [Google Scholar] [CrossRef]
- van Hoppe, S.; Sparidans, R.W.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. Breast cancer resistance protein (BCRP/ABCG2) and P-glycoprotein (P-gp/ABCB1) transport afatinib and restrict its oral availability and brain accumulation. Pharmacol. Res. 2017, 120, 43–50. [Google Scholar] [CrossRef]
- Li, W.; Sparidans, R.W.; Wang, Y.; Lebre, M.C.; Beijnen, J.H.; Schinkel, A.H. P-glycoprotein and breast cancer resistance protein restrict brigatinib brain accumulation and toxicity, and, alongside CYP3A, limit its oral availability. Pharmacol. Res. 2018, 137, 47–55. [Google Scholar] [CrossRef]
- van Hoppe, S.; Jamalpoor, A.; Rood, J.J.; Wagenaar, E.; Sparidans, R.W.; Beijnen, J.H.; Schinkel, A.H. Brain accumulation of osimertinib and its active metabolite AZ5104 is restricted by ABCB1 (P-glycoprotein) and ABCG2 (breast cancer resistance protein). Pharmacol. Res. 2019, 146, 104297. [Google Scholar] [CrossRef]
- Wu, X.; Guo, Q.; Li, Q.; Wan, S.; Li, Z.; Zhang, J. Molecular mechanism study of EGFR allosteric inhibitors using molecular dynamics simulations and free energy calculations. J. Biomol. Struct. Dyn. 2022, 40, 5848–5857. [Google Scholar] [CrossRef]
- Unnisa, A.; Chettupalli, A.K.; Hussain, T.; Kamal, M.A. Recent Advances in Epidermal Growth Factor Receptor Inhibitors (EGFRIs) and their role in the Treatment of Cancer: A Review. Anticancer Agents Med. Chem. 2022. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.F.; Zhu, M.L.; Liu, M.M.; Xu, Y.T.; Yuan, L.L.; Bian, J.; Xia, Y.Z.; Kong, L.Y. EGFR mutation mediates resistance to EGFR tyrosine kinase inhibitors in NSCLC: From molecular mechanisms to clinical research. Pharmacol. Res. 2021, 167, 105583. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gan, C.; Sparidans, R.W.; Wagenaar, E.; van Hoppe, S.; Beijnen, J.H.; Schinkel, A.H. P-glycoprotein (MDR1/ABCB1) and breast cancer resistance protein (BCRP/ABCG2) affect brain accumulation and intestinal disposition of encorafenib in mice. Pharmacol. Res. 2018, 129, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gan, C.; Retmana, I.A.; Sparidans, R.W.; Li, W.; Lebre, M.C.; Beijnen, J.H.; Schinkel, A.H. P-glycoprotein (MDR1/ABCB1) and Breast Cancer Resistance Protein (BCRP/ABCG2) limit brain accumulation of the FLT3 inhibitor quizartinib in mice. Int. J. Pharm. 2019, 556, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Bakos, É.; Német, O.; Patik, I.; Kucsma, N.; Várady, G.; Szakács, G.; Özvegy-Laczka, C. A novel fluorescence-based functional assay for human OATP1A2 and OATP1C1 identifies interaction between third-generation P-gp inhibitors and OATP1A2. FEBS J. 2020, 287, 2468–2485. [Google Scholar] [CrossRef]
- van Waterschoot, R.A.; van Herwaarden, A.E.; Lagas, J.S.; Sparidans, R.W.; Wagenaar, E.; van der Kruijssen, C.M.; Goldstein, J.A.; Zeldin, D.C.; Beijnen, J.H.; Schinkel, A.H. Midazolam metabolism in cytochrome P450 3A knockout mice can be attributed to up-regulated CYP2C enzymes. Mol. Pharmacol. 2008, 73, 1029–1036. [Google Scholar] [CrossRef]
- Schinkel, A.H.; Mayer, U.; Wagenaar, E.; Mol, C.A.; Van Deemter, L.; Smit, J.J.; Van Der Valk, M.A.; Voordouw, A.C.; Spits, H.; Van Tellingen, O. Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proc. Natl. Acad. Sci. USA 1997, 94, 4028–4033. [Google Scholar] [CrossRef]
- Jonker, J.W.; Buitelaar, M.; Wagenaar, E.; Van Der Valk, M.A.; Scheffer, G.L.; Scheper, R.J.; Plösch, T.; Kuipers, F.; Elferink, R.P.O.; Rosing, H. The breast cancer resistance protein protects against a major chlorophyll-derived dietary phototoxin and protoporphyria. Proc. Natl. Acad. Sci. USA 2002, 99, 15649–15654. [Google Scholar] [CrossRef]
- Jonker, J.W.; Freeman, J.; Bolscher, E.; Musters, S.; Alvi, A.J.; Titley, I.; Schinkel, A.H.; Dale, T.C. Contribution of the ABC transporters Bcrp1 and Mdr1a/1b to the side population phenotype in mammary gland and bone marrow of mice. Stem Cells 2005, 23, 1059–1065. [Google Scholar] [CrossRef]
- van Herwaarden, A.E.; Wagenaar, E.; van der Kruijssen, C.M.; van Waterschoot, R.A.; Smit, J.W.; Song, J.-Y.; van der Valk, M.A.; van Tellingen, O.; van der Hoorn, J.W.; Rosing, H. Knockout of cytochrome P450 3A yields new mouse models for understanding xenobiotic metabolism. J. Clin. Investig. 2007, 117, 3583–3592. [Google Scholar] [CrossRef]
- van de Steeg, E.; Wagenaar, E.; van der Kruijssen, C.M.; Burggraaff, J.E.; de Waart, D.R.; Elferink, R.P.O.; Kenworthy, K.E.; Schinkel, A.H. Organic anion transporting polypeptide 1a/1b–knockout mice provide insights into hepatic handling of bilirubin, bile acids, and drugs. J. Clin. Investig. 2010, 120, 2942–2952. [Google Scholar] [CrossRef] [PubMed]
- Susam, M.M.; Wang, J.; Schinkel, A.H.; Beijnen, J.H.; Sparidans, R.W. Bioanalytical assay for the quantification of the tyrosine kinase inhibitor EAI045 and its major metabolite PIA in mouse plasma and tissue homogenates using liquid chromatography-tandem mass spectrometry. Biomed. Chromatogr. 2022, e5457. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huo, M.; Zhou, J.; Xie, S. PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput. Methods Programs Biomed. 2010, 99, 306–314. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter (EAI045) | Genotype | ||||
|---|---|---|---|---|---|
| WT | Abcg2-/- | Abcb1a/1b-/- | Abcb1a/1b;Abcg2-/- | WT + Ela | |
| Plasma AUC0–30min (h*ng/mL) | 136 ± 80 | 110 ± 41 | 106 ± 57 + | 225 ± 91 | 549 ± 82 ***. +++ |
| Fold change AUC | 1.0 | 0.81 | 0.78 | 1.7 | 4.0 |
| tmax (min) | 5–30 | 10–30 | 10–30 | 10–30 | 10–30 |
| Cmax (ng/mL) | 354 ± 225 | 310 ± 114 | 201 ± 79 | 603 ± 277 | 1512 ± 260 |
| Cbrain (ng/g) | 23.3 ± 21.3 | 25.2 ± 17.6 +++ | 82.0 ± 29.7 ++ | 166.9 ± 58.4 *** | 395.9 ± 123.1 ***. +++ |
| Fold change Cbrain | 1.0 | 1.1 | 3.5 | 7.2 | 17.0 |
| Kp.brain | 0.103 ± 0.059 | 0.094 ± 0.046 +++ | 0.401 ± 0.149 *** | 0.492 ± 0.112 *** | 0.560 ± 0.200 *** |
| Fold change Kp.brain | 1.0 | 0.92 | 3.9 | 4.8 | 5.4 |
| Ctestis (ng/g) | 22.8 ± 8.9 | 22.8 ± 20.2 ++ | 70.9 ± 36.2 | 80.9 ± 39.4 ** | 198.7 ± 58.9 ***. +++ |
| Fold change Ctestis | 1.0 | 1.0 | 3.1 | 3.5 | 8.7 |
| Kp.testis | 0.107 ± 0.052 | 0.074 ± 0.015 ++ | 0.329 ± 0.114 *** | 0.231 ± 0.080 ** | 0.280 ± 0.096 ** |
| Fold change Kp.testis | 1.0 | 0.69 | 3.1 | 2.2 | 2.6 |
| SIWC% | 29.9 ± 11.3 | 29.5 ± 9.1 | 27.6 ± 9.2 | 14.2 ± 8.8 | 4.00 ± 2.57 *** |
| Fold change SIWC% | 1.0 | 0.99 | 0.92 | 0.48 (2.1-fold) | 0.13 (7.5-fold) |
| Kp.SIWC% | 158 ± 86 | 164 ± 70 + | 148 ± 82 + | 44.4 ± 28.3 * | 5.13 ± 2.63 *** |
| Fold change Kp.SIWC% | 1.0 | 1.04 | 0.94 | 0.28 (3.6) | 0.032 (30.8) |
| Parameter (Glucuronide) | Genotype | |
|---|---|---|
| WT | Oatp1a/1b-/- | |
| Plasma AUC0–30min (h*ng/mL) | 0.483 ± 0.168 | 7.86 ± 2.91 *** |
| Fold change AUC | 1.0 | 16 |
| tmax (min) | 5−30 | 5–30 |
| Cmax (ng/mL) | 1.53 ± 0.55 | 22.2 ± 8.9 |
| Kidney | 17.6 ± 7.3 | 622 ± 475 * |
| Fold change kidney | 1.0 | 35 |
| Kp.Kidney | 14.8 ± 6.5 | 34.8 ± 11.1 ** |
| Fold change Kp.Kidney | 1.0 | 2.4 |
| PKidney (*10−6h−1) | 36.0 ± 6.8 | 54.6 ± 14.3 * |
| Fold change PKidney | 1.0 | 1.5 |
| Liver | 24.7 ± 15.0 | 15.2 ± 9.4 |
| Fold change liver | 1.0 | 0.62 |
| Kp.Liver | 20.7 ± 13.3 | 0.915 ± 0.315 ** |
| Fold change Kp.Liver | 1.0 | 0.044 |
| PLiver (*10−6h−1) | 50.3 ± 18.0 | 1.54 ± 0.87 ** |
| Fold change PLiver | 1.0 | 0.031 |
| SIWC% | 52.3 ± 36.1 | 117 ± 211 |
| Fold change SIWC% | 1.0 | 2.2 |
| Kp.SIWC% | 46.2 ± 35.7 | 4.08 ± 5.72 ** |
| Fold change Kp.SIWC% | 1.0 | 0.088 |
| PSIWC% (*10−6h−1) | 102 ± 45 | 5.80 ± 7.84 ** |
| Fold change PSIWC% | 1.0 | 0.057 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Susam, M.M.; Gan, C.; Sparidans, R.W.; Lebre, M.C.; Beijnen, J.H.; Schinkel, A.H. P-Glycoprotein (MDR1/ABCB1) Restricts Brain Accumulation of the Novel EGFR Inhibitor EAI045 and Oral Elacridar Coadministration Enhances Its Brain Accumulation and Oral Exposure. Pharmaceuticals 2022, 15, 1124. https://doi.org/10.3390/ph15091124
Wang J, Susam MM, Gan C, Sparidans RW, Lebre MC, Beijnen JH, Schinkel AH. P-Glycoprotein (MDR1/ABCB1) Restricts Brain Accumulation of the Novel EGFR Inhibitor EAI045 and Oral Elacridar Coadministration Enhances Its Brain Accumulation and Oral Exposure. Pharmaceuticals. 2022; 15(9):1124. https://doi.org/10.3390/ph15091124
Chicago/Turabian StyleWang, Jing, M. Merve Susam, Changpei Gan, Rolf W. Sparidans, Maria C. Lebre, Jos H. Beijnen, and Alfred H. Schinkel. 2022. "P-Glycoprotein (MDR1/ABCB1) Restricts Brain Accumulation of the Novel EGFR Inhibitor EAI045 and Oral Elacridar Coadministration Enhances Its Brain Accumulation and Oral Exposure" Pharmaceuticals 15, no. 9: 1124. https://doi.org/10.3390/ph15091124
APA StyleWang, J., Susam, M. M., Gan, C., Sparidans, R. W., Lebre, M. C., Beijnen, J. H., & Schinkel, A. H. (2022). P-Glycoprotein (MDR1/ABCB1) Restricts Brain Accumulation of the Novel EGFR Inhibitor EAI045 and Oral Elacridar Coadministration Enhances Its Brain Accumulation and Oral Exposure. Pharmaceuticals, 15(9), 1124. https://doi.org/10.3390/ph15091124