
From Density Functional Theory to Conceptual Density Functional Theory and Biosystems
{kind=link}
Abstract
:1. From DFT to Conceptual DFT
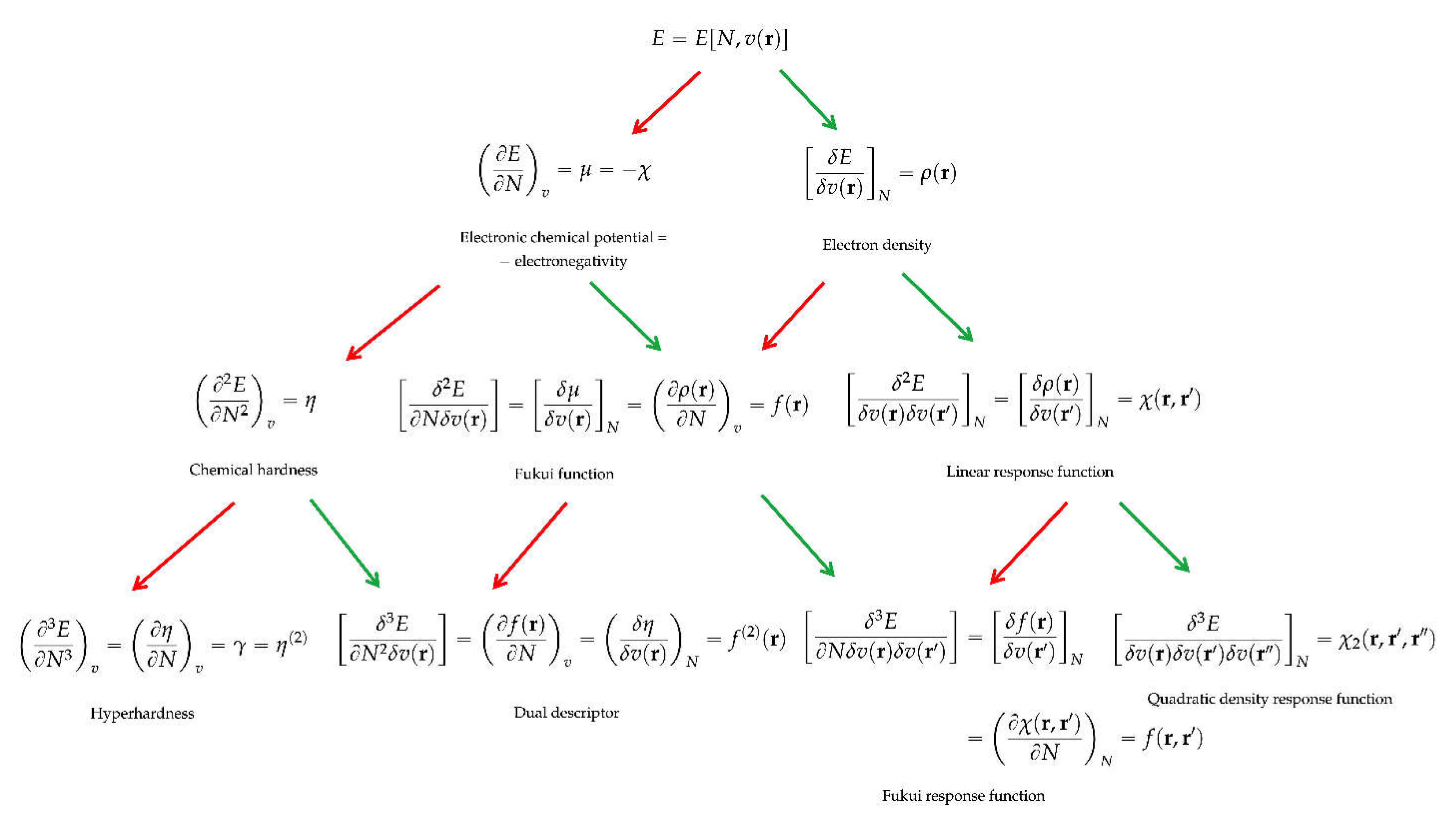
2. The Basics of Conceptual DFT
3. Response Functions and Derived Descriptors
4. Principles
5. Applications
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Koch, W.; Holthausen, M.C. A Chemist’s Guide to Density Functional Theory, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2002. [Google Scholar]
- Burke, K. Perspective on density functional theory. J. Chem. Phys. 2012, 136, 150901. [Google Scholar] [CrossRef]
- Fermi, Z.E. Eine statistische Methode zur Bestimmung einiger Eigenschaften des Atoms und ihre Anwendung auf die Theorie des periodischen Systems der Elemente. Phys.A 1928, 48, 73–79. [Google Scholar] [CrossRef]
- Thomas, L.H. The calculation of atomic fields. Proc. Camb. Phil. Soc. 1927, 23, 542–548. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. B 1964, 136, 864–871. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Kohn, W.; Sham, L.J. Self consistent equations including exchange and correlation effects. Phys. Rev. A 1965, 140, 1133–1138. [Google Scholar] [CrossRef]
- Parr, R.G.; Donnelly, R.A.; Levy, M.; Palke, W.E. Electronegativity: The density functional viewpoint. J. Chem. Phys. 1978, 68, 3801–3807. [Google Scholar] [CrossRef]
- Einhorn, M.B.; Blankenbecler, R. Bounds on scattering amplitudes. Ann. Phys. 1971, 67, 480–517. [Google Scholar] [CrossRef]
- Iczkowski, R.P.; Margrave, J.L. Electronegativity. J. Am. Chem. Soc. 1961, 83, 3547–3551. [Google Scholar] [CrossRef]
- Mulliken, R.S. A new electroaffinity scale; together with data on valence states and on valence ionization potentials and electron affinities. J. Chem. Phys. 1934, 2, 782–793. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Pearson, R.G. Chemical Hardness: Applications from Molecules to the Solid State; Wiley-VCH, Verlag: Weinheim, Germany, 1993. [Google Scholar]
- Geerlings, P. Conceptual Density Functional Theory; Liu, S., Ed.; Wiley-VCH GmbH, Verlag: Weinheim, Germany, 2022; Chapter 1. [Google Scholar]
- Parr, R.G.; Yang, W. Density functional approach to the frontier-electron theory of chemical reactivity. J. Am. Chem. Soc. 1984, 106, 4049–4050. [Google Scholar] [CrossRef]
- Fukui, K.; Yonezawa, T.; Shingu, H. A molecular orbital theory of reactivity in aromatic hydrocarbons. J. Chem. Phys. 1952, 20, 722–725. [Google Scholar] [CrossRef]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density-functional theory of the electronic structure of molecules. Ann. Rev. Phys. Chem. 1995, 46, 701–728. [Google Scholar] [CrossRef]
- Geerlings, P.; Chamorro, E.; Chattaraj, P.K.; De Proft, F.; Gázquez, J.L.; Liu, S.; Ayers, P. Conceptual density functional theory: Status, prospects, issues. Theoret. Chem. Acc. 2020, 139, i36. [Google Scholar] [CrossRef]
- Chermette, H. Chemical reactivity indexes in density functional theory. J. Comput. Chem. 1999, 20, 129–154. [Google Scholar] [CrossRef]
- Ayers, P.W.; Anderson, J.M.; Bartolotti, L.J. Perturbative perspectives on the chemical reaction prediction problem. Int. J. Quant. Chem. 2005, 101, 520–534. [Google Scholar] [CrossRef]
- Geerlings, P.; De Proft, F. Conceptual DFT: The chemical relevance of higher response functions. Phys. Chem. Chem. Phys. 2008, 10, 3028–3042. [Google Scholar] [CrossRef]
- Gazquez, J.L. Properties of the density functional theory of chemical reactivity. J. Mex. Chem. Soc. 2008, 52, 8–10. [Google Scholar]
- Liu, S.B. Conceptual density functional theory and some recent developments. Acta Physico-Chim. Sin. 2009, 25, 590–600. [Google Scholar]
- Conceptual Density Functional Theory; Liu, S. (Ed.) Wiley VCH GmbH, Verlag: Weinheim, Germany, 2022. [Google Scholar]
- Heidar-Zadeh, F.; Richer, M.; Fias, S.; Miranda-Quintana, R.A.; Chan, M.; Franco-Pérez, M.; Ayers, P.W. An explicit approach to conceptual density functional theory descriptors of arbitrary order. Chem. Phys. Lett. 2016, 660, 307–312. [Google Scholar] [CrossRef]
- Klopman, G. Chemical Reactivity and Reaction Paths; Klopman, G., Ed.; Wiley: New York, NY, USA, 1974; Chapter 1. [Google Scholar]
- Senet, P. Kohn-Sham orbital formulation of the chemical electronic responses, including the hardness. J. Chem. Phys. 1997, 107, 2516–2524. [Google Scholar] [CrossRef]
- De Proft, F. Conceptual Density Functional Theory; Liu, S., Ed.; Wiley VCH GmbH, Verlag: Weinheim, Germany, 2022; Chapter 2. [Google Scholar]
- Mulliken, R.S. Electronic population analysis on LCAO-MO molecular wavefunctions. J. Chem. Phys. 1955, 23, 1833. [Google Scholar] [CrossRef]
- Yang, W.; Mortier, W.J. The use of global and local molecular parameters for the analysis of the gas-phase basicity of amines. J. Am. Chem. Soc. 1986, 108, 5708–5711. [Google Scholar] [CrossRef]
- De Proft, F.; Langenaeker, W.; Geerlings, P. Ab initio determination of substituent constants in a density functional theory formalism: Calculation of intrinsic group electronegativity, hardness, and softness. J. Phys. Chem. 1997, 97, 1826–1831. [Google Scholar] [CrossRef]
- Geerlings, P.; Fias, S.; Boisdenghien, Z.; De Proft, F. Conceptual DFT: Chemistry from the linear response function. Chem. Soc. Rev. 2014, 43, 4989–5008. [Google Scholar] [CrossRef]
- Geerlings, P.; Fias, S.; Stuyver, T.; Ayers, P.; Balawender, R.; De Proft, F. Density Functional Theory; Glossman Mitnik, D., Ed.; IntechOpen: London, UK, 2019; Chapter 1. [Google Scholar]
- Stuyver, T.; Fias, S.; De Proft, F. Conduction of molecular electronic devices: Qualitative insights through atom-atom polarizabilities. J. Chem. Phys. 2015, 142, 094103. [Google Scholar] [CrossRef]
- Fias, S.; Heidar-Zadeh, F.; Geerlings, P.; Ayers, P.W. Chemical transferability of functional groups follows from the nearsightedness of electronic matter. Proc. Nat. Acad. Sci. USA 2017, 114, 11633–11638. [Google Scholar] [CrossRef]
- Fuentealba, P.; Parr, R.G. Higher-order derivatives in density-functional theory, especially the hardness derivative ∂ η/∂ N. J. Chem. Phys. 1991, 94, 5559–5564. [Google Scholar] [CrossRef]
- Morell, C.; Grand, A.; Toro-Labbé, A. New dual descriptor for chemical reactivity. J. Phys. Chem. A 2005, 109, 205–212. [Google Scholar] [CrossRef]
- Morell, C.; Grand, A.; Toro-Labbé, A. Theoretical support for using the Δf (r) descriptor. Chem. Phys. Lett. 2006, 425, 342–346. [Google Scholar] [CrossRef]
- Nalewajski, R.F.; Parr, R.G. Legendre transforms and Maxwell relations in density functional theory. J. Chem. Phys. 1982, 77, 393–407. [Google Scholar] [CrossRef]
- Yang, W.; Parr, R.G. Hardness, softness, and the fukui function in the electronic theory of metals and catalysis. Proc. Nat. Acad. Sci. USA 1985, 82, 6723–6726. [Google Scholar] [CrossRef]
- Ghosh, S.K.; Berkowitz, M. A classical fluid-like approach to the density-functional formalism of many-electron systems. J. Chem. Phys. 1985, 83, 2976–2983. [Google Scholar] [CrossRef]
- Berkowitz, M.; Ghosh, S.K.; Parr, R.G. On the concept of local hardness in chemistry. J. Am. Chem. Soc. 1985, 107, 6811–6814. [Google Scholar] [CrossRef]
- Berkowitz, M.; Parr, R.G. Molecular hardness and softness, local hardness and softness, hardness and softness kernels, and relations among these quantities. J. Chem. Phys. 1988, 88, 2554–2557. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Roy, D.R.; Geerlings, P.; Torrent-Sucarrat, M. Local hardness: A critical account. Theoret. Chem. Acc. 2007, 118, 923–930. [Google Scholar] [CrossRef]
- Torrent-Sucarrat, M.; De Proft, F.; Geerlings, P.; Ayers, P.W. Do the local softness and hardness indicate the softest and hardest regions of a molecule? Chem. A Eur. J. 2008, 14, 8652–8660. [Google Scholar] [CrossRef]
- Galván, M.; Gázquez, J.L.; Vela, A. Fukui function: Spin-density and chemical reactivity. J. Chem. Phys. 1986, 85, 2337–2338. [Google Scholar] [CrossRef]
- Ghanty, T.K.; Ghosh, S.K. Spin-polarized generalization of the concepts of electronegativity and hardness and the description of chemical binding. J. Am. Chem. Soc. 1994, 116, 3943–3948. [Google Scholar] [CrossRef]
- Gázquez, J.L.; Franco-Pérez, M.; Ayers, P.W.; Vela, A. Temperature-dependent approach to chemical reactivity concepts in density functional theory. Int. J. Quant. Chem. 2019, 119, e25797. [Google Scholar] [CrossRef]
- Perdew, J.P.; Parr, R.G.; Levy, M.; Balduz, J.L., Jr. Density-functional theory for fractional particle number: Derivative discontinuities of the energy. Phys. Rev. Lett. 1982, 49, 1691–1694. [Google Scholar] [CrossRef]
- Franco-Pérez, M.; Ayers, P.W.; Gázquez, J.L.; Vela, A. Local and linear chemical reactivity response functions at finite temperature in density functional theory. J. Chem. Phys. 2015, 143, 244117. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathi, R.; Subramanian, V.; Chattaraj, P.K. Effect of electric field on the global and local reactivity indices. Chem. Phys. Lett. 2003, 382, 48–56. [Google Scholar] [CrossRef]
- Khatua, M.; Sarkar, U.; Chattaraj, P.K. Reactivity dynamics of confined atoms in the presence of an external magnetic field. Eur. J. Phys. D 2014, 68, 2. [Google Scholar]
- Geerlings, P.; De Proft, F. External fields in conceptual density functional theory. J. Comput. Chem. 2022, 43. in press. [Google Scholar] [CrossRef]
- Clarys, T.; Stuyver, T.; De Proft, F.; Geerlings, P. Extending conceptual DFT to include additional variables: Oriented external electric field. Phys. Chem. Chem. Phys. 2021, 23, 990–1005. [Google Scholar] [CrossRef]
- Francotte, R.; Irons, T.J.; Teale, A.M.; De Proft, F.; Geerlings, P. Extending conceptual DFT to include external variables: The influence of magnetic fields. Chem. Sci. 2022, 13, 5311–5324. [Google Scholar] [CrossRef]
- Bettens, T.; Alonso, M.; Geerlings, P. Implementing the mechanical force into the conceptual DFT framework: Understanding and predicting molecular mechanochemical properties. Phys. Chem. Chem. Phys. 2019, 21, 7378–7388. [Google Scholar] [CrossRef]
- Bettens, T.; Alonso, M.; Geerlings, P. The hunt for reactive alkynes in bio-orthogonal click reactions: Insights from mechanochemical and conceptual DFT calculations. Chem. Sci. 2020, 11, 1431–1439. [Google Scholar] [CrossRef]
- Eeckhoudt, J.P.; Bettens, T.; Geerlings, P.; Cammi, R.; Chen, B.; Alonso, M.; DeProft, F. Conceptual density functional theory under pressure. Part 1 XP-PCM method applied to atoms. Chem. Sci. 2022, 13, 9329–9350. [Google Scholar] [CrossRef]
- Borgoo, A.; Tozer, D.J.; Geerlings, P. Confinement effects on excitation energies and regioselectivity as probed by the Fukui function and the molecular electrostatic potential. Phys. Chem. Chem. Phys. 2009, 11, 2862–2868. [Google Scholar] [CrossRef]
- Geerlings, P.; Tozer, D.J.; De Proft, F. Chemical Reactivity in Confined Systems: Theory Modelling and Applications; Chattaraj, P.K., Chakraborty, D., Eds.; John Wiley: New York, NY, USA, 2021; Chapter 3. [Google Scholar]
- Stauch, T.; Dreuw, A. Advances in quantum mechanochemistry: Electronic structure methods and force analysis. Chem. Rev. 2016, 116, 14137–14180. [Google Scholar] [CrossRef]
- Grochala, W.; Hoffmann, R.; Feng, J. The chemical imagination at work in very tight places. Angewand. Chem. Int. Ed. 2007, 46, 3620–3642. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpály, L.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Roy, D.R. Electrophilicity index. Chem. Rev. 2006, 106, 2065–2091. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Roy, D.R. Update 1 of: Electrophilicity index. Chem. Rev. 2007, 107, PR46–PR74. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Maiti, B.; Sarkar, U. Philicity: A unified teratment of chemical reactivity and selectivity. J. Phys. Chem. A 2003, 107, 4973–4975. [Google Scholar] [CrossRef]
- Domingo, L.R.; Aurell, M.J.; Pérez, P. Quantitative characterization of the local electrophilicity of organic molecules. Understanding the regioselectivity on Diels− Alder reactions. J. Phys. Chem. A 2002, 106, 6871–6875. [Google Scholar] [CrossRef]
- Padmanabhan, J.; Parthasarathi, R.; Elango, M.; Subramanian, V.; Krishnamoorty, B.S.; Guttierrez-Oliva, S.; Toro-Labbé, A.; Roy, D.R.; Chattaraj, P.K. Multiphilic descriptor for chemical reactivity and selectivity. J. Phys. Chem. A 2007, 111, 9130–9138. [Google Scholar] [CrossRef]
- Sanderson, R.T. An interpretation of bond lengths and a classification of bonds. Science 1951, 114, 670–672. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, R.T. Chemical Bonds and Bond Energies; Academic Press: New York, NY, USA, 1976. [Google Scholar]
- Donnelly, R.A.; Parr, R.G. Elementary properties of an energy functional of the first-order reduced density matrix. J. Chem. Phys. 1978, 69, 4431–4439. [Google Scholar] [CrossRef]
- Parr, R.G.; Bartolotti, L.J. On the geometric mean principle for electronegativity equation, J. Am. Chem. Soc. 1982, 104, 3801–3803. [Google Scholar] [CrossRef]
- Mortier, W.J.; Van Genechten, K.; Gasteiger, J. Electronegativity equalization: Application and parametrization. J. Am. Chem. Soc. 1985, 107, 829–835. [Google Scholar] [CrossRef]
- Mortier, W.J.; Ghosh, S.K.; Shankar, S. Electronegativity-equalization method for the calculation of atomic charges in molecules. J. Am. Chem. Soc. 1986, 108, 4315–4320. [Google Scholar] [CrossRef]
- Bultinck, P.; Langenaeker, W.; Lahorte, P.; De Proft, F.; Geerlings, P.; Waroquier, M.; Tollenaere, J.P. The electronegativity equalization method I: Parametrization and validation for atomic charge calculations. J. Phys. Chem. A 2002, 106, 7887–7894. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Lee, H.; Parr, R.G. HSAB principle J. Am. Chem. Soc. 1991, 113, 1855–1856. [Google Scholar] [CrossRef]
- Mendez, F.; Gazquez, J.L. Chemical reactivity of enolate ions: The local hard and soft acids and bases principle viewpoint. J. Am. Chem. Soc. 1994, 116, 9298–9301. [Google Scholar] [CrossRef]
- Pearson, R.G.; Chattaraj, P.K. The hard-soft, acid-base principle. Chemtracts-Inorg. Chem. 2008, 21, 1–7. [Google Scholar]
- Pearson, R.G. Recent advances in the concept of hard and soft acids and bases. J. Chem. Educ. 1987, 64, 561–567. [Google Scholar] [CrossRef]
- Parr, R.G.; Chattaraj, P.K. Principle of maximum hardness. J. Am. Chem. Soc. 1991, 113, 1854–1855. [Google Scholar] [CrossRef]
- Miranda-Quintana, R.A.; Heidar-Zadeh, F.; Fias, S.; Chapman, A.E.A.; Liu, S.; Morell, C.; Gomez, T.; Cardenas, C.; Ayers, P.W. Molecular interactions from the density functional theory for chemical reactivity: Interaction chemical potential, hardness, and reactivity principles. Front. Chem. 2022, 10, 929464. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Sengupta, S. Popular electronic structure principles in a dynamical context. J. Phys. Chem. 1996, 100, 16126–16130. [Google Scholar] [CrossRef]
- Khandogin, J.; York, D.M. Quantum descriptors for biological macromolecules from linear-scaling electronic structure methods. Prot. Struct. Funct. Bioinform. 2004, 56, 724–737. [Google Scholar] [CrossRef]
- Bonaccorsi, R.; Scrocco, E.; Tomasi, J. Molecular SCF calculations for the ground state of some three-membered ring molecules:(CH2) 3,(CH2) 2NH,(CH2) 2NH2+,(CH2) 2O,(CH2) 2S,(CH) 2CH2, and N2CH2. J. Chem. Phys. 1970, 52, 5270–5284. [Google Scholar] [CrossRef]
- Molecular Electrostatic Potentials Concepts and Applications; Murray, J.S.; Sen, K. (Eds.) Elsevier: Amsterdam, The Netherlands, 1996. [Google Scholar]
- Rivas, P.; Zapata-Torres, G.; Melin, J.; Contreras, R. Probing the hydride transfer process in the lumiflavine–1-methylnicotinamide model system using group softness. Tetrahedron 2004, 60, 4189–4196. [Google Scholar] [CrossRef]
- Roos, G.; Messens, J.; Loverix, S. A computational and conceptual DFT study on the Michaelis complex of pI258 arsenate reductase. Structural aspects and activation of the electrophile and nucleophile. J. Phys. Chem. B 2004, 108, 17216–17225. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Loverix, S.; Geerlings, P.; Tourwé, D. DFT-based ranking of zinc-binding groups in histone deacetylase inhibitors. Bioorg.Med. Chem. 2005, 13, 6070–6082. [Google Scholar] [CrossRef]
- Roos, G.; Loverix, S.; Brosens, E. The activation of electrophile, nucleophile and leaving group during the reaction catalysed by pI258 arsenate reductase. ChemBioChem 2006, 7, 981–989. [Google Scholar] [CrossRef]
- Roos, G.; Foloppe, N.; Van Laer, K. How thioredoxin dissociates its mixed disulfide. PLoS Comput. Biol. 2009, 5, e1000461. [Google Scholar] [CrossRef]
- Roos, G.; Geerlings, P.; Messens, J. Enzymatic catalysis: The emerging role of conceptual density functional theory. J. Phys. Chem. B 2009, 113, 13465–13475. [Google Scholar] [CrossRef] [PubMed]
- Faver, J.; Merz, K.M., Jr. Utility of the hard/soft acid−base principle via the fukui function in biological systems. J. Chem. Ther. Comput. 2010, 6, 548–559. [Google Scholar] [CrossRef]
- Grillo, I.B.; Urquiza-Carvalho, G.A.; Chaves, E.J.F.; Rocah, G.B. Semiempirical methods do Fukui functions: Unlocking a modeling framework for biosystems. J. Comput. Chem. 2020, 41, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Grillo, I.B.; Urquiza-Carvalho, G.A.; Bachega, J.F.R.; Rocha, G.B. Elucidating enzymatic catalysis using fast quantum chemical descriptors. J. Chem. Inform. Model. 2020, 60, 578–591. [Google Scholar] [CrossRef] [PubMed]
- Oller, J.; Saez, D.A.; Vohringer-Martinez, E. Atom-condensed fukui function in condensed phases and biological systems and its application to enzymatic fixation of carbon dioxide. J. Phys. Chem. A 2020, 124, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, U.; Roy, D.R.; Chattaraj, P.K.; Parthasarathi, R.; Padmanabhan, J.; Subramanian, V. Conceptual DFT approach towards analysing toxicity. J. Chem. Sci. 2005, 117, 599–612. [Google Scholar] [CrossRef]
- Roy, D.R.; Sarkar, U.; Chattaraj, P.K. Analyzing toxicity through electrophilicity. Mol. Divers. 2006, 10, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Pan, S.; Chattaraj, P.K. Biological activity and toxicity: A conceptual DFT approach. In Applications of Density Functional Theory in Biological and Bio-inorganic Chemistry; Putz, M.V., Mingos, M.P., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; p. 150. [Google Scholar]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. Computational pharmacokinetics report, ADMET study and conceptual DFT-based estimation of the chemical reactivity properties of marine cyclopeptides. ChemistryOpen 2021, 10, 1142–1149. [Google Scholar] [CrossRef]
- Flores-Holguín, N.; Ortega-Castro, J.; Frau, J. Conceptual DFT-Based Computational Peptidology, Pharmacokinetics Study and ADMET Report of the Veraguamides A–G Family of Marine Natural Drugs. Marine Drugs 2022, 20, 97. [Google Scholar] [CrossRef]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. Virtual Prospection of Marine Cyclopeptides as Therapeutics by Means of Conceptual DFT and Computational ADMET. Pharmaceuticals 2022, 15, 509. [Google Scholar] [CrossRef]
- Shreevatsa, B.; Dharmashekara, C.; Jain, A.S.; Amachawadi, R.; Achar, R.R.; Syed, A.; Shivamallu, C.; Kollur, S.P.; Frau, J.; Flores-Hoguin, N.; et al. An insight into reactivity and bioactivity properties of quorum sensing peptides against PDE10A: A computational peptidology approach. J. Mol. Model. 2022, 28, 209. [Google Scholar] [CrossRef]
- Pujal, F.; Tehrani, F.; Heidar-Zadeh, F. Conceptual Density Functional Theory; Liu, S., Ed.; Wiley-VCH GmbH, Verlag: Weinheim, Germany, 2022; Chapter 32. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geerlings, P. From Density Functional Theory to Conceptual Density Functional Theory and Biosystems. Pharmaceuticals 2022, 15, 1112. https://doi.org/10.3390/ph15091112
Geerlings P. From Density Functional Theory to Conceptual Density Functional Theory and Biosystems. Pharmaceuticals. 2022; 15(9):1112. https://doi.org/10.3390/ph15091112
Chicago/Turabian StyleGeerlings, Paul. 2022. "From Density Functional Theory to Conceptual Density Functional Theory and Biosystems" Pharmaceuticals 15, no. 9: 1112. https://doi.org/10.3390/ph15091112
APA StyleGeerlings, P. (2022). From Density Functional Theory to Conceptual Density Functional Theory and Biosystems. Pharmaceuticals, 15(9), 1112. https://doi.org/10.3390/ph15091112