
Alzheimer’s Disease Severity Is Associated with an Imbalance in Serum Levels of Enzymes Regulating Plasmin Synthesis

 ,
,
Abstract
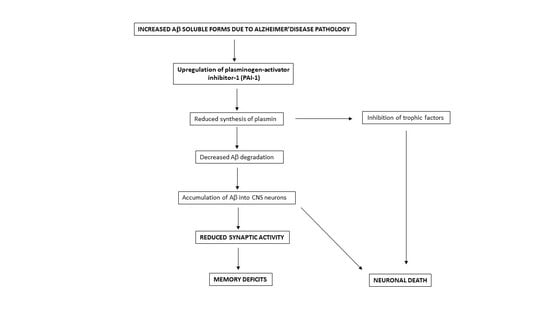
:
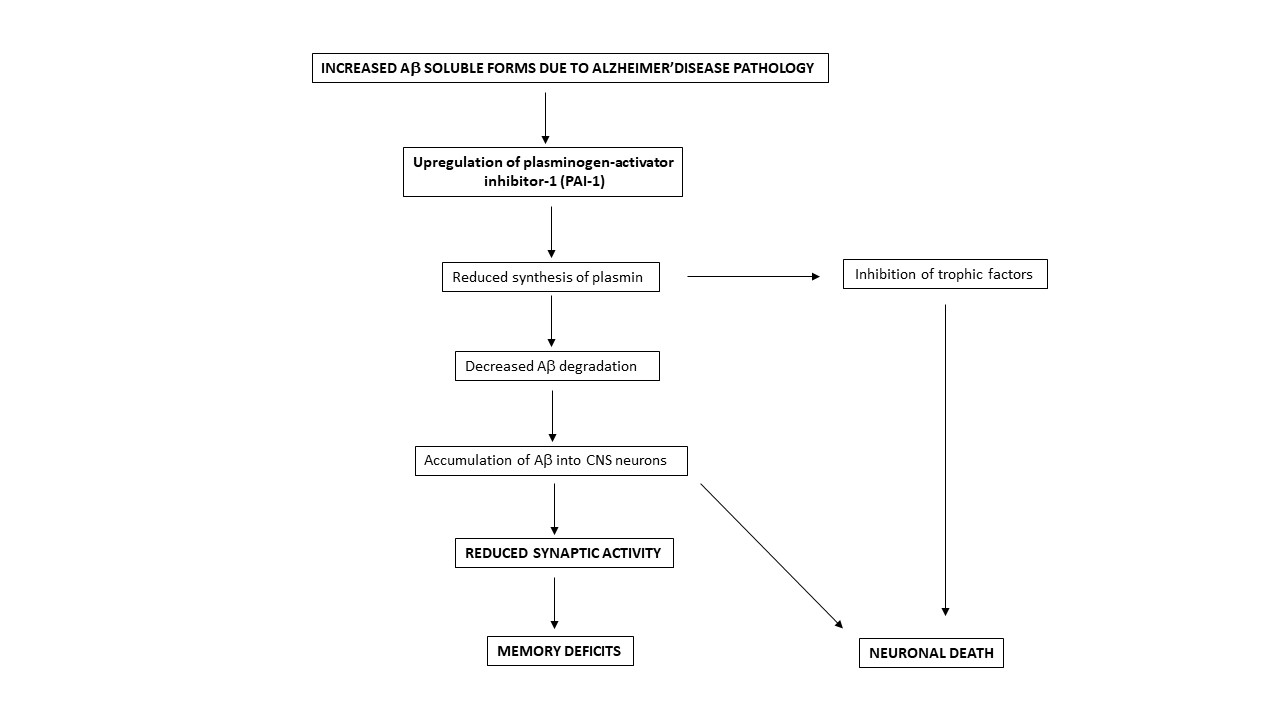
1. Introduction
2. Results
2.1. Demographic Characteristics
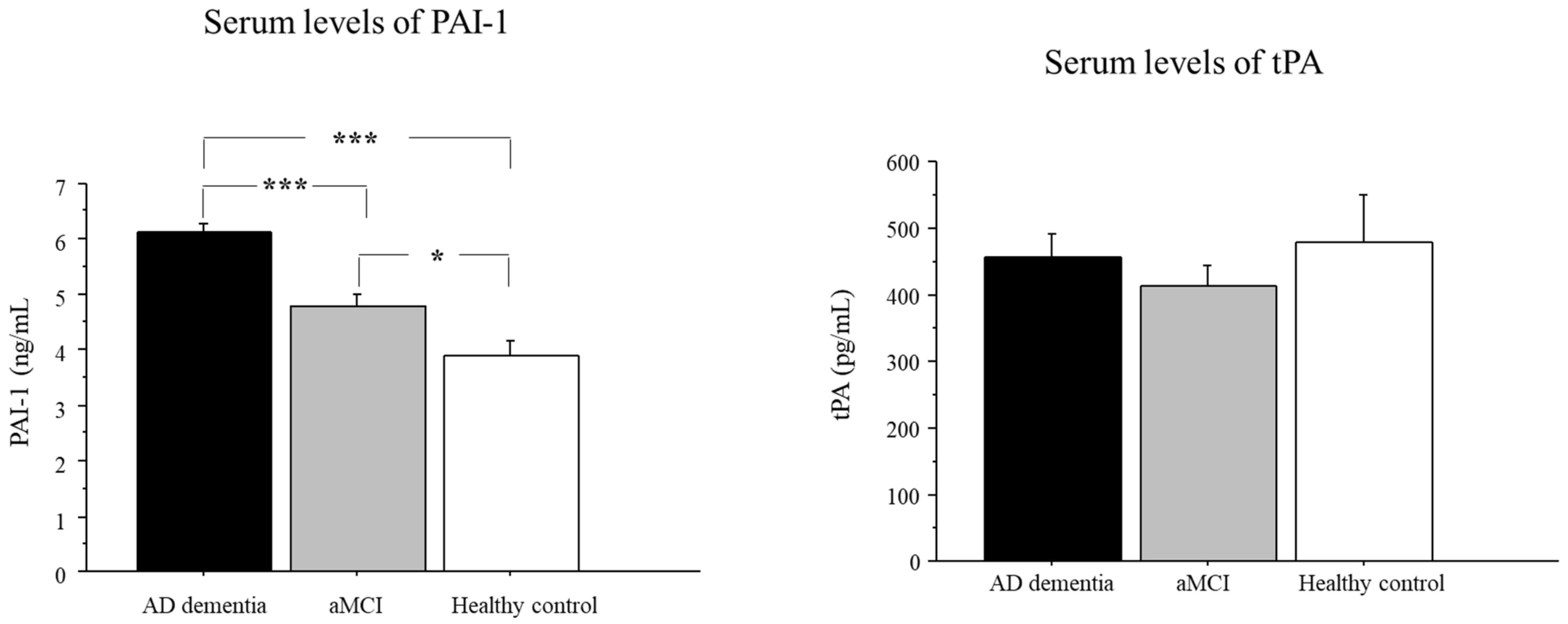
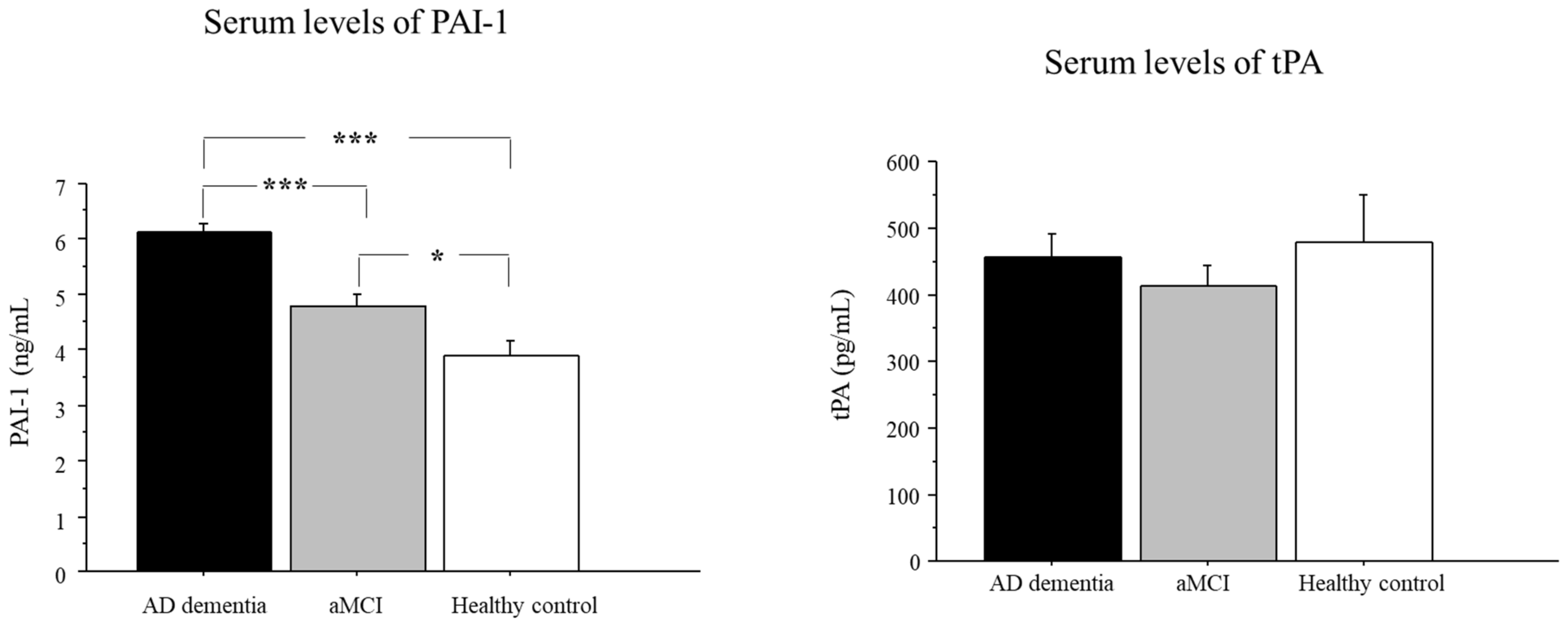
2.2. Serum Levels of PAI-1 in AD, aMCI, and Controls
2.3. Serum Levels of tPA in AD, aMCI, and Controls
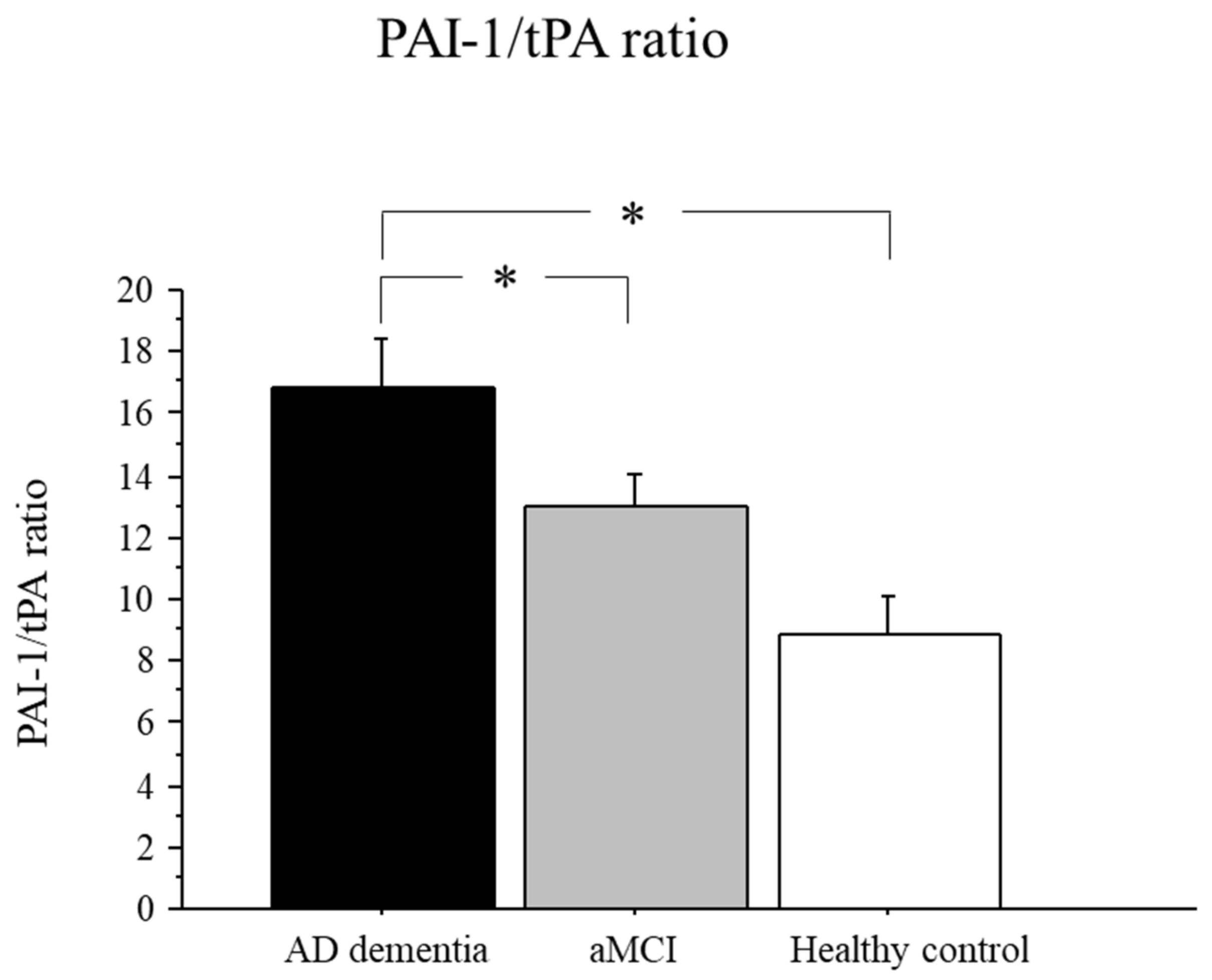
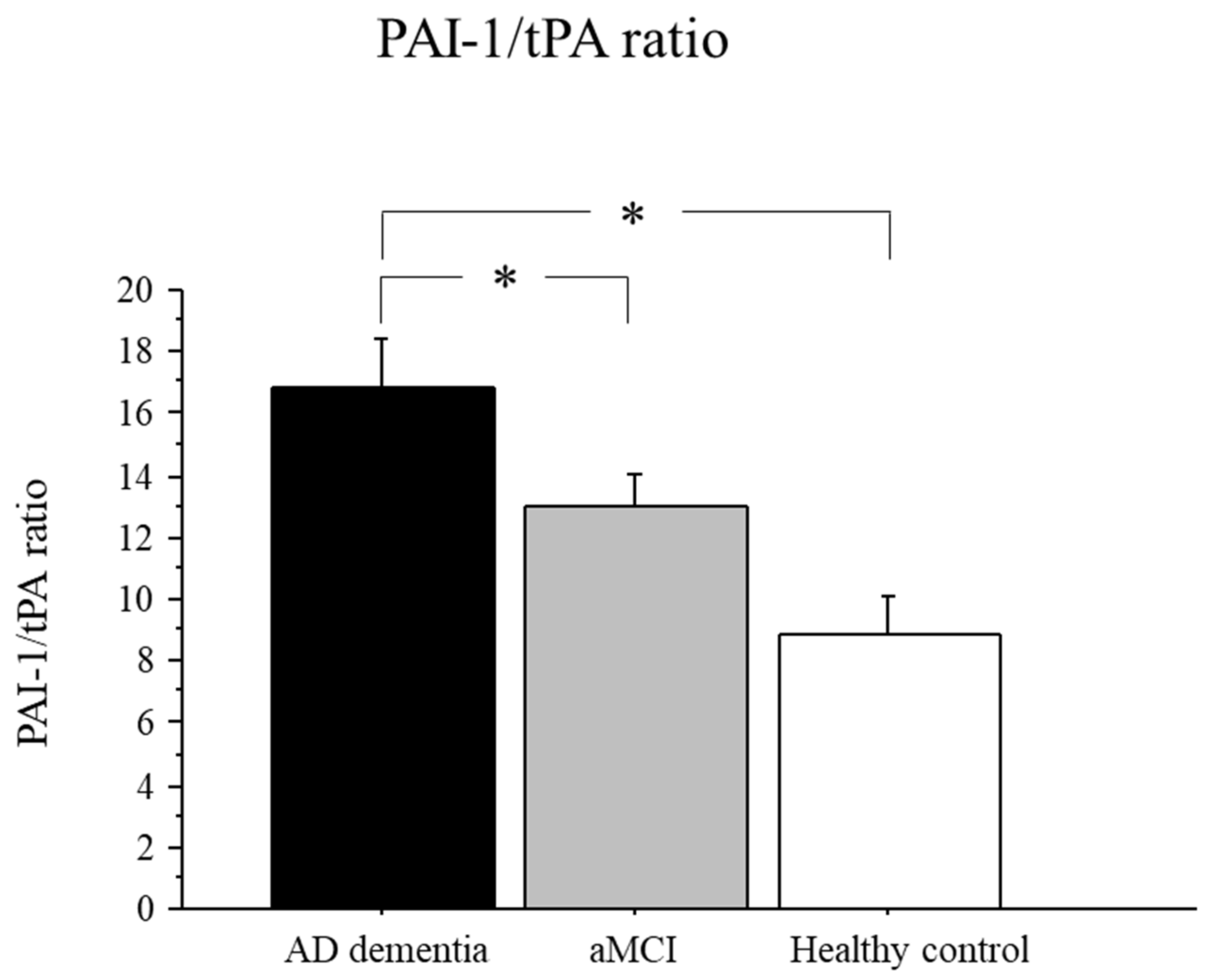
2.4. PAI-1/tPA Ratio in AD, aMCI, and Controls
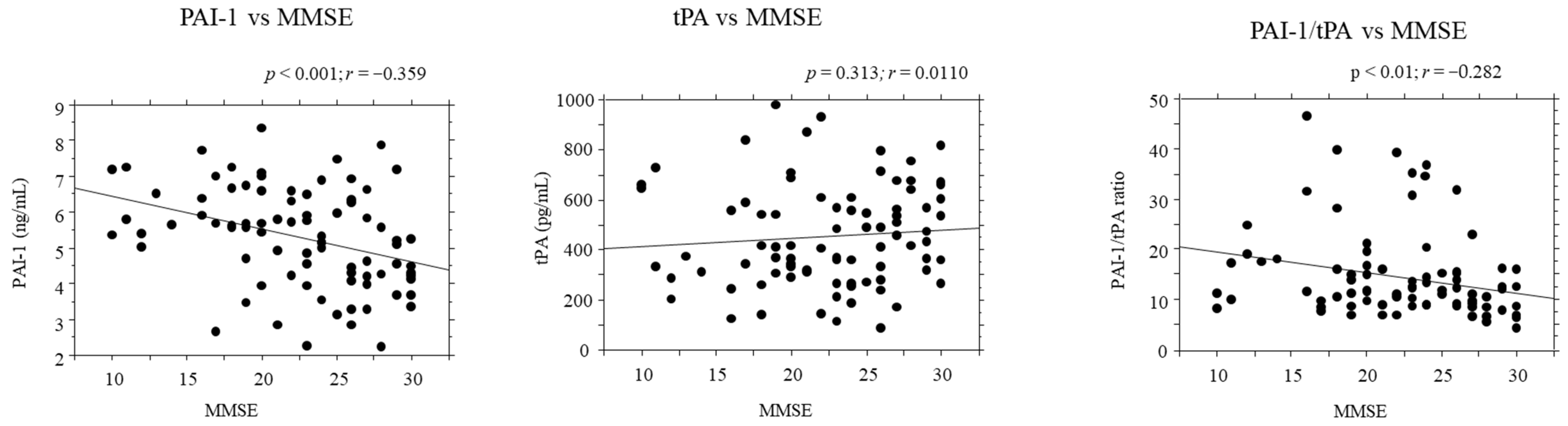
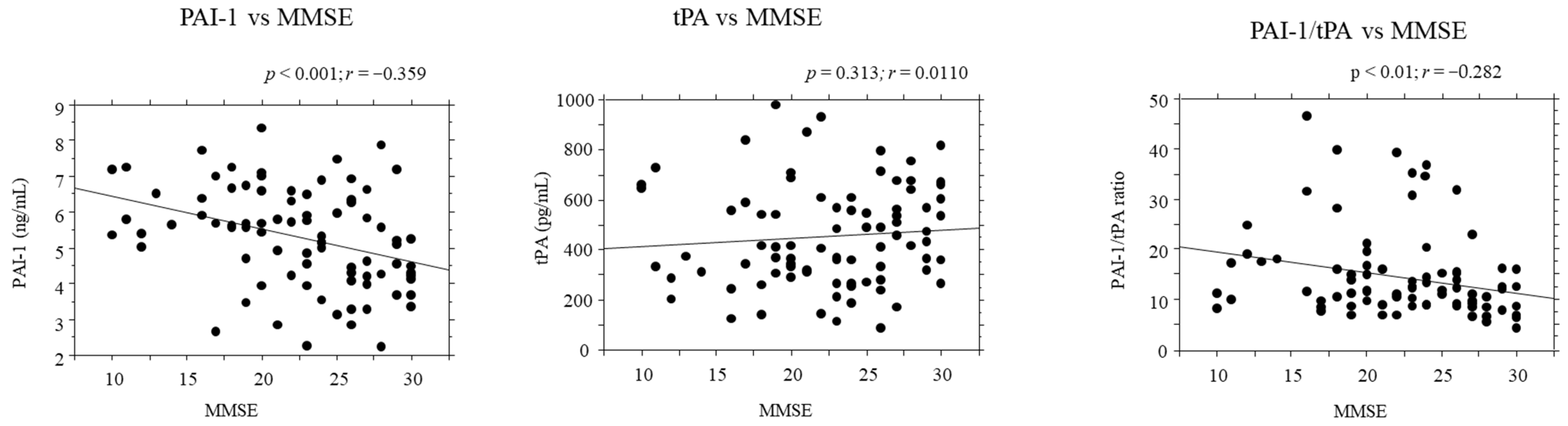
2.5. Correlations between PAI-1/tPA Serum Levels and Disease Severity
2.6. Correlations between PAI-1/tPA Serum Levels and Demographic Data
3. Discussion
4. Materials and Methods
4.1. Participants
4.2. Exclusion Criteria
4.3. Blood Sampling
4.4. PAI-1 and tPA Determination
4.5. PAI-1/tPA Ratio Determination
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Jagust, W. Imaging the evolution and pathophysiology of Alzheimer disease. Nat. Rev. Neurosci. 2018, 19, 687–700. [Google Scholar] [CrossRef]
- Counts, S.E.; He, B.; Prout, J.G.; Michalski, B.; Farotti, L.; Fahnestock, M.; Mufson, E.J. Cerebrospinal Fluid proNGF: A Putative Biomarker for Early Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 800–808. [Google Scholar]
- Yang, T.; Li, S.; Xu, H.; Walsh, D.M.; Selkoe, D.J. Large Soluble Oligomers of Amyloid β-Protein from Alzheimer Brain Are Far Less Neuroactive Than the Smaller Oligomers to Which They Dissociate. J. Neurosci. 2016, 37, 152–163. [Google Scholar] [CrossRef]
- Alifragis, P.; Marsh, J. Synaptic dysfunction in Alzheimer’s disease: The effects of amyloid beta on synaptic vesicle dynamics as a novel target for therapeutic intervention. Neural Regen. Res. 2018, 13, 616. [Google Scholar] [CrossRef]
- Angelucci, F.; Čechová, K.; Průša, R.; Hort, J. Amyloid beta soluble forms and plasminogen activation system in Alzheimer’s disease: Consequences on extracellular maturation of brain-derived neurotrophic factor and therapeutic implications. CNS Neurosci. Ther. 2018, 25, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Miszta, A.; Huskens, D.; Donkervoort, D.; Roberts, M.J.M.; Wolberg, A.S.; de Laat, B. Assessing Plasmin Generation in Health and Disease. Int. J. Mol. Sci. 2021, 22, 2758. [Google Scholar] [CrossRef]
- Rosso, M. Del The plasminogen activation system in inflammation. Front. Biosci. 2008, 13, 4667. [Google Scholar] [CrossRef]
- Barker, R.; Love, S.; Kehoe, P.G. Plasminogen and plasmin in Alzheimer’s disease. Brain Res. 2010, 1355, 7–15. [Google Scholar] [CrossRef]
- Ledesma, M.D.; Da Silva, J.S.; Crassaerts, K.; Delacourte, A.; De Strooper, B.; Dotti, C.G. Brain plasmin enhances APP α-cleavage and Aβ degradation and is reduced in Alzheimer’s disease brains. EMBO Rep. 2000, 1, 530–535. [Google Scholar] [CrossRef]
- Nicole, O.; Docagne, F.; Ali, C.; Margaill, I.; Carmeliet, P.; MacKenzie, E.T.; Vivien, D.; Buisson, A. The proteolytic activity of tissue-plasminogen activator enhances NMDA receptor-mediated signaling. Nat. Med. 2001, 7, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Gerenu, G.; Martisova, E.; Ferrero, H.; Carracedo, M.; Rantamäki, T.; Ramirez, M.J.; Gil-Bea, F.J. Modulation of BDNF cleavage by plasminogen-activator inhibitor-1 contributes to Alzheimer’s neuropathology and cognitive deficits. Biochim. Biophys. Acta-Mol. Basis Dis. 2017, 1863, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Leal, G.; Bramham, C.R.; Duarte, C.B. BDNF and Hippocampal Synaptic Plasticity. Vitam. Horm. 2017, 104, 153–195. [Google Scholar]
- Samson, A.L.; Medcalf, R.L. Tissue-Type Plasminogen Activator: A Multifaceted Modulator of Neurotransmission and Synaptic Plasticity. Neuron 2006, 50, 673–678. [Google Scholar] [CrossRef]
- Ploplis, V.; Castellino, F. Structure and function of the plasminogen/plasmin system. Thromb. Haemost. 2005, 93, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Salles, F.J.; Strickland, S. Localization and regulation of the tissue plasminogen activator-plasmin system in the hippocampus. J. Neurosci. 2002, 22, 2125–2134. [Google Scholar] [CrossRef] [PubMed]
- Yepes, M.; Roussel, B.D.; Ali, C.; Vivien, D. Tissue-type plasminogen activator in the ischemic brain: More than a thrombolytic. Trends Neurosci. 2009, 32, 48–55. [Google Scholar] [CrossRef]
- Barker, R.; Kehoe, P.G.; Love, S. Activators and inhibitors of the plasminogen system in Alzheimer’s disease. J. Cell. Mol. Med. 2012, 16, 865–876. [Google Scholar] [CrossRef]
- Ledesma, M.D.; Abad-Rodriguez, J.; Galvan, C.; Biondi, E.; Navarro, P.; Delacourte, A.; Dingwall, C.; Dotti, C.G. Raft disorganization leads to reduced plasmin activity in Alzheimer’s disease brains. EMBO Rep. 2003, 4, 1190–1196. [Google Scholar] [CrossRef]
- Jacobsen, J.S.; Comery, T.A.; Martone, R.L.; Elokdah, H.; Crandall, D.L.; Oganesian, A.; Aschmies, S.; Kirksey, Y.; Gonzales, C.; Xu, J.; et al. Enhanced clearance of A in brain by sustaining the plasmin proteolysis cascade. Proc. Natl. Acad. Sci. USA 2008, 105, 8754–8759. [Google Scholar] [CrossRef]
- Tucker, H.M.; Kihiko, M.; Caldwell, J.N.; Wright, S.; Kawarabayashi, T.; Price, D.; Walker, D.; Scheff, S.; McGillis, J.P.; Rydel, R.E.; et al. The Plasmin System Is Induced by and Degrades Amyloid-β Aggregates. J. Neurosci. 2000, 20, 3937–3946. [Google Scholar] [CrossRef]
- Tucker, H.M.; Kihiko-Ehmann, M.; Wright, S.; Rydel, R.E.; Estus, S. Tissue Plasminogen Activator Requires Plasminogen to Modulate Amyloid-β Neurotoxicity and Deposition. J. Neurochem. 2002, 75, 2172–2177. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Lee, H.-J.; Song, J.-H.; Park, S.I.; Kim, H. Plasminogen activator inhibitor-1 as an early potential diagnostic marker for Alzheimer’s disease. Exp. Gerontol. 2014, 60, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yuan, Y.; Cai, R.; Huang, R.; Tian, S.; Lin, H.; Guo, D.; Wang, S. Association between Plasma Levels of PAI-1, tPA/PAI-1 Molar Ratio, and Mild Cognitive Impairment in Chinese Patients with Type 2 Diabetes Mellitus. J. Alzheimer’s Dis. 2018, 63, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.-M.; van Groen, T.; Katre, A.; Cao, D.; Kadisha, I.; Ballinger, C.; Wang, L.; Carroll, S.L.; Li, L. Knockout of plasminogen activator inhibitor 1 gene reduces amyloid beta peptide burden in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2011, 32, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Buisson, A.; Nicole, O.; Docagne, F.; Sartelet, H.; Mackenzie, E.T.; Vivien, D. Up-regulation of a serine protease inhibitor in astrocytes mediates the neuroprotective activity of transforming growth factor β1. FASEB J. 1998, 12, 1683–1691. [Google Scholar] [CrossRef]
- Cacquevel, M.; Launay, S.; Castel, H.; Benchenane, K.; Chéenne, S.; Buée, L.; Moons, L.; Delacourte, A.; Carmeliet, P.; Vivien, D. Ageing and amyloid-beta peptide deposition contribute to an impaired brain tissue plasminogen activator activity by different mechanisms. Neurobiol. Dis. 2007, 27, 164–173. [Google Scholar] [CrossRef]
- Melchor, J.P.; Pawlak, R.; Strickland, S. The Tissue Plasminogen Activator-Plasminogen Proteolytic Cascade Accelerates Amyloid-β (Aβ) Degradation and Inhibits Aβ-Induced Neurodegeneration. J. Neurosci. 2003, 23, 8867–8871. [Google Scholar] [CrossRef]
- Sawdey, M.S.; Loskutoff, D.J. Regulation of murine type 1 plasminogen activator inhibitor gene expression in vivo. Tissue specificity and induction by lipopolysaccharide, tumor necrosis factor-alpha, and transforming growth factor-beta. J. Clin. Investig. 1991, 88, 1346–1353. [Google Scholar] [CrossRef]
- Podor, T.J.; Joshua, P.; Butcher, M.; Seiffert, D.; Loskutoff, D.; Gauldie, J. Accumulation of Type 1 Plasminogen Activator Inhibitor and Vitronectin at Sites of Cellular Necrosis and Inflammation. Ann. N. Y. Acad. Sci. 1992, 667, 173–177. [Google Scholar] [CrossRef]
- Kosaka, K.; Iseki, E.; Hino, H.; Akiyama, H.; Kondo, H.; Kato, M.; Ikeda, K. Immunohistochemical localization of plasminogen activator inhibitor-1 in rat and human brain tissues. Neurosci. Lett. 2002, 297, 105–108. [Google Scholar] [CrossRef]
- Oh, S.B.; Byun, C.J.; Yun, J.-H.; Jo, D.-G.; Carmeliet, P.; Koh, J.-Y.; Lee, J.-Y. Tissue plasminogen activator arrests Alzheimer’s disease pathogenesis. Neurobiol. Aging 2014, 35, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Bi Oh, S.; Suh, N.; Kim, I.; Lee, J.-Y.Y. Impacts of aging and amyloid-β deposition on plasminogen activators and plasminogen activator inhibitor-1 in the Tg2576 mouse model of Alzheimer’s disease. Brain Res. 2015, 1597, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Fabbro, S.; Seeds, N.W. Plasminogen activator activity is inhibited while neuroserpin is up-regulated in the Alzheimer disease brain. J. Neurochem. 2009, 109, 303–315. [Google Scholar] [CrossRef]
- Medina, M.G.; Ledesma, M.D.; Domínguez, J.E.; Medina, M.; Zafra, D.; Alameda, F.; Dotti, C.G.; Navarro, P. Tissue plasminogen activator mediates amyloid-induced neurotoxicity via Erk1/2 activation. EMBO J. 2005, 24, 1706–1716. [Google Scholar] [CrossRef]
- Kanno, Y. The Role of Fibrinolytic Regulators in Vascular Dysfunction of Systemic Sclerosis. Int. J. Mol. Sci. 2019, 20, 619. [Google Scholar] [CrossRef]
- Huang, Y.Y.; Bach, M.E.; Lipp, H.P.; Zhuo, M.; Wolfer, D.P.; Hawkins, R.D.; Schoonjans, L.; Kandel, E.R.; Godfraind, J.M.; Mulligan, R.; et al. Mice lacking the gene encoding tissue-type plasminogen activator show a selective interference with late-phase long-term potentiation in both Schaffer collateral and mossy fiber pathways. Proc. Natl. Acad. Sci. USA 1996, 93, 8699–8704. [Google Scholar] [CrossRef]
- Mossiat, C.; Prigent-Tessier, A.; Garnier, P.; Marie, C.; Jacquin, A.; Rodier, M.; Béjot, Y. Exogenous t-PA Administration Increases Hippocampal Mature BDNF Levels. Plasmin- or NMDA-Dependent Mechanism? PLoS ONE 2014, 9, e92416. [Google Scholar] [CrossRef]
- Sheardova, K.; Vyhnalek, M.; Nedelska, Z.; Laczo, J.; Andel, R.; Marciniak, R.; Cerman, J.; Lerch, O.; Hort, J. Czech Brain Aging Study (CBAS): Prospective Multicentre Cohort Study on Risk and Protective Factors for Dementia in the Czech Republic. BMJ Open 2019, 9, e030379. [Google Scholar] [CrossRef]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers. Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers. Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Yesavage, J.A. Geriatric Depression Scale. Psychopharmacol. Bull. 1988, 24, 709–711. [Google Scholar] [PubMed]
- Fazekas, F.; Chawluk, J.; Alavi, A.; Hurtig, H.; Zimmerman, R. MR signal abnormalities at 1.5 T in Alzheimer’s dementia and normal aging. Am. J. Roentgenol. 1987, 149, 351–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | AD Patients (n = 40) | aMCI Patients (n = 40) | Controls (n = 10) | Statistics |
|---|---|---|---|---|
| Age (years) | 70.5 ± 9.44 * | 69.8 ± 6.83 * | 61.2 ± 12.4 | * p < 0.01 vs. Controls |
| Sex (male/female) | 12 M/28 F | 17 M/23 F | 2 M/8 F | chi-square p = 0.297 |
| Years of education | 13.5 ± 2.9 * | 14.8 ± 3.1 * | 17 ± 1.65 | * p < 0.001 vs. Controls |
| MMSE | 19 ± 3.84 **,† | 25 ± 2.9 * | 29.9 ± 0.33 | * p < 0.001 vs. Controls ** p < 0.0001 vs. Controls † p < 0.001 vs. aMCI |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angelucci, F.; Veverova, K.; Katonová, A.; Piendel, L.; Vyhnalek, M.; Hort, J. Alzheimer’s Disease Severity Is Associated with an Imbalance in Serum Levels of Enzymes Regulating Plasmin Synthesis. Pharmaceuticals 2022, 15, 1074. https://doi.org/10.3390/ph15091074
Angelucci F, Veverova K, Katonová A, Piendel L, Vyhnalek M, Hort J. Alzheimer’s Disease Severity Is Associated with an Imbalance in Serum Levels of Enzymes Regulating Plasmin Synthesis. Pharmaceuticals. 2022; 15(9):1074. https://doi.org/10.3390/ph15091074
Chicago/Turabian StyleAngelucci, Francesco, Katerina Veverova, Alžbeta Katonová, Lydia Piendel, Martin Vyhnalek, and Jakub Hort. 2022. "Alzheimer’s Disease Severity Is Associated with an Imbalance in Serum Levels of Enzymes Regulating Plasmin Synthesis" Pharmaceuticals 15, no. 9: 1074. https://doi.org/10.3390/ph15091074
APA StyleAngelucci, F., Veverova, K., Katonová, A., Piendel, L., Vyhnalek, M., & Hort, J. (2022). Alzheimer’s Disease Severity Is Associated with an Imbalance in Serum Levels of Enzymes Regulating Plasmin Synthesis. Pharmaceuticals, 15(9), 1074. https://doi.org/10.3390/ph15091074