
Why Multitarget Vasodilatory (Endo)cannabinoids Are Not Effective as Antihypertensive Compounds after Chronic Administration: Comparison of Their Effects on Systemic and Pulmonary Hypertension

Abstract
:1. Introduction
2. Systemic Hypertension
3. Pulmonary Hypertension
4. Animal Models of Hypertension
5. Cannabinoids as a Potential New Therapy against Systemic and Pulmonary Hypertension
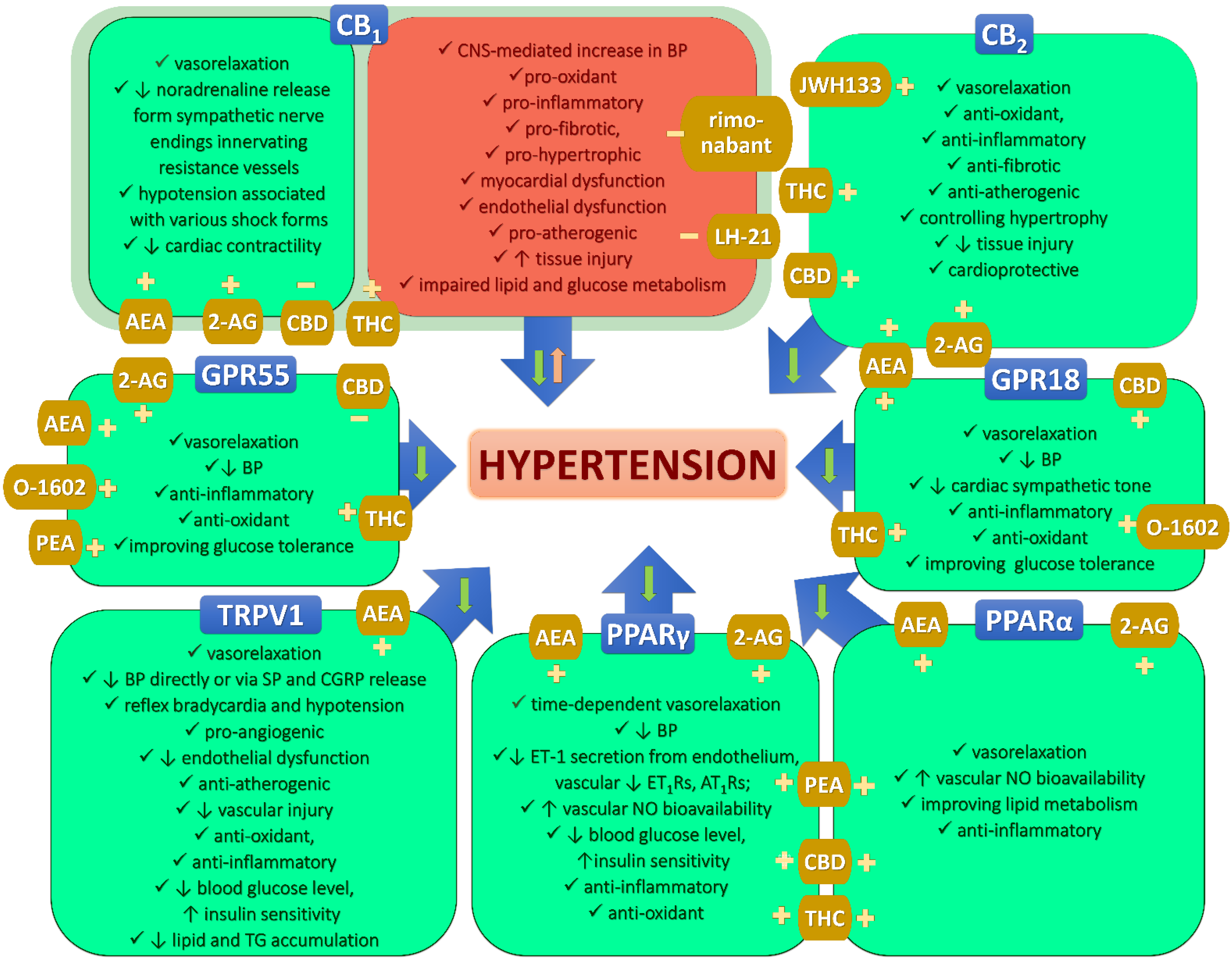
6. Cannabinoids and the Endocannabinoid System
7. Vasodilatory Effects of Chosen (Endo)cannabinoids

{kind=link}
{kind=link}
{kind=link}
| Compound | Model | Artery | Emax (%) (in Parentheses Concentrations in µM for Which Emax Was Obtained) | pEC50 | Suggested Mechanism of Action in Hypertension | Ref. | ||
|---|---|---|---|---|---|---|---|---|
| N | H | N | H | |||||
| AEA | WKY vs. SHR | perfused mesenteric bed | ~100 1 (10) | ~100 1 (10) | 7.1 | 6.3 * | ↓ NO-dependent relaxation; TRPV1-dependent | [96] |
| G3 mesenteric | 98 (3) | 70 * (10) | 6.5 | 6.8 * | sex-dependent (stronger in female); TRPV1- and endothelium-dependent | [97] | ||
| thoracic aorta | 13 (30) | 48 * (30) | 8.1 | 7.9 | endothelium-dependent; CB1R- and TRPV1-independent | [96] | ||
| L-NAME-induced | perfused mesenteric bed | 100 (10) | 107 (10) | 6.5 | 7.1 * | - | [98] | |
| ~90 1 (10) | ~90 1 (10) | 6.3 | 6.4 | ↑ sensory nerve-mediated activity | [96] | |||
| G3 mesenteric | ~70 1,2 (30) | ~70 1,2 (30) | 5.7 | 5.6 | - | [98] | ||
| thoracic aorta | 25 (30) | 33 (30) | 6.7 | 6.6 | CB1R-, TRPV1-, NO- and PG-independent | [96] | ||
| 2K1C | thoracic aorta | 4 (30) | 44 * (30) | - | 5.2 | CB1R-, CB2R-, NO- and endothelium-dependent | [100] | |
| hypoxia 3 | isolated perfused lung | - | ↑ pulmonary arterial tone (10) | - | - | FAAH-dependent metabolites;sex-dependent (stronger in females) | [102] | |
| large pulmonary | - | no effect (10) | - | no effect | - | |||
| MethAEA | DOCA-salt | G3 mesenteric | 84 (30) | 85 (30) | 4.9 | 5.6 * | TRPV1-dependent in N and H;CB1R-dependent in H only | [101] |
| aorta | 84 (30) | 41 * (30) | 6.1 | n.d. | - | |||
| SHR | G3 mesenteric | 97 (30) | 98 (30) | 6.1 | 5.6 * | CB1R-dependent in H only | [95] | |
| hypoxia 3 | isolated perfused lung | - | no effect (10) | - | - | - | [102] | |
| CBD | DOCA-salt | G3 mesenteric | 92 (30) | 91 (30) | 5.5 | 5.9 * | CB1R-, CB2R- and endothelium-dependent | [93] |
| SHR | 93 (30) | 82 (30) | 6.0 | 5.6 * | CB1R-dependent; CB2R- and endothelium-independent | |||
| Hypertension 4 | pulmonary | 94 (30) | 93 (30) | 4.9 | 4.1 * | endothelium, PG- and TRPV1-dependent;CB1R-, CB2R-independent | ||
| THC | L-NAME-induced | G3 mesenteric | ~60 1 (100) | ~70 1 (100) | 5.6 | 6.1 * | CB1R-independent; ↑ sensory nerve-mediated activity and PG-dependent | [99] |
| G0 mesenteric | 16 (100) | 38 * (100) | - | - | - | |||
| aorta | 5—constriction (100) | 4—relaxation (100) | - | - | - | |||
8. Acute In Vivo Cardiovascular Effects of (Endo)cannabinoids
9. Cardiovascular Effects of Chronic (Endo)cannabinoid Administration in Hypertension
| Compound, Dose, and Protocol | Model | BP and HR Effects | Influence on Changes Induced by Hypertension | References | |
|---|---|---|---|---|---|
| Cardiac Effects/Expression in Heart (If Not Stated Otherwise) | Vascular Effects | ||||
| PEA 30 mg/kg, s.c., once daily, 5 weeks | SHR | - ↓SBP (only in the 5th week of the treatment; by ~50–60 mmHg) - ↔HR | n.d. | vasodilatory effects in mesenteric or carotid arteries: - ↑EDHF-mediated relaxation to Ach; - ↑synthesis/release of vasodilatory EETs, NO, and PGI2 and/or ↓EETs degradation; - ↓RAAS activity (↓ACE and AT1R signaling pathway); anti-inflammatory effects: ↓NF-κB signaling pathway | [107,108] |
| AEA 3 mg/kg, i.v., once daily, 14 days | Dahl salt-sensitive + high salt (8%) diet | - consistent trend to ↑MBP at the 2nd week of the treatment (by ~20 mmHg) | n.d. | n.d. | [109] |
| nf-AEA 5 mg/kg, i.p., once weekly, 4 weeks | SHR | - ↓SBP after 4 weeks (by 35 mmHg) 1 | anti-hypertrophic effects: ↓ventricular mass and LV hypertrophy indexes | n.d. | [110,111] |
| URB597 1 mg/kg, i.p., twice daily, 14 days | DOCA-salt | - ↓SBP (after 2 weeks by ~30–60 mmHg) - ↔ HR | anti-hypertrophic effects: - ↓cardiac (only in younger) and LV hypertrophy - ↓medium and large coronary artery thickness in LV cardiac functional effects: - ↓diastolic stiffness - tendency to ↑cardiostimulatory effects of isoprenaline: contractility, cardiac work and inotropism - normalization of (-) inotropic effect of CB1R agonism anti-oxidant effects: ↓ROS, 4-HNE, CO gr., XO, NADPH oxidase activity and ↑GSH, GSH/GSSG, vit. C, ↑Nrf2, p21, ↓Keap1 pro-oxidant effects: ↓GSH-Px, GSSG-R, Cu-Zn-SOD, Trx-R activity and ↑MDA, 8-OHdG, ↓Trx, vit. A, ERK1/2, HO-1, MAPK pro-inflammatory effects: ↑TNFα endocannabinoid effects: - ↑FAAH in LV, ↓FAAH, MAGL activity - tendency to ↓CB1R and CB2R in LV but ↑CB1R and CB2R in whole heart - ↑TRPV1, GPR55, PPARα, ↓PPARγ - ↑NADA and 2-AG other effects: - ↑heart availability of energy substrates - ↑intramyocardial glycogen storage - ↓apoptosis (↓ Bax, caspase 3, 9) | vasodilatory effects: ↓response to phenylephrine in sMAs anti-hypertrophic effects: ↓medial thoracic aorta hypertrophy endocannabinoid effects: ↓FAAH in sMAs other effects: ↑KCa3.1 sMAs | [101,113,114,115,116,117,118,119,120] |
| URB597 1 mg/kg, i.p., twice daily, 14 days | SHR | - ↔SBP or slight ↓SBP (by ~20 mmHg after 2 weeks) and HR | hypertrophic effects: ↑heart hypertrophy but ↓LV hypertrophy cardiac functional effects: - ↑(+) chronotropic effect of isoprenaline - normalization of (+) inotropic effect of isoprenaline in atria anti-oxidant effects: ↓XO, ↑CAT, Trx-R activity, ↑GSH, GSH/GSSG, vit. E, C, Trx, ↓Keap1, Bach1, ↑ERK1/2, MAPK pro-oxidant effects: ↓GSH-Px activity and ↑MDA, 4-HNE, 8-isoprostanes, 8-OHdG, CO gr., ↓Nrf2, Keap1, HO-1 anti-inflammatory effects: ↓TNFα endocannabinoid effects: ↑CB1R, CB2R, GPR55, PPARγ and ↓TRPV1, PPARα - translocation of CB1R immunoreactivity to the intercalated discs in LV - tendency to ↑FAAH in LV - ↓FAAH and MAGL activity - ↑AEA, NADA, and 2-AG other effects: - ↑cardioprotective LV sphingolipid (S1P) - ↑palmitate uptake by LV cardiomyocytes - protection from DAG and CER accumulation in LV - improvement of insulin signaling in LV - ↓free AA - ↓apoptosis (↑Bcl-2, ↓Bax, caspase 3, 8, 9) | vasodilatory effects - ↓phenylephrine-mediated CB1R-independent vasoconstriction in sMAs - ↑potency of Ach-mediated endothelium-dependent vasorelaxation in sMAs and aorta - ↑potency of MethAEA-mediated CB1R-independent vasorelaxation vasoconstrictive effects: ↑vasoconstrictive potency of U46619 (thromboxane analog) in sMAs anti-hypertrophic effects: tendency to ↓sMAs wall hypertrophy endocannabinoid effects - ↑2-AG in aorta, ↑AEA in sMAs and aorta - ↓CB1R in aorta | [95,116,117,120,121,122] |
| JZL195 10 mg/kg, i.p., once daily, 14 days | SHR | - tendency to ↓BP (by ~20 mmHg after 2 weeks) - ↔HR | - no changes in cardiac hypertrophy | n.d. | [123] |
| rimonabant 20 mg, oral, once daily, 12 months | hypertension 2 | - ↓SBP by ~13 and 7 mmHg and DBP by ~6 and 2 mmHg in H. and N. patients, respectively | n.d. | n.d. | [124] |
| rimonabant 20 mg, oral, once daily, 12 months | hypertension 2 | - ↓SBP by ~3 and 0.5 mmHg and DBP by ~2 and 0.5 mmHg in H. and N. patients, respectively | - reductions more evident in patients with higher cardiometabolic risk (e.g., dyslipidemia and type 2 diabetes) - the hypotensive effect seems to be mediated by weight loss | [125] | |
| rimonabant 20 mg, oral, once daily, 24 months | hypertension 2 | - ↓SBP by ~1.5 and 0.5 mmHg and DBP by ~2 and 0.5 mmHg in H. and N. patients, respectively | - changes not significantly different from placebo | [126] | |
| rimonabant 10 mg/kg, oral, once daily, 3 weeks | (mRen2)27 higher RAAS activity | - ↓SBP (by ~25 mmHg within 24 h and remained lower through 3 weeks); ↔HR - better sympathetic and parasympathetic baroreflex sensitivity | n.d. | n.d. | [127] |
| LH-21 1 mg/kg, 3 mg/kg, i.p., 3 weeks | KKAγ mice (BP was ↑ by about 10 mmHg only) 3 | - normalization of SBP, DBP, MBP (only for 3 mg/kg) - ↔HR | n.d. | anti-inflammatory effects on aorta: - ↓ICAM-1, MCP-1, TNFα mRNA - ↓lipocalin-2 | [128] |
| JWH133 1 mmol/l, 10 µL, i.c.v., once daily, 4 weeks | SHR (conscious and anesthetized) | - ↓MBP and HR by ~35 mmHg and 70 beats/min respectively after 2 weeks of administration | n.d. | n.d. | [106] |
| O-1602 0.25 mg/kg, i.a., once daily, 14 days | SHR 3 | - ↓MBP by ~30 mmHg 1 - ↑HR by ~50 beats/min 1 | n.d. | other effects: ↓RhoA/Rho-kinase signaling in aorta | [87] |
| CBD 10 mg/kg, i.p., once daily, 14 days | DOCA-salt | - ↔HR, SBP, DBP, and MBP | anti-hypertrophic effects: ↓width of LV cardiomyocytes cardiac functional effects - ↓carbachol-induced vasoconstriction of coronary arteries - ↑(-) inotropic effect of CB1R agonism - ↑lusitropic effects: (+) isoprenaline and (-) carbachol anti-oxidant effects: ↓MDA, ↓GSSG, ↑GSH and small ↓4-HNE pro-oxidant effects: small ↓vit. A and E endocannabinoid effects: - ↓2-AG, OEA, DEA, DGLEA - ↓FAAH activity - small ↓CB1R, CB2R, and GPR18 other effects: - ↑FFA LA and ↓ FFA AA - ↓β1-adrenoceptor in LV | vasodilatory effects: - ↑Ach-induced endothelium-dependent vasorelaxation in aortas (NO-dependent) and sMAs - ↑eNOS in aortas and sMAs, ↑NOS3 in sMAs, ↑PGIS in sMAs anti-hypertrophic effects: ↓aorta and sMAs hypertrophy endocannabinoid effects: - ↓CB1R in sMAs but ↑Cnr1 in aortas - ↑Cnr2 in aortas and sMAs - ↑AEA, 2-AG, PEA, and DEA; tendency to ↑OEA, HEA, POEA, LEA, and 2-LG; ↓EPEA, DHEA, and NAGly in aorta other effects: - ↓vWF in aortas and sMAs - ↑KCNN4 in aortas and sMAs - ↑KCNN3 in sMAs | [94,129,130] |
| CBD 10 mg/kg, i.p., once daily, 14 days | SHR | - ↔HR, SBP, DBP, and MBP | anti-hypertrophic effects: ↓width of LV and RV myocytes and ↓RV hypertrophy cardiac functional effects - small ↓diastolic stiffness - ↓carbachol-induced vasoconstriction of coronary arteries - ↓(-) inotropic effect of CB1R agonism - ↑lusitropic effects: (+) isoprenaline and (-) carbachol anti-oxidant effects: ↓4-HHE and tendency to ↓4-HNE, ↑GSH, and ↓GSSG pro-oxidant effects: ↓vit. A and E endocannabinoid effects: - small ↓FAAH activity - ↓GPR55 and small ↓CB1R and GPR18 other effects: ↑FFA LA, FFA AA | vasodilatory effects: - ↑Ach-induced endothelium-dependent vasorelaxation in aortas and sMAs (COX dependent) - ↑eNOS in aortas and sMAs, ↑NOS3 in aortas and sMAs, ↑PGIS in sMAs vasoconstrictive effects: ↓potency of SNP-induced vasorelaxation in sMAs anti-hypertrophic effects: ↓aorta and sMAs hypertrophy pro-inflammatory effects: ↑COX-1 in aorta endocannabinoid effects: - ↑CB1R in sMAs and tendency to ↑Cnr1 in aortas and sMAs - ↑Cnr2 in aortas and sMAs - ↑TRPV1 in aortas - ↓AEA and small ↓2-AG, PEA, HEA, DEA, EPEA, DHEA, LEA, 2-LG, and NAGly in aortas other effects: - ↓vWF in aortas and sMAs - ↑KCNN4 in aortas and sMAs - ↑KCNN3 in sMAs | [94,129,130] |
| CBD 200 mg/kg, oral, 4 weeks | OLETF rats with metabolic syndrome | - ↔BP 1 | - loss of visceral adiposity was not associated with reduced BP | [131] | |
| Δ8-THC 3 mg/kg, i.p., once daily, 14 days | ARH unilaterally adrenalectomized +1% NaCl 3 | - ↓BP (by ~13 and 15 mmHg at the end of the 1st and 2nd week) | n.d. | n.d. | [132] |
| Δ9-THC 3 mg/kg, i.p., once daily, 7 or 14 days | - ↓BP (by ~18 and 13 mmHg at the end of the 1st and 2nd week); - tolerance to the acute hypotensive effect of the compound (in a shorter protocol) | n.d. | n.d. | ||
| Δ9-THC 1 mg/kg 2 mg/kg, s.c., once daily, 3–5 weeks | metacorticoid and renal hypertension | - ↔BP and HR | n.d. | n.d. | [133] |
| Δ9-THC 5–25 mg/kg (increasing dosing), oral, once daily, 5 or 10 days | SHR | - transient ↓BP after increasing the dose (tolerance developed) - ↓SBP after highest dose chronic treatment (with no tolerance effect) | n.d. | n.d. | [134,135] |
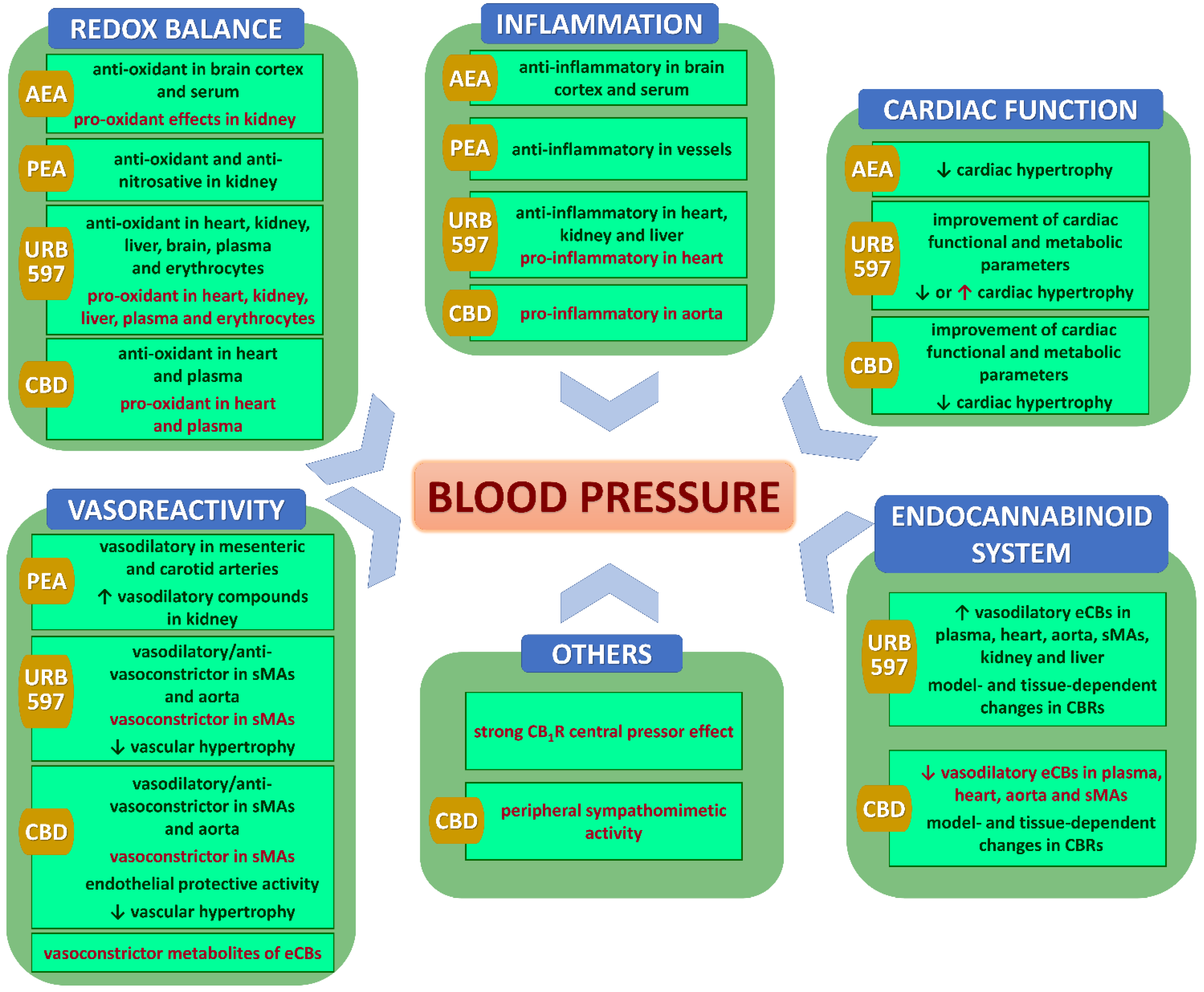
10. Potential Mechanisms of Cardiovascular Effects of Chronic (Endo)cannabinoid Administration in Hypertension
10.1. Vasodilatation
10.2. Cardiac Functional Antihypertensive Effects
10.3. Changes in Endocannabinoid System Components
10.4. Anti- and Pro-Oxidative Effects
10.5. Anti-Inflammatory Effects
10.6. Other Pro-Hypertensive Effects
11. Why Multitarget Vasodilatory (Endo)cannabinoids Are Not Effective as Antihypertensive Compounds
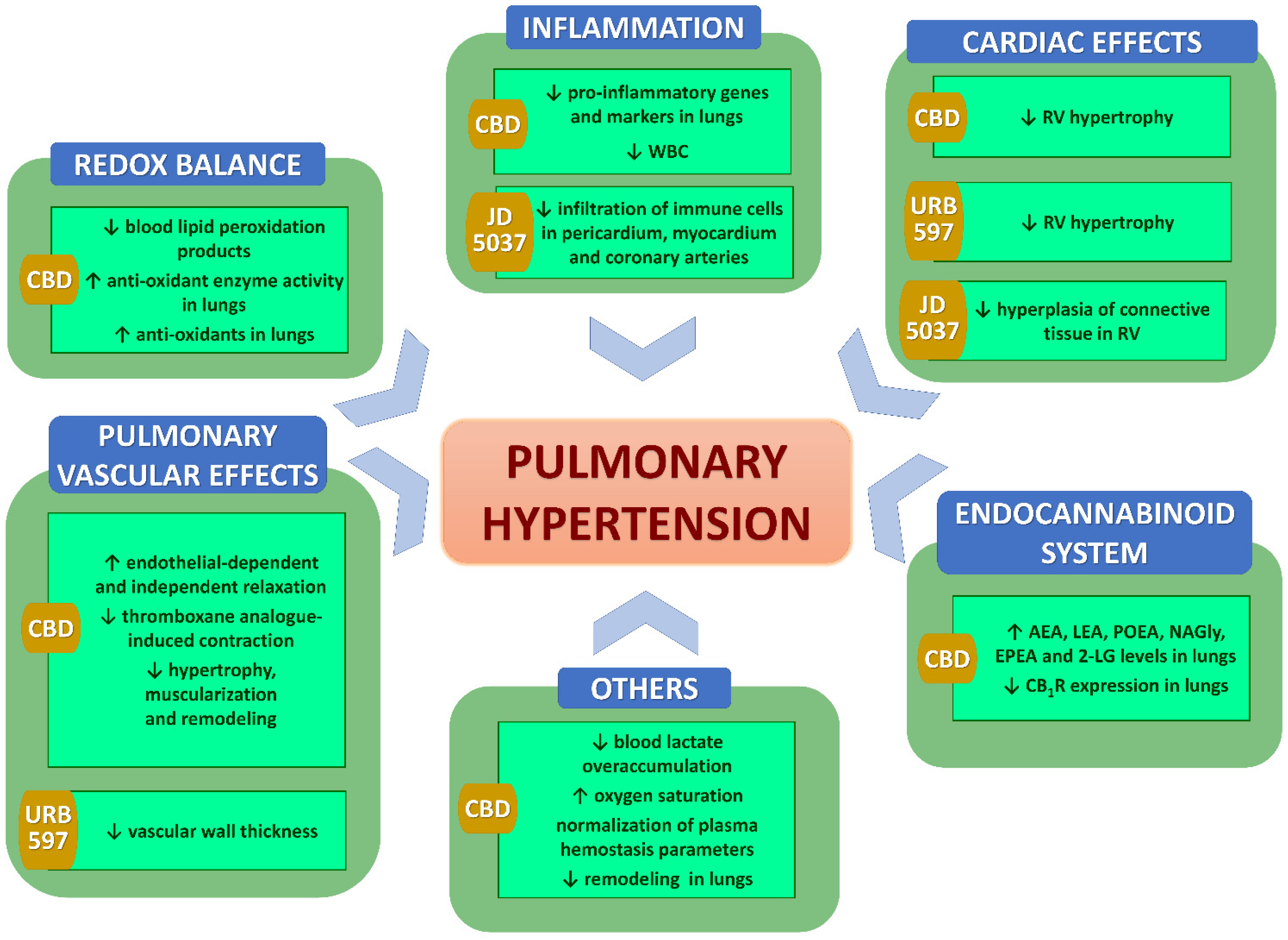
12. In Vivo Effects of Chronic (Endo)cannabinoids in PH
| Compound, Dose, and Protocol | Model | Effects | Ref. | ||
|---|---|---|---|---|---|
| CARDIOVASCULAR | |||||
| BP and HR Effects | Influence on Changes Induced by Hypertension | ||||
| Cardiac Effects/Expression in Heart (If Not Stated Otherwise) | Vascular Effects | ||||
| FAAH−/− in comparison to WT | hypoxia (mice) 1 | - no ↑RVSP | hypertrophic effects: no ↑Fulton index | hypertrophic effects: no ↑vascular wall thickness | [102] |
| URB597 5 mg/kg, i.p., once daily, 3 days or 3 weeks | - ↓RVSP (in longer procedure) (by ~5 mmHg) | anti-hypertrophic effects: ↓Fulton index (in longer procedure) | anti-hypertrophic effects: ↓vascular wall thickness (in longer procedure) | ||
| JD5037 3 mg/kg, oral, once daily, 3 weeks | MCT (rat) | - intensification of the metformin-induced ↓RVSP - ↔BP; ↔HR | anti-hypertrophic effects: ↓hyperplasia of connective tissue in myocardium anti-inflammatory effects: - ↓infiltration of immune cells in pericardium, myocardium, and coronary arteries other effects: - ↓vacuolization of tunica media of coronary arteries | - | [151] |
| CBD 10 mg/kg, 20 mg/kg, i.g., once daily, 14 days (treatment) or 3 weeks (preventive) | SuHx/ SuHx Cnr2-/- (mice) | - ↓RVSP (by ~10 mmHg) | anti-hypertrophic effects: ↓Fulton index | anti-hypertrophic effects: - ↓PA hypertrophy - ↓PA muscularization - ↓remodeling (PCNA+/nuclei) | [152] |
| CBD 10 mg/kg, i.p., once daily, 3 weeks (preventive) | MCT (rat) | - ↓RVSP (by ~15 mmHg) - ↔BP; ↔HR | anti-hypertrophic effects: small ↓Fulton index | vasodilatory effects in PA: - ↑endothelial-dependent (Ach) and endothelial-independent (SNP) relaxation - ↓thromboxane analog-induced contraction anti-hypertrophic effects in PA: - ↓hypertrophy - ↓muscularization - ↓remodeling (PCNA+/nuclei) | [152,153] |
| BLOOD | |||||
| CBD 10 mg/kg, i.g., once daily, 3 weeks (preventive) | SuHx (mice) | anti-oxidant effects: ↓blood MDA other effects: ↓blood lactate overaccumulation | [152] | ||
| CBD 10 mg/kg, i.p., once daily, 3 weeks (preventive) | MCT (rat) | anti-inflammatory effects: ↓WBC other effects: - ↑oxygen saturation - normalization of plasma hemostasis parameters (↓PAI-1 and t-PA levels) | [153] | ||
| LUNGS | |||||
| CBD 10 mg/kg, i.g., once daily, 3 weeks (preventive) | SuHx (mice) | anti-oxidant effects: ↑GSSG-R and GSH-Px activity anti-inflammatory effects: ↓Il6 and Tnfα other effects: ↓lactate accumulation (↓Pfkfb3) | [152] | ||
| CBD 10 mg/kg, i.p., once daily, 3 weeks (preventive) | MCT (rat) | anti-oxidant effects: ↑TAC, GSH, GSSG-R activity anti-inflammatory effects: ↓NFκB, TNFα, MCP-1, IL-1β, CD68 endocannabinoid effects: - ↑AEA, LEA, POEA, NAGLy, EPEA, and 2-LG; ↓CB1R other effects: ↓Gal-3 | [153,155,156] | ||
13. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Oparil, S.; Acelajado, M.C.; Bakris, G.L.; Berlowitz, D.R.; Cífková, R.; Dominiczak, A.F.; Grassi, G.; Jordan, J.; Poulter, N.R.; Rodgers, A.; et al. Hypertension. Nat. Rev. Dis. Primers 2018, 4, 18014. [Google Scholar] [CrossRef]
- Ott, C.; Schmieder, R.E. Diagnosis and treatment of arterial hypertension 2021. Kidney Int. 2022, 101, 36–46. [Google Scholar] [CrossRef]
- Rossier, B.C.; Bochud, M.; Devuyst, O. The hypertension pandemic: An evolutionary perspective. Physiology 2017, 32, 112–125. [Google Scholar] [CrossRef]
- Mills, K.T.; Stefanescu, A.; He, J. The global epidemiology of hypertension. Nat. Rev. Nephrol. 2020, 16, 223–237. [Google Scholar] [CrossRef]
- Jordan, J.; Kurschat, C.; Reuter, H. Arterial hypertension. Dtsch. Arztebl. Int. 2018, 115, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Seravalle, G.; Grassi, G. Sympathetic nervous system and hypertension: New evidences. Auton. Neurosci. 2022, 238, 102954. [Google Scholar] [CrossRef]
- Almeida, L.F.; Tofteng, S.S.; Madsen, K.; Jensen, B.L. Role of the renin-angiotensin system in kidney development and programming of adult blood pressure. Clin. Sci. 2020, 134, 641–656. [Google Scholar] [CrossRef]
- Harrison, D.G.; Coffman, T.M.; Wilcox, C.S. Pathophysiology of hypertension: The mosaic theory and beyond. Circ. Res. 2021, 128, 847–863. [Google Scholar] [CrossRef]
- Brant, L.C.C.; Passaglia, L.G.; Pinto-Filho, M.M.; de Castilho, F.M.; Ribeiro, A.L.P.; Nascimento, B.R. The burden of resistant hypertension across the world. Curr. Hypertens. Rep. 2022, 24, 55–66. [Google Scholar] [CrossRef]
- Stewart, S.; Strange, G.A.; Playford, D. The challenge of an expanded therapeutic window in pulmonary hypertension. Nat. Rev. Cardiol. 2020, 17, 195–197. [Google Scholar] [CrossRef]
- Kovacs, G.; Maron, B.A. The assessment of pulmonary arterial pressure and its clinical relevance: A 100-year journey from Europe, over the United States to Australia. Eur. Respir. J. 2022, 59, 2102064. [Google Scholar] [CrossRef]
- Fernandes, C.J.; Calderaro, D.; Assad, A.P.L.; Salibe-Filho, W.; Kato-Morinaga, L.T.; Hoette, S.; Piloto, B.; Castro, M.A.; Lisboa, R.P.; Silva, T.; et al. Update on the treatment of pulmonary arterial hypertension. Arq. Bras. Cardiol. 2021, 117, 750–764. [Google Scholar] [CrossRef]
- Beshay, S.; Sahay, S.; Humbert, M. Evaluation and management of pulmonary arterial hypertension. Respir. Med. 2020, 171, 106099. [Google Scholar] [CrossRef]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Ruopp, N.F.; Cockrill, B.A. Diagnosis and treatment of pulmonary arterial hypertension: A review. JAMA 2022, 327, 1379–1391. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.A.; Abman, S.H.; Elliott, C.G.; Frantz, R.P.; Hopper, R.K.; Horn, E.M.; Nicolls, M.R.; Shlobin, O.A.; Shah, S.J.; Kovacs, G.; et al. Pulmonary arterial hypertension: Diagnosis, treatment, and novel advances. Am. J. Respir. Crit. Care Med. 2021, 203, 1472–1487. [Google Scholar] [CrossRef]
- Mandras, S.A.; Mehta, H.S.; Vaidya, A. Pulmonary hypertension: A brief guide for clinicians. Mayo Clin. Proc. 2020, 95, 1978–1988. [Google Scholar] [CrossRef]
- Hassoun, P.M. Pulmonary arterial hypertension. N. Engl. J. Med. 2021, 385, 2361–2376. [Google Scholar] [CrossRef]
- Leber, L.; Beaudet, A.; Muller, A. Epidemiology of pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension: Identification of the most accurate estimates from a systematic literature review. Pulm. Circ. 2021, 11, 2045894020977300. [Google Scholar] [CrossRef]
- Hoeper, M.M.; Humbert, M. The new haemodynamic definition of pulmonary hypertension: Evidence prevails, finally! Eur. Respir. J. 2019, 53, 1900038. [Google Scholar] [CrossRef] [Green Version]
- Bisserier, M.; Pradhan, N.; Hadri, L. Current and emerging therapeutic approaches to pulmonary hypertension. Rev. Cardiovasc. Med. 2020, 21, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Kylhammar, D.; Kjellström, B.; Hjalmarsson, C.; Jansson, K.; Nisell, M.; Söderberg, S.; Wikström, G.; Rådegran, G. A comprehensive risk stratification at early follow-up determines prognosis in pulmonary arterial hypertension. Eur. Heart J. 2018, 39, 4175–4181. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Kramer, T.; Pan, Z.; Eichstaedt, C.A.; Spiesshoefer, J.; Benjamin, N.; Olsson, K.M.; Meyer, K.; Vizza, C.D.; Vonk-Noordegraaf, A.; et al. Mortality in pulmonary arterial hypertension: Prediction by the 2015 European pulmonary hypertension guidelines risk stratification model. Eur. Respir. J. 2017, 50, 1700740. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Chen, P.P.; Zhou, R.L.; Zhang, Y.; Tian, Z.; Zhang, S.Y. Pathological mechanisms and potential therapeutic targets of pulmonary arterial hypertension: A review. Aging Dis. 2020, 11, 1623–1639. [Google Scholar] [CrossRef]
- Hu, Y.; Chi, L.; Kuebler, W.M.; Goldenberg, N.M. Perivascular inflammation in pulmonary arterial hypertension. Cells 2020, 9, 2338. [Google Scholar] [CrossRef] [PubMed]
- Prisco, S.Z.; Thenappan, T.; Prins, K.W. Treatment targets for right ventricular dysfunction in pulmonary arterial hypertension. JACC Basic Transl. Sci. 2020, 5, 1244–1260. [Google Scholar] [CrossRef]
- Sommer, N.; Ghofrani, H.A.; Pak, O.; Bonnet, S.; Provencher, S.; Sitbon, O.; Rosenkranz, S.; Hoeper, M.M.; Kiely, D.G. Current and future treatments of pulmonary arterial hypertension. Br. J. Pharmacol. 2021, 178, 6–30. [Google Scholar] [CrossRef]
- Jama, H.A.; Muralitharan, R.R.; Xu, C.; O’Donnell, J.A.; Bertagnolli, M.; Broughton, B.R.S.; Head, G.A.; Marques, F.Z. Rodent models of hypertension. Br. J. Pharmacol. 2022, 179, 918–937. [Google Scholar] [CrossRef]
- Lerman, L.O.; Kurtz, T.W.; Touyz, R.M.; Ellison, D.H.; Chade, A.R.; Crowley, S.D.; Mattson, D.L.; Mullins, J.J.; Osborn, J.; Eirin, A.; et al. Animal models of hypertension: A scientific statement from the American Heart Association. Hypertension 2019, 73, e87–e120. [Google Scholar] [CrossRef]
- Moreno, K.G.T.; Marques, A.A.M.; da Silva, G.P.; Lourençone, B.R.; Fortini, C.S.; Leite, P.R.T.; Dos Santos, A.C.; Souza, R.I.C.; da Siva, L.I.; Gasparotto, A., Jr. A new approach for the development of multiple cardiovascular risk factors in two rat models of hypertension. Pharmaceuticals 2022, 15, 853. [Google Scholar] [CrossRef]
- Polak, A.; Harasim-Symbor, E.; Chabowski, A. Animal models of hypertension—Revisited. Prog. Health Sci. 2018, 8, 167–175. [Google Scholar] [CrossRef]
- Lin, H.Y.; Lee, Y.T.; Chan, Y.W.; Tse, G. Animal models for the study of primary and secondary hypertension in humans. Biomed. Rep. 2016, 5, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Jasińska-Stroschein, M. Toward better reproducibility in experimental research on new agents for pulmonary hypertension. An analysis of data from four hundred animal studies. Cardiovasc. Drugs Ther. 2021, 35, 707–718. [Google Scholar] [CrossRef]
- Dignam, J.P.; Scott, T.E.; Kemp-Harper, B.K.; Hobbs, A.J. Animal models of pulmonary hypertension: Getting to the heart of the problem. Br. J. Pharmacol. 2022, 179, 811–837. [Google Scholar] [CrossRef]
- Suparmi, S.; Wesseling, S.; Rietjens, I. Monocrotaline-induced liver toxicity in rat predicted by a combined in vitro physiologically based kinetic modeling approach. Arch. Toxicol. 2020, 94, 3281–3295. [Google Scholar] [CrossRef]
- Sztuka, K.; Jasińska-Stroschein, M. Animal models of pulmonary arterial hypertension: A systematic review and meta-analysis of data from 6126 animals. Pharmacol. Res. 2017, 125, 201–214. [Google Scholar] [CrossRef]
- Jasińska-Stroschein, M. A review of genetically-driven rodent models of pulmonary hypertension. Vascul. Pharmacol. 2022, 144, 106970. [Google Scholar] [CrossRef]
- Toczek, M.; Malinowska, B. Enhanced endocannabinoid tone as a potential target of pharmacotherapy. Life Sci. 2018, 204, 20–45. [Google Scholar] [CrossRef]
- Cinar, R.; Iyer, M.R.; Kunos, G. The therapeutic potential of second and third generation CB1R antagonists. Pharmacol. Ther. 2020, 208, 107477. [Google Scholar] [CrossRef]
- Pisanti, S.; Malfitano, A.M.; Ciaglia, E.; Lamberti, A.; Ranieri, R.; Cuomo, G.; Abate, M.; Faggiana, G.; Proto, M.C.; Fiore, D.; et al. Cannabidiol: State of the art and new challenges for therapeutic applications. Pharmacol. Ther. 2017, 175, 133–150. [Google Scholar] [CrossRef]
- Fowler, C.J. The endocannabinoid system—Current implications for drug development. J. Intern. Med. 2021, 290, 2–26. [Google Scholar] [CrossRef] [PubMed]
- Stasiulewicz, A.; Znajdek, K.; Grudzień, M.; Pawiński, T.; Sulkowska, A.J.I. A guide to targeting the endocannabinoid system in drug design. Int. J. Mol. Sci. 2020, 21, 2778. [Google Scholar] [CrossRef] [PubMed]
- Lowe, H.; Toyang, N.; Steele, B.; Bryant, J.; Ngwa, W. The endocannabinoid system: A potential target for the treatment of various diseases. Int. J. Mol. Sci. 2021, 22, 9472. [Google Scholar] [CrossRef] [PubMed]
- Fraguas-Sanchez, A.I.; Torres-Suarez, A.I. Medical use of cannabinoids. Drugs 2018, 78, 1665–1703. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Steffens, S.; Hasko, G.; Schindler, T.H.; Kunos, G. Cardiovascular effects of marijuana and synthetic cannabinoids: The good, the bad, and the ugly. Nat. Rev. Cardiol. 2018, 15, 151–166. [Google Scholar] [CrossRef]
- Malinowska, B.; Toczek, M.; Pędzińska-Betiuk, A.; Schlicker, E. Cannabinoids in arterial, pulmonary and portal hypertension—Mechanisms of action and potential therapeutic significance. Br. J. Pharmacol. 2019, 176, 1395–1411. [Google Scholar] [CrossRef]
- Garza-Cervantes, J.A.; Ramos-González, M.; Lozano, O.; Jerjes-Sánchez, C.; García-Rivas, G. Therapeutic applications of cannabinoids in cardiomyopathy and heart failure. Oxid. Med. Cell Longev. 2020, 2020, 4587024. [Google Scholar] [CrossRef]
- Krzyżewska, A.; Baranowska-Kuczko, M.; Mińczuk, K.; Kozłowska, H. Cannabinoids-a new perspective in adjuvant therapy for pulmonary hypertension. Int. J. Mol. Sci. 2021, 22, 10048. [Google Scholar] [CrossRef]
- Kicman, A.; Toczek, M. The effects of cannabidiol, a non-intoxicating compound of Cannabis, on the cardiovascular system in health and disease. Int. J. Mol. Sci. 2020, 21, 6740. [Google Scholar] [CrossRef]
- Rabino, M.; Mallia, S.; Castiglioni, E.; Rovina, D.; Pompilio, G.; Gowran, A. The endocannabinoid system and cannabidiol: Past, present, and prospective for cardiovascular diseases. Pharmaceuticals 2021, 14, 936. [Google Scholar] [CrossRef]
- O’Keefe, E.L.; Peterson, T.M.; Lavie, C.J. Reevaluating America’s latest pharmaceutical trend: The cardiovascular risk of Cannabis. Curr. Opin. Psychol. 2021, 38, 31–37. [Google Scholar] [CrossRef]
- Puhl, S.L. Cannabinoid-sensitive receptors in cardiac physiology and ischaemia. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118462. [Google Scholar] [CrossRef]
- Ghosh, M.; Naderi, S. Cannabis and cardiovascular disease. Curr. Atheroscler. Rep. 2019, 21, 21. [Google Scholar] [CrossRef]
- Sierra, S.; Luquin, N.; Navarro-Otano, J. The endocannabinoid system in cardiovascular function: Novel insights and clinical implications. Clin. Auton. Res. 2018, 28, 35–52. [Google Scholar] [CrossRef]
- Eid, B.G. Cannabinoids for treating cardiovascular disorders: Putting together a complex puzzle. J. Microsc. Ultrastruct. 2018, 6, 171–176. [Google Scholar] [CrossRef]
- Martín Giménez, V.M.; Noriega, S.E.; Kassuha, D.E.; Fuentes, L.B.; Manucha, W. Anandamide and endocannabinoid system: An attractive therapeutic approach for cardiovascular disease. Ther. Adv. Cardiovasc. Dis. 2018, 12, 177–190. [Google Scholar] [CrossRef]
- Dimmito, M.P.; Stefanucci, A.; Della Valle, A.; Scioli, G.; Cichelli, A.; Mollica, A. An overview on plants cannabinoids endorsed with cardiovascular effects. Biomed. Pharmacother. 2021, 142, 111963. [Google Scholar] [CrossRef]
- Stanley, C.; O’Sullivan, S.E. Vascular targets for cannabinoids: Animal and human studies. Br. J. Pharmacol. 2014, 171, 1361–1378. [Google Scholar] [CrossRef]
- Bondarenko, A.I. Cannabinoids and cardiovascular system. Adv. Exp. Med. Biol. 2019, 1162, 63–87. [Google Scholar] [CrossRef]
- Nwabuo, C.C.; Vasan, R.S. Pathophysiology of hypertensive heart disease: Beyond left ventricular hypertrophy. Curr. Hypertens. Rep. 2020, 22, 11. [Google Scholar] [CrossRef]
- Ambrosino, P.; Bachetti, T.; D’Anna, S.E.; Galloway, B.; Bianco, A.; D’Agnano, V.; Papa, A.; Motta, A.; Perrotta, F.; Maniscalco, M. Mechanisms and clinical implications of endothelial dysfunction in arterial hypertension. J. Cardiovasc. Dev. Dis. 2022, 9, 136. [Google Scholar] [CrossRef]
- Tanase, D.M.; Apostol, A.G.; Costea, C.F.; Tarniceriu, C.C.; Tudorancea, I.; Maranduca, M.A.; Floria, M.; Serban, I.L. Oxidative stress in arterial hypertension (HTN): The nuclear factor erythroid factor 2-related factor 2 (Nrf2) pathway, implications and future perspectives. Pharmaceutics 2022, 14, 534. [Google Scholar] [CrossRef] [PubMed]
- Hengel, F.E.; Benitah, J.P.; Wenzel, U.O. Mosaic theory revised: Inflammation and salt play central roles in arterial hypertension. Cell. Mol. Immunol. 2022, 19, 561–576. [Google Scholar] [CrossRef] [PubMed]
- Morales, P.; Hurst, D.P.; Reggio, P.H. Molecular targets of the phytocannabinoids: A complex picture. Prog. Chem. Org. Nat. Prod. 2017, 103, 103–131. [Google Scholar] [CrossRef] [PubMed]
- Im, D.S. GPR119 and GPR55 as receptors for fatty acid ethanolamides, oleoylethanolamide and palmitoylethanolamide. Int. J. Mol. Sci. 2021, 22, 1034. [Google Scholar] [CrossRef]
- Hillard, C.J. Circulating endocannabinoids: From whence do they come and where are they going? Neuropsychopharmacology 2018, 43, 155–172. [Google Scholar] [CrossRef]
- Kilaru, A.; Chapman, K.D. The endocannabinoid system. Essays Biochem. 2020, 64, 485–499. [Google Scholar] [CrossRef]
- Clayton, P.; Hill, M.; Bogoda, N.; Subah, S.; Venkatesh, R. Palmitoylethanolamide: A natural compound for health management. Int. J. Mol. Sci. 2021, 22, 5305. [Google Scholar] [CrossRef]
- Malinowska, B.; Baranowska-Kuczko, M.; Schlicker, E. Triphasic blood pressure responses to cannabinoids: Do we understand the mechanism? Br. J. Pharmacol. 2012, 165, 2073–2088. [Google Scholar] [CrossRef]
- Tang, X.; Liu, Z.; Li, X.; Wang, J.; Li, L. Cannabinoid receptors in myocardial injury: A brother born to rival. Int. J. Mol. Sci. 2021, 22, 6886. [Google Scholar] [CrossRef]
- Mińczuk, K.; Baranowska-Kuczko, M.; Krzyżewska, A.; Schlicker, E.; Malinowska, B. Cross-talk between the (endo)cannabinoid and renin-angiotensin systems: Basic evidence and potential therapeutic significance. Int. J. Mol. Sci. 2022, 23, 6350. [Google Scholar] [CrossRef] [PubMed]
- Han, J.H.; Kim, W. Peripheral CB1R as a modulator of metabolic inflammation. FASEB J. 2021, 35, e21232. [Google Scholar] [CrossRef] [PubMed]
- Cristino, L.; Bisogno, T.; Di Marzo, V. Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat. Rev. Neurol. 2020, 16, 9–29. [Google Scholar] [CrossRef]
- Pertwee, R.G.; Howlett, A.C.; Abood, M.E.; Alexander, S.P.; Di Marzo, V.; Elphick, M.R.; Greasley, P.J.; Hansen, H.S.; Kunos, G.; Mackie, K.; et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: Beyond CB1 and CB2. Pharmacol. Rev. 2010, 62, 588–631. [Google Scholar] [CrossRef]
- O’Sullivan, S.E. An update on PPAR activation by cannabinoids. Br. J. Pharmacol. 2016, 173, 1899–1910. [Google Scholar] [CrossRef] [PubMed]
- Latif, Z.; Garg, N. The impact of marijuana on the cardiovascular system: A review of the most common cardiovascular events associated with marijuana use. J. Clin. Med. 2020, 9, 1925. [Google Scholar] [CrossRef]
- Ramírez-Orozco, R.E.; García-Ruiz, R.; Morales, P.; Villalón, C.M.; Villafán-Bernal, J.R.; Marichal-Cancino, B.A. Potential metabolic and behavioural roles of the putative endocannabinoid receptors GPR18, GPR55 and GPR119 in feeding. Curr. Neuropharmacol. 2019, 17, 947–960. [Google Scholar] [CrossRef]
- Weresa, J.; Pędzińska-Betiuk, A.; Mińczuk, K.; Malinowska, B.; Schlicker, E. Why do marijuana and synthetic cannabimimetics induce acute myocardial infarction in healthy young people? Cells 2022, 11, 1142. [Google Scholar] [CrossRef]
- Alexander, S.P.; Christopoulos, A.; Davenport, A.P.; Kelly, E.; Mathie, A.; Peters, J.A.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; et al. The concise guide to pharmacology 2021/22: G protein-coupled receptors. Br. J. Pharmacol. 2021, 178 (Suppl. S1), S27–S156. [Google Scholar] [CrossRef]
- Di Marzo, V. New approaches and challenges to targeting the endocannabinoid system. Nat. Rev. Drug Discov. 2018, 17, 623–639. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Rosti, V.; Antognazza, M.R.; Lodola, F.; Moccia, F. Endothelial TRPV1 as an emerging molecular target to promote therapeutic angiogenesis. Cells 2020, 9, 1341. [Google Scholar] [CrossRef] [PubMed]
- Randhawa, P.K.; Jaggi, A.S. TRPV1 channels in cardio.ovascular system: A double edged sword? Int. J. Cardiol. 2017, 228, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Ye, L.; Zhang, Q.; Wu, F.; Wang, L. The role of TRPV1 channels in atherosclerosis. Channels 2020, 14, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Morales, P.; Lago-Fernandez, A.; Hurst, D.P.; Sotudeh, N.; Brailoiu, E.; Reggio, P.H.; Abood, M.E.; Jagerovic, N. Therapeutic exploitation of GPR18: Beyond the cannabinoids? J. Med. Chem. 2020, 63, 14216–14227. [Google Scholar] [CrossRef] [PubMed]
- Matouk, A.I.; Taye, A.; El-Moselhy, M.A.; Heeba, G.H.; Abdel-Rahman, A.A. The effect of chronic activation of the novel endocannabinoid receptor GPR18 on myocardial function and blood pressure in conscious rats. J. Cardiovasc. Pharmacol. 2017, 69, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Kotańska, M.; Kubacka, M.; Bednarski, M.; Nicosia, N.; Szafarz, M.; Jawień, W.; Müller, C.E.; Kieć-Kononowicz, K. The GPR18 agonist PSB-KD-107 exerts endothelium-dependent vasorelaxant effects. Pharmaceuticals 2021, 14, 799. [Google Scholar] [CrossRef]
- Wróbel, A.; Serefko, A.; Szopa, A.; Poleszak, E. Stimulation of atypical cannabinoid receptor GPR55 abolishes the symptoms of detrusor overactivity in spontaneously hypertensive rats. Eur. J. Pharm. Sci. 2020, 150, 105329. [Google Scholar] [CrossRef]
- Apweiler, M.; Saliba, S.W.; Streyczek, J.; Hurrle, T.; Gräßle, S.; Bräse, S.; Fiebich, B.L. Targeting oxidative stress: Novel coumarin-based inverse agonists of GPR55. Int. J. Mol. Sci. 2021, 22, 11665. [Google Scholar] [CrossRef]
- Botta, M.; Audano, M.; Sahebkar, A.; Sirtori, C.R.; Mitro, N.; Ruscica, M. PPAR agonists and metabolic syndrome: An established role? Int. J. Mol. Sci. 2018, 19, 1197. [Google Scholar] [CrossRef] [PubMed]
- Ertuglu, L.A.; Elijovich, F.; Laffer, C.L.; Kirabo, A. Salt-sensitivity of blood pressure and insulin resistance. Front. Physiol. 2021, 12, 793924. [Google Scholar] [CrossRef]
- Fang, S.; Livergood, M.C.; Nakagawa, P.; Wu, J.; Sigmund, C.D. Role of the peroxisome proliferator activated receptors in hypertension. Circ. Res. 2021, 128, 1021–1039. [Google Scholar] [CrossRef] [PubMed]
- Iannotti, F.A.; Vitale, R.M. The endocannabinoid system and PPARs: Focus on their signalling crosstalk, action and transcriptional regulation. Cells 2021, 10, 586. [Google Scholar] [CrossRef] [PubMed]
- Baranowska-Kuczko, M.; Kozłowska, H.; Kloza, M.; Sadowska, O.; Kozłowski, M.; Kusaczuk, M.; Kasacka, I.; Malinowska, B. Vasodilatory effects of cannabidiol in human pulmonary and rat small mesenteric arteries: Modification by hypertension and the potential pharmacological opportunities. J. Hypertens. 2020, 38, 896–911. [Google Scholar] [CrossRef] [PubMed]
- Baranowska-Kuczko, M.; Kozłowska, H.; Kloza, M.; Kusaczuk, M.; Harasim-Symbor, E.; Biernacki, M.; Kasacka, I.; Malinowska, B. Vasoprotective endothelial effects of chronic cannabidiol treatment and its influence on the endocannabinoid system in rats with primary and secondary hypertension. Pharmaceuticals 2021, 14, 1120. [Google Scholar] [CrossRef]
- Baranowska-Kuczko, M.; Kozłowska, H.; Kloza, M.; Harasim-Symbor, E.; Biernacki, M.; Kasacka, I.; Malinowska, B. Beneficial changes in rat vascular endocannabinoid system in primary hypertension and under treatment with chronic inhibition of fatty acid amide hydrolase by URB597. Int. J. Mol. Sci. 2021, 22, 4833. [Google Scholar] [CrossRef]
- Wheal, A.J.; Randall, M.D. Effects of hypertension on vasorelaxation to endocannabinoids in vitro. Eur. J. Pharmacol. 2009, 603, 79–85. [Google Scholar] [CrossRef]
- Ho, W.S. Modulation by 17β-estradiol of anandamide vasorelaxation in normotensive and hypertensive rats: A role for TRPV1 but not fatty acid amide hydrolase. Eur. J. Pharmacol. 2013, 701, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Wheal, A.J.; Bennett, T.; Randall, M.D.; Gardiner, S.M. Effects of chronic nitric oxide synthase inhibition on the cardiovascular responses to cannabinoids in vivo and in vitro. Br. J. Pharmacol. 2007, 150, 662–671. [Google Scholar] [CrossRef]
- O’Sullivan, S.E.; Randall, M.D.; Gardiner, S.M. The in vitro and in vivo cardiovascular effects of Δ9-tetrahydrocannabinol in rats made hypertensive by chronic inhibition of nitric-oxide synthase. J. Pharmacol. Exp. Ther. 2007, 321, 663–672. [Google Scholar] [CrossRef]
- Guo, Z.; Liu, Y.X.; Yuan, F.; Ma, H.J.; Maslov, L.; Zhang, Y. Enhanced vasorelaxation effect of endogenous anandamide on thoracic aorta in renal vascular hypertension rats. Clin. Exp. Pharmacol. Physiol. 2015, 42, 950–955. [Google Scholar] [CrossRef]
- Baranowska-Kuczko, M.; Kozłowska, H.; Kloza, M.; Karpińska, O.; Toczek, M.; Harasim, E.; Kasacka, I.; Malinowska, B. Protective role of cannabinoid CB1 receptors and vascular. ef.ffects of chronic administration of FAAH inhibitor URB597 in DOCA-salt hypertensive rats. Life Sci. 2016, 151, 288–299. [Google Scholar] [CrossRef]
- Wenzel, D.; Matthey, M.; Bindila, L.; Lerner, R.; Lutz, B.; Zimmer, A.; Fleischmann, B.K. Endocannabinoid anandamide mediates hypoxic pulmonary vasoconstriction. Proc. Natl. Acad. Sci. USA 2013, 110, 18710–18715. [Google Scholar] [CrossRef]
- Carnevale, L.N.; Das, A. Novel anti-inflammatory and vasodilatory omega-3 endocannabinoid epoxide regioisomers. Adv. Exp. Med. Biol. 2019, 1161, 219–232. [Google Scholar] [CrossRef]
- Ho, W.S.; Barrett, D.A.; Randall, M.D. ‘Entourage’ effects of N-palmitoylethanolamide and N-oleoylethanolamide on vasorelaxation to anandamide occur through TRPV1 receptors. Br. J. Pharmacol. 2008, 155, 837–846. [Google Scholar] [CrossRef]
- Stanke-Labesque, F.; Mallaret, M.; Lefebvre, B.; Hardy, G.; Caron, F.; Bessard, G. 2-Arachidonoyl glycerol induces contraction of isolated rat aorta: Role of cyclooxygenase-derived products. Cardiovasc. Res. 2004, 63, 155–160. [Google Scholar] [CrossRef]
- Shi, H.K.; Guo, H.C.; Liu, H.Y.; Zhang, Z.L.; Hu, M.Y.; Zhang, Y.; Li, Q. Cannabinoid type 2 receptor agonist JWH133 decreases blood pressure of spontaneously hypertensive rats through relieving inflammation in the rostral ventrolateral medulla of the brain. J. Hypertens. 2020, 38, 886–895. [Google Scholar] [CrossRef]
- Raso, G.M.; Simeoli, R.; Russo, R.; Santoro, A.; Pirozzi, C.; d’Emmanuele di Villa Bianca, R.; Mitidieri, E.; Paciello, O.; Pagano, T.B.; Orefice, N.S.; et al. N-Palmitoylethanolamide protects the kidney from hypertensive injury in spontaneously hypertensive rats via inhibition of oxidative stress. Pharmacol. Res. 2013, 76, 67–76. [Google Scholar] [CrossRef]
- Mattace Raso, G.; Pirozzi, C.; d’Emmanuele di Villa Bianca, R.; Simeoli, R.; Santoro, A.; Lama, A.; Di Guida, F.; Russo, R.; De Caro, C.; Sorrentino, R.; et al. Palmitoylethanolamide treatment reduces blood pressure in spontaneously hypertensive rats: Involvement of cytochrome p450-derived eicosanoids and renin angiotensin system. PLoS ONE 2015, 10, e0123602. [Google Scholar] [CrossRef]
- Golosova, D.; Levchenko, V.; Kravtsova, O.; Palygin, O.; Staruschenko, A. Acute and long-term effects of cannabinoids on hypertension and kidney injury. Sci. Rep. 2022, 12, 6080. [Google Scholar] [CrossRef]
- Martín Giménez, V.M.; Díaz-Rodríguez, P.; Sanz, R.L.; Vivero-Lopez, M.; Concheiro, A.; Diez, E.; Prado, N.; Enrique Kassuha, D.; Alvarez-Lorenzo, C.; Manucha, W. Anandamide-nanoformulation obtained by electrospraying for cardiovascular therapy. Int. J. Pharm. 2019, 566, 1–10. [Google Scholar] [CrossRef]
- Martín Giménez, V.M.; Mocayar Marón, F.J.; García, S.; Mazzei, L.; Guevara, M.; Yunes, R.; Manucha, W. Central nervous system, peripheral and hemodynamic effects of nanoformulated anandamide in hypertension. Adv. Med. Sci. 2021, 66, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Rajesh, M.; Batkai, S.; Patel, V.; Kashiwaya, Y.; Liaudet, L.; Evgenov, O.V.; Mackie, K.; Hasko, G.; Pacher, P. CB1 cannabinoid receptors promote oxidative stress and cell death in murine models of doxorubicin-induced cardiomyopathy and in human cardiomyocytes. Cardiovasc. Res. 2010, 85, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Biernacki, M.; Łuczaj, W.; Gęgotek, A.; Toczek, M.; Bielawska, K.; Skrzydlewska, E. Crosstalk between liver antioxidant and the endocannabinoid systems after chronic administration of the FAAH inhibitor, URB597, to hypertensive rats. Toxicol. Appl. Pharmacol. 2016, 301, 31–41. [Google Scholar] [CrossRef]
- Kloza, M.; Baranowska-Kuczko, M.; Malinowska, B.; Karpińska, O.; Harasim-Symbor, E.; Kasacka, I.; Kozłowska, H. The influence of DOCA-salt hypertension and chronic administration of the FAAH inhibitor URB597 on KCa2.3/KCa3.1-EDH-type relaxation in rat small mesenteric arteries. Vascul. Pharmacol. 2017, 99, 65–73. [Google Scholar] [CrossRef]
- Polak, A.; Harasim-Symbor, E.; Malinowska, B.; Kasacka, I.; Pędzińska-Betiuk, A.; Weresa, J.; Chabowski, A. The effects of chronic FAAH inhibition on myocardial lipid metabolism in normotensive and DOCA-salt hypertensive rats. Life Sci. 2017, 183, 1–10. [Google Scholar] [CrossRef]
- Pędzińska-Betiuk, A.; Weresa, J.; Toczek, M.; Baranowska-Kuczko, M.; Kasacka, I.; Harasim-Symbor, E.; Malinowska, B. Chronic inhibition of fatty acid amide hydrolase by URB597 produces differential effects on cardiac performance in normotensive and hypertensive rats. Br. J. Pharmacol. 2017, 174, 2114–2129. [Google Scholar] [CrossRef]
- Biernacki, M.; Malinowska, B.; Timoszuk, M.; Toczek, M.; Jastrząb, A.; Remiszewski, P.; Skrzydlewska, E. Hypertension and chronic inhibition of endocannabinoid degradation modify the endocannabinoid system and redox balance in rat heart and plasma. Prostaglandins Other Lipid. Mediat. 2018, 138, 54–63. [Google Scholar] [CrossRef]
- Polak, A.; Harasim-Symbor, E.; Malinowska, B.; Kasacka, I.; Lewandowska, A.; Chabowski, A. The endocannabinoid system affects myocardial glucose metabolism in the DOCA-salt model of hypertension. Cell Physiol. Biochem. 2018, 46, 727–739. [Google Scholar] [CrossRef]
- Toczek, M.; Baranowska-Kuczko, M.; Grzęda, E.; Pędzińska-Betiuk, A.; Weresa, J.; Malinowska, B. Age-specific influences of chronic administration of the fatty acid amide hydrolase inhibitor URB597 on cardiovascular parameters and organ hypertrophy in DOCA-salt hypertensive rats. Pharmacol. Rep. 2016, 68, 363–369. [Google Scholar] [CrossRef]
- Biernacki, M.; Łuczaj, W.; Jarocka-Karpowicz, I.; Ambrożewicz, E.; Toczek, M.; Skrzydlewska, E. The effect of long-term administration of fatty acid amide hydrolase inhibitor URB597 on oxidative metabolism in the heart of rats with primary and secondary hypertension. Molecules 2018, 23, 2350. [Google Scholar] [CrossRef] [Green Version]
- Harasim-Symbor, E.; Polak, A.; Pedzinska-Betiuk, A.; Weresa, J.; Malinowska, B.; Lewandowska, A.; Kasacka, I.; Chabowski, A. Fatty acid amide hydrolase inhibitor (URB597) as a regulator of myocardial lipid metabolism in spontaneously hypertensive rats. Chem. Phys. Lipids 2019, 218, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Harasim-Symbor, E.; Polak-Iwaniuk, A.; Konstantynowicz-Nowicka, K.; Bielawiec, P.; Malinowska, B.; Kasacka, I.; Chabowski, A. Experimental activation of endocannabinoid system reveals antilipotoxic effects on cardiac myocytes. Molecules 2020, 25, 1932. [Google Scholar] [CrossRef] [PubMed]
- Toczek, M.; Kicman, A.; Malinowska, B. The effects of enhanced endocannabinoid tone induced by chronic administration of dual FAAH/MAGL inhibitor JZL195 in spontanously hypertensive rats. In Proceedings of the 28th Congress of the Polish Physiological Society, Gdańsk, Poland, 15–17 September 2021; p. 87. [Google Scholar]
- Després, J.P.; Golay, A.; Sjöström, L.; Rimonabant in Obesity-Lipids Study, G. Effects of rimonabant on metabolic risk factors in overweight patients with dyslipidemia. N. Engl. J. Med. 2005, 353, 2121–2134. [Google Scholar] [CrossRef] [PubMed]
- Ruilope, L.M.; Després, J.P.; Scheen, A.; Pi-Sunyer, X.; Mancia, G.; Zanchetti, A.; Van Gaal, L. Effect of rimonabant on blood pressure in overweight/obese patients with/without co-morbidities: Analysis of pooled RIO study results. J. Hypertens. 2008, 26, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Van Gaal, L.F.; Scheen, A.J.; Rissanen, A.M.; Rössner, S.; Hanotin, C.; Ziegler, O.; Group, R.I.-E.S. Long-term effect of CB1 blockade with rimonabant on cardiometabolic risk factors: Two year results from the RIO-Europe Study. Eur. Heart J. 2008, 29, 1761–1771. [Google Scholar] [CrossRef]
- Schaich, C.L.; Shaltout, H.A.; Brosnihan, K.B.; Howlett, A.C.; Diz, D.I. Acute and chronic systemic CB1 cannabinoid receptor blockade improves blood pressure regulation and metabolic profile in hypertensive (mRen2)27 rats. Physiol. Rep. 2014, 2, e12108. [Google Scholar] [CrossRef]
- Dong, Z.; Gong, H.; Chen, Y.; Wu, H.; Wu, J.; Deng, Y.; Song, X. LH-21, a peripheral cannabinoid receptor 1 antagonist, exerts favorable metabolic modulation including antihypertensive effect in KKAy mice by regulating inflammatory cytokines and adipokines on adipose tissue. Front. Endocrinol. 2018, 9, 167. [Google Scholar] [CrossRef]
- Remiszewski, P.; Jarocka-Karpowicz, I.; Biernacki, M.; Jastrząb, A.; Schlicker, E.; Toczek, M.; Harasim-Symbor, E.; Pędzińska-Betiuk, A.; Malinowska, B. Chronic cannabidiol administration fails to diminish blood pressure in rats with primary and secondary hypertension despite its effects on cardiac and plasma endocannabinoid system, oxidative stress and lipid metabolism. Int. J. Mol. Sci. 2020, 21, 1295. [Google Scholar] [CrossRef]
- Pędzińska-Betiuk, A.; Weresa, J.; Schlicker, E.; Harasim-Symbor, E.; Toczek, M.; Kasacka, I.; Gajo, B.; Malinowska, B. Chronic cannabidiol treatment reduces the carbachol-induced coronary constriction and left ventricular cardiomyocyte width of the isolated hypertensive rat heart. Toxicol. Appl. Pharmacol. 2021, 411, 115368. [Google Scholar] [CrossRef]
- Wilson, J.N.; Mendez, D.; Mascal, M.; Fitzgerald, R.; Ortiz, R.M. Synthetic cannabidiol reduced body mass and visceral adiposity but not blood pressure in rats with advanced metabolic syndrome. In Proceedings of the Experimental Biology, Philadelphia, PA, USA, 2–5 April 2022; p. R2910. [Google Scholar]
- Birmingham, M.K.; Bartova, A. Effects of cannabinol derivatives on blood pressure, body weight, pituitary-adrenal function, and mitochondrial respiration in the rat. In Marihuana: Chemistry, Biochemistry, and Cellular Effects; Nahas, G.G., Paton, W.D.M., Idänpään-Heikkilä, J.E., Eds.; Springer: Berlin/Heidelberg, Germany, 1976; pp. 425–438. [Google Scholar]
- Varma, D.R.; Goldbaum, D. Effect of Δ9-tetrahydrocannabinol on experimental hypertension in rats. J. Pharm. Pharmacol. 1975, 27, 790–791. [Google Scholar] [CrossRef]
- Nahas, G.G.; Schwartz, I.W.; Adamec, J.; Manger, W.M. Tolerance to delta-9-tetrahydrocannabinol in the spontaneously hypertensive rat. Proc. Soc. Exp. Biol. Med. 1973, 142, 58–60. [Google Scholar] [CrossRef] [PubMed]
- Kosersky, D.S. Antihypertensive effects of Δ9-tetrahydrocannabinol. Arch. Int. Pharmacodyn. Ther. 1978, 233, 76–81. [Google Scholar] [PubMed]
- Bátkai, S.; Pacher, P.; Osei-Hyiaman, D.; Radaeva, S.; Liu, J.; Harvey-White, J.; Offertáler, L.; Mackie, K.; Rudd, M.A.; Bukoski, R.D.; et al. Endocannabinoids acting at cannabinoid-1 receptors regulate cardiovascular function in hypertension. Circulation 2004, 110, 1996–2002. [Google Scholar] [CrossRef]
- Spindle, T.R.; Bonn-Miller, M.O.; Vandrey, R. Changing landscape of cannabis: Novel products, formulations, and methods of administration. Curr. Opin. Psychol. 2019, 30, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Stella, B.; Baratta, F.; Della Pepa, C.; Arpicco, S.; Gastaldi, D.; Dosio, F. Cannabinoid formulations and delivery systems: Current and future options to treat pain. Drugs 2021, 81, 1513–1557. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.T.; Chandy, M.; Nishiga, M.; Zhang, A.; Kumar, K.K.; Thomas, D.; Manhas, A.; Rhee, S.; Justesen, J.M.; Chen, I.Y.; et al. Cannabinoid receptor 1 antagonist genistein attenuates marijuana-induced vascular inflammation. Cell 2022, 185, 1676–1693 e1623. [Google Scholar] [CrossRef]
- Biernacki, M.; Baranowska-Kuczko, M.; Niklinska, G.N.; Skrzydlewska, E. The FAAH inhibitor URB597 modulates lipid mediators in the brain of rats with spontaneous hypertension. Biomolecules 2020, 10, 1022. [Google Scholar] [CrossRef] [PubMed]
- Dobrzyńska, I.; Szachowicz-Petelska, B.; Pędzińska-Betiuk, A.; Figaszewski, Z.A.; Skrzydlewska, E. Effects of hypertension and FAAH inhibitor treatment of rats with primary and secondary hypertension considering the physicochemical properties of erythrocytes. Toxicol. Mech. Methods 2020, 30, 297–305. [Google Scholar] [CrossRef]
- Biernacki, M.; Ambrozewicz, E.; Gegotek, A.; Toczek, M.; Bielawska, K.; Skrzydlewska, E. Redox system and phospholipid metabolism in the kidney of hypertensive rats after FAAH inhibitor URB597 administration. Redox. Biol. 2018, 15, 41–50. [Google Scholar] [CrossRef]
- Dobrzynska, I.; Szachowicz-Petelska, B.; Weresa, J.; Figaszewski, Z.A.; Skrzydlewska, E. Changes in physicochemical properties of kidney cells membrane as a consequence of hypertension and treatment of hypertensive rats with FAAH inhibitor. Chem. Biol. Interact. 2019, 299, 52–58. [Google Scholar] [CrossRef]
- Biernacki, M.; Ambrozewicz, E.; Gegotek, A.; Toczek, M.; Skrzydlewska, E. Long-term administration of fatty acid amide hydrolase inhibitor (URB597) to rats with spontaneous hypertension disturbs liver redox balance and phospholipid metabolism. Adv. Med. Sci. 2019, 64, 15–23. [Google Scholar] [CrossRef]
- Atalay, S.; Jarocka-Karpowicz, I.; Skrzydlewska, E. Antioxidative and anti-inflammatory properties of cannabidiol. Antioxidants 2019, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Kossakowski, R.; Schlicker, E.; Toczek, M.; Weresa, J.; Malinowska, B. Cannabidiol affects the Bezold-Jarisch reflex via TRPV1 and 5-HT3 receptors and has peripheral sympathomimetic effects in spontaneously hypertensive and normotensive rats. Front. Pharmacol. 2019, 10, 500. [Google Scholar] [CrossRef] [PubMed]
- Schloss, M.J.; Horckmans, M.; Guillamat-Prats, R.; Hering, D.; Lauer, E.; Lenglet, S.; Weber, C.; Thomas, A.; Steffens, S. 2-Arachidonoylglycerol mobilizes myeloid cells and worsens heart function after acute myocardial infarction. Cardiovasc. Res. 2019, 115, 602–613. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zheng, L.; Chen, G. 2-Arachidonoylglycerol attenuates myocardial fibrosis in diabetic mice via the TGF-β1/Smad pathway. Cardiovasc. Drugs Ther. 2022. [Google Scholar] [CrossRef]
- Wróbel, A.; Szopa, A.; Serefko, A.; Poleszak, E. A novel alternative in the treatment of detrusor overactivity? In vivo activity of O-1602, the newly synthesized agonist of GPR55 and GPR18 cannabinoid receptors. Molecules 2020, 25, 1384. [Google Scholar] [CrossRef]
- Austrich-Olivares, A.; García-Gutiérrez, M.S.; Illescas, L.; Gasparyan, A.; Manzanares, J. Cannabinoid CB1 receptor involvement in the actions of CBD on anxiety and coping behaviors in mice. Pharmaceuticals 2022, 15, 473. [Google Scholar] [CrossRef]
- Remiszewski, P.; Pędzińska-Betiuk, A.; Mińczuk, K.; Schlicker, E.; Klimek, J.; Dzięcioł, J.; Malinowska, B. Effects of the peripheral CB1 receptor antagonist JD5037 in mono- and polytherapy with the AMPK activator metformin in a monocrotaline-induced rat model of pulmonary hypertension. Front. Pharmacol. 2022, 13, 965613. [Google Scholar] [CrossRef]
- Lu, X.; Zhang, J.; Liu, H.; Ma, W.; Yu, L.; Tan, X.; Wang, S.; Ren, F.; Li, X.; Li, X. Cannabidiol attenuates pulmonary arterial hypertension by improving vascular smooth muscle cells mitochondrial function. Theranostics 2021, 11, 5267–5278. [Google Scholar] [CrossRef]
- Sadowska, O.; Baranowska-Kuczko, M.; Gromotowicz-Popławska, A.; Biernacki, M.; Kicman, A.; Malinowska, B.; Kasacka, I.; Krzyżewska, A.; Kozłowska, H. Cannabidiol ameliorates monocrotaline-induced pulmonary hypertension in rats. Int. J. Mol. Sci. 2020, 21, 7077. [Google Scholar] [CrossRef]
- Abdallah, S.J.; Smith, B.M.; Ware, M.A.; Moore, M.; Li, P.Z.; Bourbeau, J.; Jensen, D. Effect of vaporized Cannabis on exertional breathlessness and exercise endurance in advanced chronic bbstructive pulmonary disease. A randomized controlled trial. Ann. Am. Thorac. Soc. 2018, 15, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Krzyzewska, A.; Baranowska-Kuczko, M.; Kasacka, I.; Kozlowska, H. Evaluation of the anti-inflammatory and anti-proliferative properties of cannabidiol in an experimental model of monocrotaline-induced pulmonary hypertension. In Proceedings of the 28th Congress of the Polish Physiological Society, Gdańsk, Poland, 15–17 September 2021; p. 81. [Google Scholar]
- Krzyżewska, A.; Baranowska-Kuczko, M.; Jastrząb, A.; Kasacka, I.; Kozłowska, H. Cannabidiol improves antioxidant capacity and reduces inflammation in the lungs of rats with monocrotaline-induced pulmonary hypertension. Molecules 2022, 27, 3327. [Google Scholar] [CrossRef] [PubMed]
| Type of Hypertension | Model | Main Characteristics | |
|---|---|---|---|
| Systemic | Primary | SHR |
|
| Dahl salt-sensitive rat |
| ||
| TGR(mRen2)27 |
| ||
| Secondary | Ang-II |
| |
| L-NAME |
| ||
| DOCA-salt |
| ||
| ARH |
| ||
| metacorticoid hypertension |
| ||
| renal hypertension (2K1C) |
| ||
| Pulmonary | MCT |
| |
| hypoxia |
| ||
| sugen/hypoxia |
| ||
| Compound, Dose, and Protocol | Model | Effects | References |
|---|---|---|---|
| CENTRAL NERVOUS SYSTEM | |||
| nf-AEA 5 mg/kg, i.p., once weekly, 4 weeks | SHR | anti-inflammatory/-oxidant effects: ↓WT-1, AT1R, iNOS, and ↑Hsp70 in brain cortex other effects: ↓apoptosis (TUNEL and caspase-3) in brain cortex | [111] |
| URB597 1 mg/kg, i.p., twice daily, 14 days | SHR | anti-oxidant effects in brain: - ↑Cu-Zn-SOD, GSH-Px, GSSG-R activity, ↓MDA, ↑vit. E - ↑Nrf2 and HO-1 and ↓Bach1 endocannabinoid effects in brain: - ↓FAAH activity and ↑AEA - ↓CB2R and ↑GPR55 other effects: ↓phospholipid but ↑free AA, DHA, and LA in brain | [140] |
| JWH133 1 mmol/l, 10 µL, i.c.v., once daily, 4 weeks | SHR | anti-inflammatory effects: ↓IL-1β, IL-6, and TNFα in RVLM | [106] |
| BLOOD | |||
| nf-AEA 5 mg/kg, i.p., once weekly, 4 weeks | SHR | anti-inflammatory effects: ↓IL-1, IL-6, TNFα, uCRP, and Hsp70 in serum anti-oxidant effects: ↓NADPH oxidase serum activity and ↑nitrites (an indirect measure of NO) in serum | [111] |
| URB597 1 mg/kg, i.p., twice daily, 14 days | DOCA-salt | anti-oxidant effects: ↑GSH, ↓MDA in plasma, and ↓MDA in erythrocytes pro-oxidant effects: ↓ plasma GSH-Px activity endocannabinoid effects: - ↑AEA and NADA but ↓2-AG in plasma - ↓CB1R, CB2R, TRPV1, GPR55 in lymphocytes other effects: - ↑plasma insulin and ↑insulin sensitivity (HOMA-IR, QUICKI, and FGIR) - ↑anti-aggregation effect (↑sialic acid in erythrocytes, sialic acid in plasma and ↑negative charge of the erythrocyte membrane) - normalization of electrochemical properties of erythrocyte; ↓erythrocyte size - ↓phospholipid AA and ↑free AA, DHA, LA in plasma - ↑phospholipids in erythrocytes membrane (PC, PS, and PE) | [115,117,118,141] |
| URB597 1 mg/kg, i.p., twice daily, 14 days | SHR | anti-oxidant effects: ↑GSSG-R plasma activity and ↓MDA in erythrocytes pro-oxidant effects: ↑plasma ROS, MDA, and ↓GSH in erythrocytes endocannabinoid effects: - ↑AEA, NADA, and 2-AG in plasma - ↑TRPV1 and ↓CB2R in lymphocytes other effects: - ↓plasma insulin and ↓ insulin sensitivity (HOMA-IR) - ↑anti-aggregation effect (↑sialic acid in erythrocytes, ↓sialic acid in plasma and ↑negative charge of the erythrocyte membrane) - normalization of electrochemical properties of erythrocyte, ↓erythrocyte size - ↓phospholipid DHA in plasma, ↑phospholipids in erythrocytes membrane (PC, PS, PE, and PI) | [117,122,141] |
| rimonabant 10 mg/kg, oral, once daily, 4 weeks | (mRen2)27 | other effects: ↓serum leptin and insulin | [127] |
| CBD 10 mg/kg¸ i.p., once daily, 14 days | DOCA-salt | anti-oxidant effects: ↑vit. E, GSH, ↓MDA, and tendency to ↓GSSG and 4-HHE in plasma pro-oxidant effects: small ↓plasma GSH-Px and GSSG-R activity endocannabinoid effects: ↓AEA and LEA in plasma | [129] |
| CBD 10 mg/kg, i.p., once daily, 14 days | SHR | anti-oxidant effects: ↓CO gr., tendency to ↑GSH, ↓GSSG, and 4-HNE in plasma pro-oxidant effects: small ↓plasma GSH-Px activity endocannabinoid effects: ↓SEA, HEA, DGLEA and tendency to ↓PEA, OEA, LEA in plasma other effects: ↓free AA in plasma | [129] |
| KIDNEY | |||
| AEA 3 mg/kg, i.v., once daily, 14 days | Dahl salt-sensitive + high salt (8%) diet | pro-oxidant effects: ↓Nrf2 in renal cortex other effects: - ↑Smad3 in renal cortex and ↑interstitial fibrosis and glomeruli damage score - ↑Ca2+ excretion on day 7 | [109] |
| PEA 30 mg/kg, s.c., once daily, 5 weeks | SHR | vasodilatory effects: - ↑vasodilatory metabolites (HETEs and EETs) synthesis and/or ↓their degradation - ↓RAAS activity (↓AT1R, ↑AT2R signaling pathway) anti-oxidant and anti-nitrosative effects: - ↓ROS, MDA and ↑Cu-Zn-SOD and p47phox - ↓iNOS and protein nitrotyrosylation - small ↓urinary MDA and nitrite other effects: ↑urinary output - ↓severity of glomerulosclerosis and tubulointerstitial fibrosis | [107] |
| URB597 1 mg/kg, i.p., twice daily, 14 days | DOCA-salt | anti-hypertrophic effects: ↓renal hypertrophy (only in younger rats) anti-oxidant effects: ↓ROS, XO, NADPH oxidase, Trp and ↑GSH-Px, GSSG-R activity, ↑GSH, vit. A, p-cJun, ↓Keap1 pro-oxidant effects: ↓Cu-Zn-SOD, CAT activity and ↑4-HNE, MDA, 8-OHdG and ↓p21 and HO-1 anti-inflammatory effects: ↓TNFα and ↓COX-1 and COX-2 activity endocannabinoid effects: ↓FAAH and MAGL activity - ↑AEA, 2-AG, and NADA; ↓CB1R, ↑ CB2R, and TRPV1 other effects: ↑free AA, DHA, and phospholipid AA - intensification of changes induced by hypertension | [119,142,143] |
| URB597 1 mg/kg, i.p., twice daily, 14 days | SHR | anti-oxidant effects: ↓ROS, XO, CO gr.; ↑Cu-Zn-SOD activity, GSH, vit. E, A, HO-1 pro-oxidant effects: ↓GSH-Px activity, ↑4-HNE, MDA, NPs, 8-OHdG, Keap1, Bach1, ↓p21 anti-inflammatory effects: ↓COX-1, COX-2 activity pro-inflammatory effects: ↑cPLA2 activity endocannabinoid effects: ↓FAAH and MAGL activity - ↑AEA, 2-AG, and NADA; ↑CB2R and CB1R other effects: ↑free AA and DHA - prevention of changes in electrical properties of the cell membrane, sialic acid, and protein content | [142,143] |
| rimonabant 10 mg/kg, oral, once daily, 4 weeks | (mRen2)27 | other effects: ↑urine osmolality (at day 21) | [127] |
| LIVER | |||
| URB597 1 mg/kg, i.p., twice daily, 14 days | DOCA-salt | anti-oxidant effects: ↓XO, NADPH oxidase, ↑Cu-Zn-SOD, GSH-T activity, ↑GSH, GSSG, vit. A, ↓Trp, Keap1, Bach1, ↑p-cJun pro-oxidant effects: ↓GSSG-R activity, vit. E, p21, ERK1/2, HO-1, ↑4-HNE, MDA, 4-ONE, 8-OHdG, dityrosine anti-inflammatory effects: ↓NFκB, TNFα endocannabinoid effects: ↓FAAH and MAGL activity - ↓2-AG, ↑ CB1R, and ↓ PPARα other effects: ↓phospholipid DHA and LA - ↓ apoptosis (↓caspase 3, 9 but ↑ caspase 8) | [113] |
| URB597 1 mg/kg, i.p., twice daily, 14 days | SHR | anti-oxidant effects: ↓XO, NADPH oxidase, ↑CAT, GSH-Px activity, p21, p-ERK1/2, HO-1, ↓ CO gr. pro-oxidant effects: ↓GSSG-R activity, ↑MDA, 8-OHdG, Keap1, Bach1, ↓ p-cJun, Trx anti-inflammatory effects: ↓NFκB, TNFα, and ↑COX-2 endocannabinoid effects: ↓FAAH activity - ↑AEA, NADA, ↓ CB2R, and ↑TRPV1 other effects: ↓phospholipid AA, free AA, and ↑ free DHA, LA | [144] |
| Δ8-THC, Δ9-THC 3 mg/kg, i.p., once daily, 14 days | ARH unilaterally adrenalectomized +1% NaCl 1 | hypertrophic effects: ↑liver hypertrophy/weight | [132] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Remiszewski, P.; Malinowska, B. Why Multitarget Vasodilatory (Endo)cannabinoids Are Not Effective as Antihypertensive Compounds after Chronic Administration: Comparison of Their Effects on Systemic and Pulmonary Hypertension. Pharmaceuticals 2022, 15, 1119. https://doi.org/10.3390/ph15091119
Remiszewski P, Malinowska B. Why Multitarget Vasodilatory (Endo)cannabinoids Are Not Effective as Antihypertensive Compounds after Chronic Administration: Comparison of Their Effects on Systemic and Pulmonary Hypertension. Pharmaceuticals. 2022; 15(9):1119. https://doi.org/10.3390/ph15091119
Chicago/Turabian StyleRemiszewski, Patryk, and Barbara Malinowska. 2022. "Why Multitarget Vasodilatory (Endo)cannabinoids Are Not Effective as Antihypertensive Compounds after Chronic Administration: Comparison of Their Effects on Systemic and Pulmonary Hypertension" Pharmaceuticals 15, no. 9: 1119. https://doi.org/10.3390/ph15091119
APA StyleRemiszewski, P., & Malinowska, B. (2022). Why Multitarget Vasodilatory (Endo)cannabinoids Are Not Effective as Antihypertensive Compounds after Chronic Administration: Comparison of Their Effects on Systemic and Pulmonary Hypertension. Pharmaceuticals, 15(9), 1119. https://doi.org/10.3390/ph15091119