
Identification of Novel Aryl Carboxamide Derivatives as Death-Associated Protein Kinase 1 (DAPK1) Inhibitors with Anti-Proliferative Activities: Design, Synthesis, In Vitro, and In Silico Biological Studies
 ,
, 
 ,
,
Abstract
:1. Introduction
2. Results and Discussion
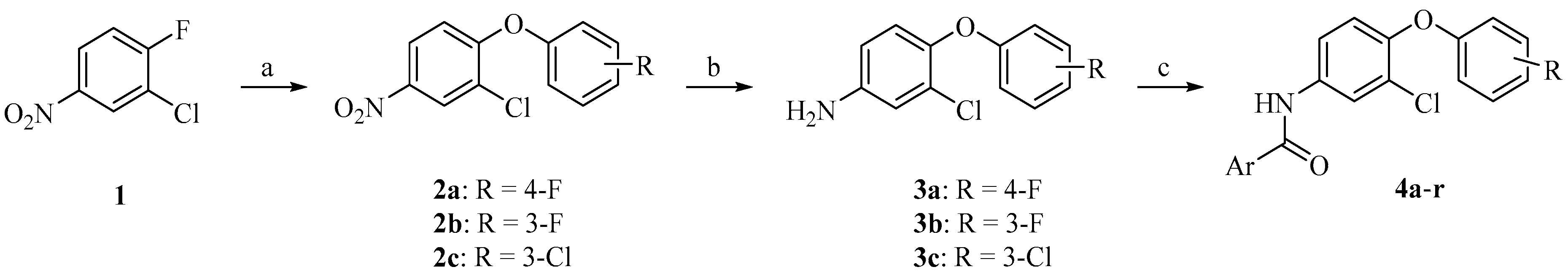
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. Evaluation of Compound 4a against a Panel of 45 Kinases
2.2.2. In Vitro DAPK1 Kinase Assay and Optimization towards Lead Development
2.2.3. Dose-Dependent Assay of Compounds 4h, 4j, 4k, and 4q with DAPK1 Kinase
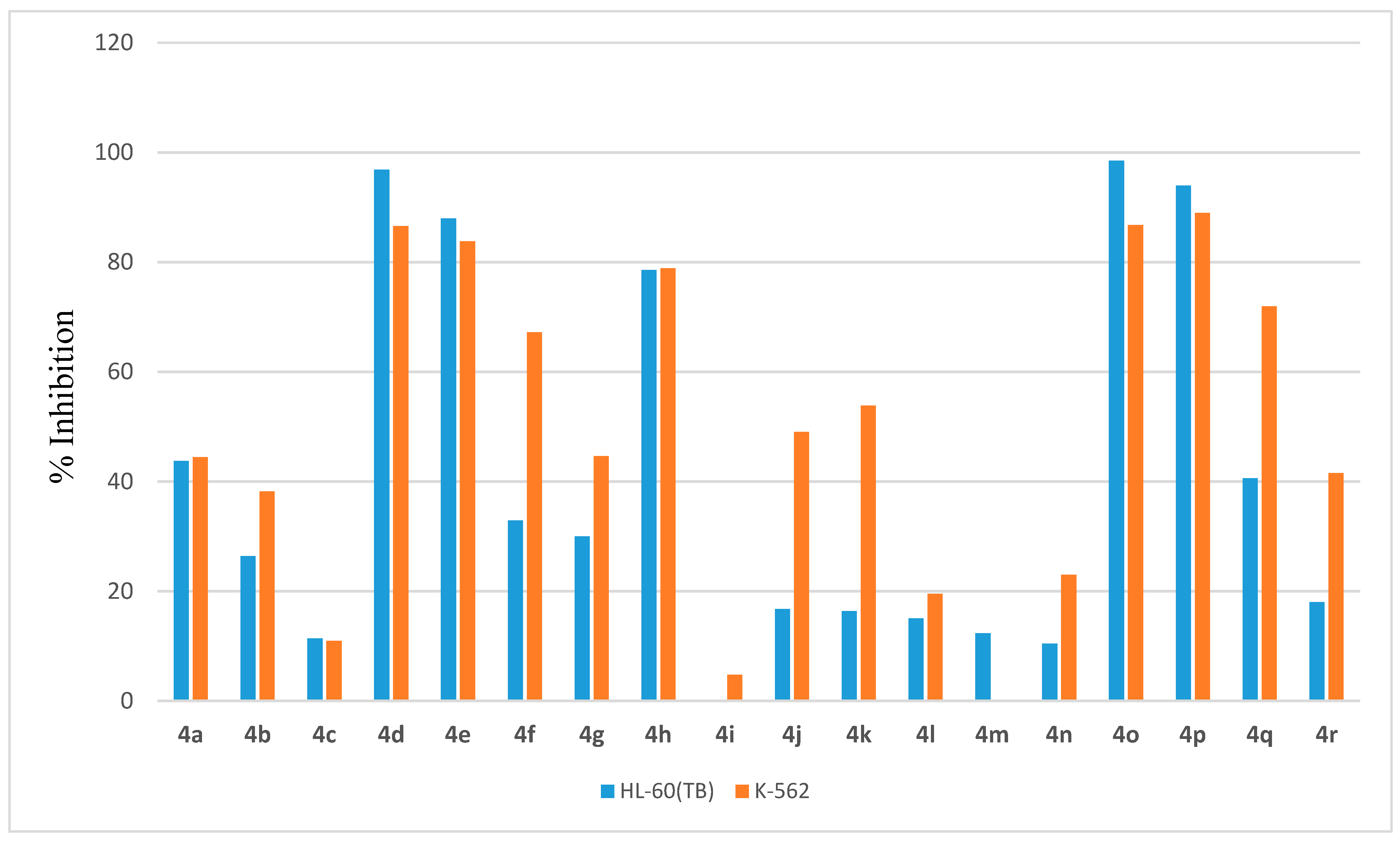
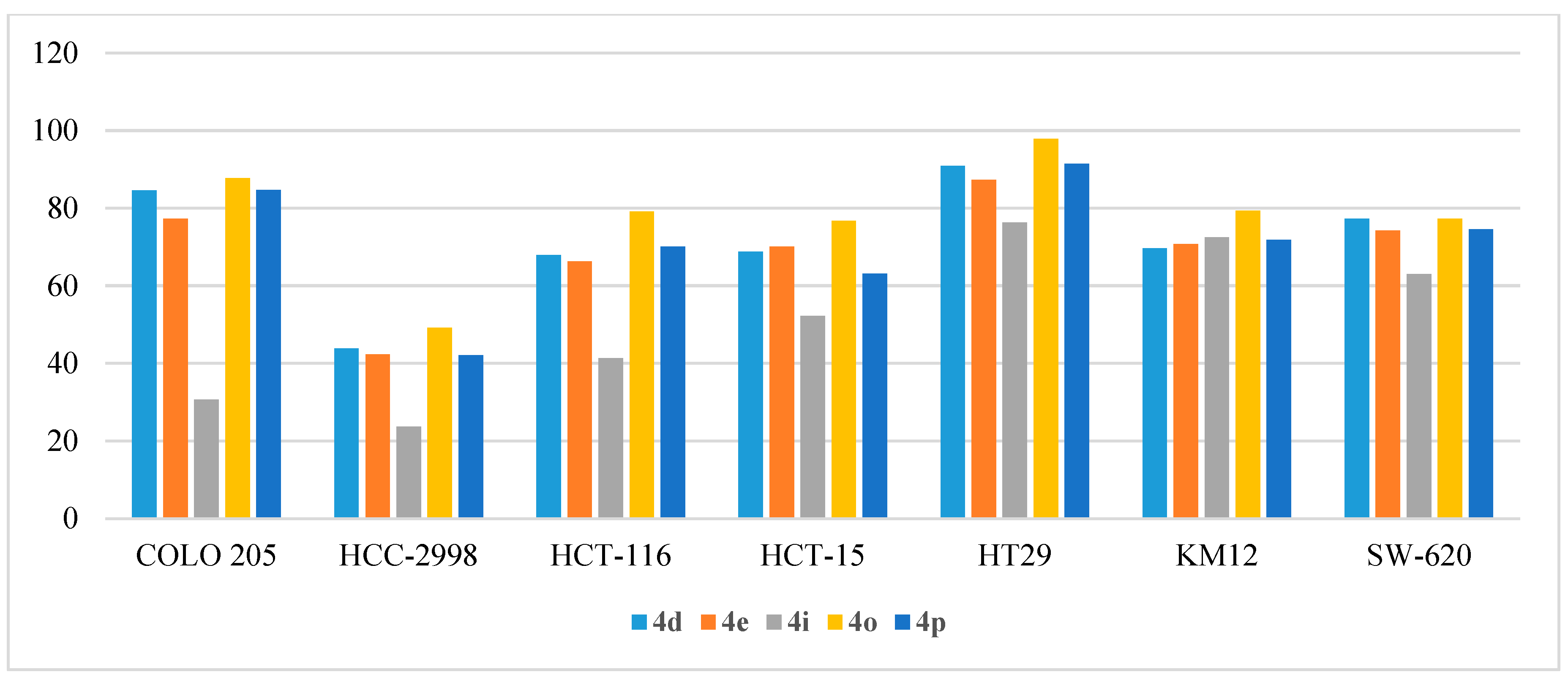
2.2.4. In Vitro Anticancer Assay
- ■
- Anti-proliferative activity against leukemia:
- ■
- Anti-proliferative activity on non-small cell lung cancer:
- ■
- Anti-proliferative activity on colon cancers:
- ■
- Anti-proliferative activity on CNS cancers:
- ■
- Anti-proliferative activity on melanoma:
- ■
- Anti-proliferative activity on ovarian cancers:
- ■
- Anti-proliferative activity on renal cancers:
- ■
- Anti-proliferative activity on prostate cancers:
- ■
- Anti-proliferative activity on breast cancers:
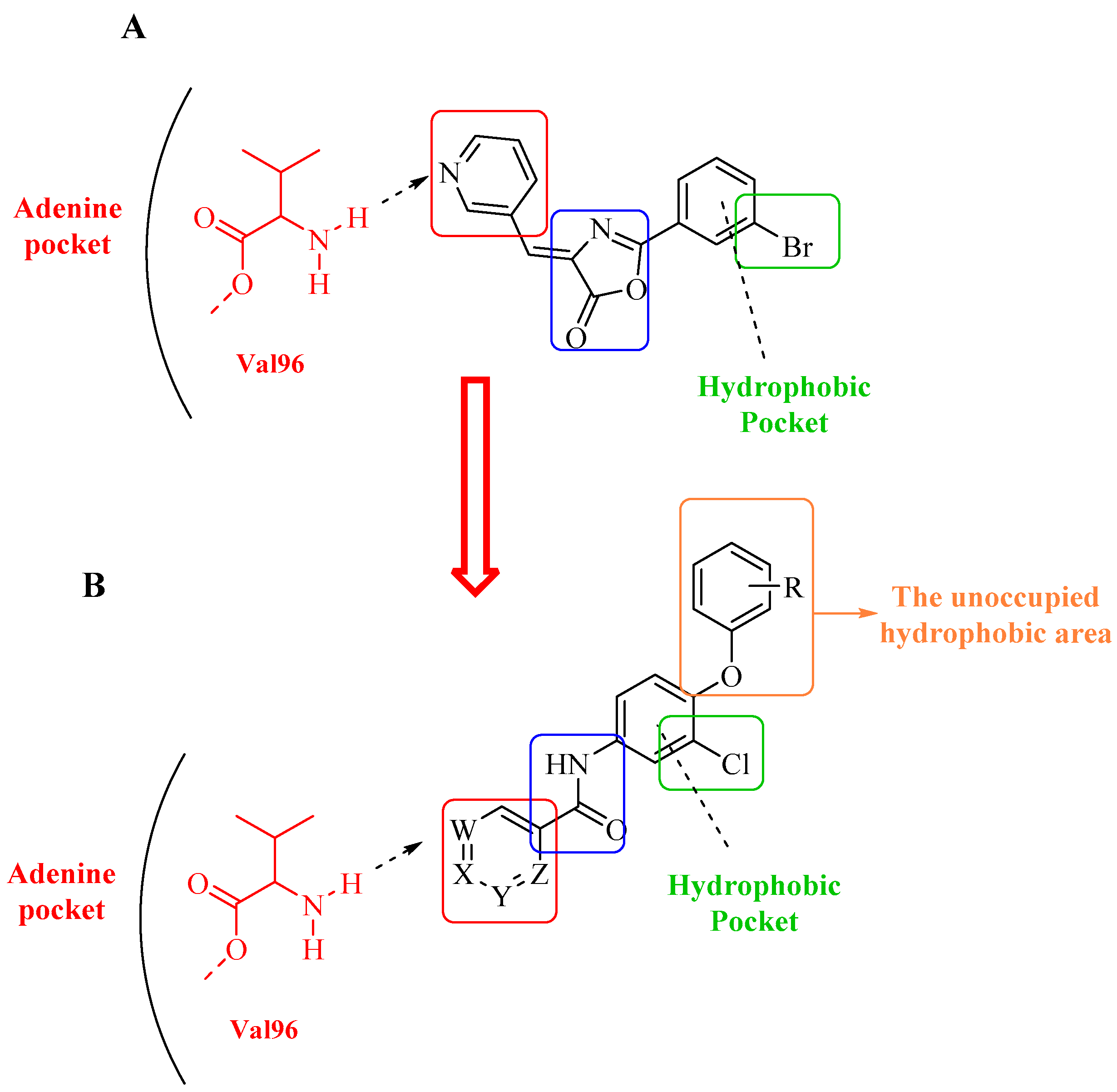
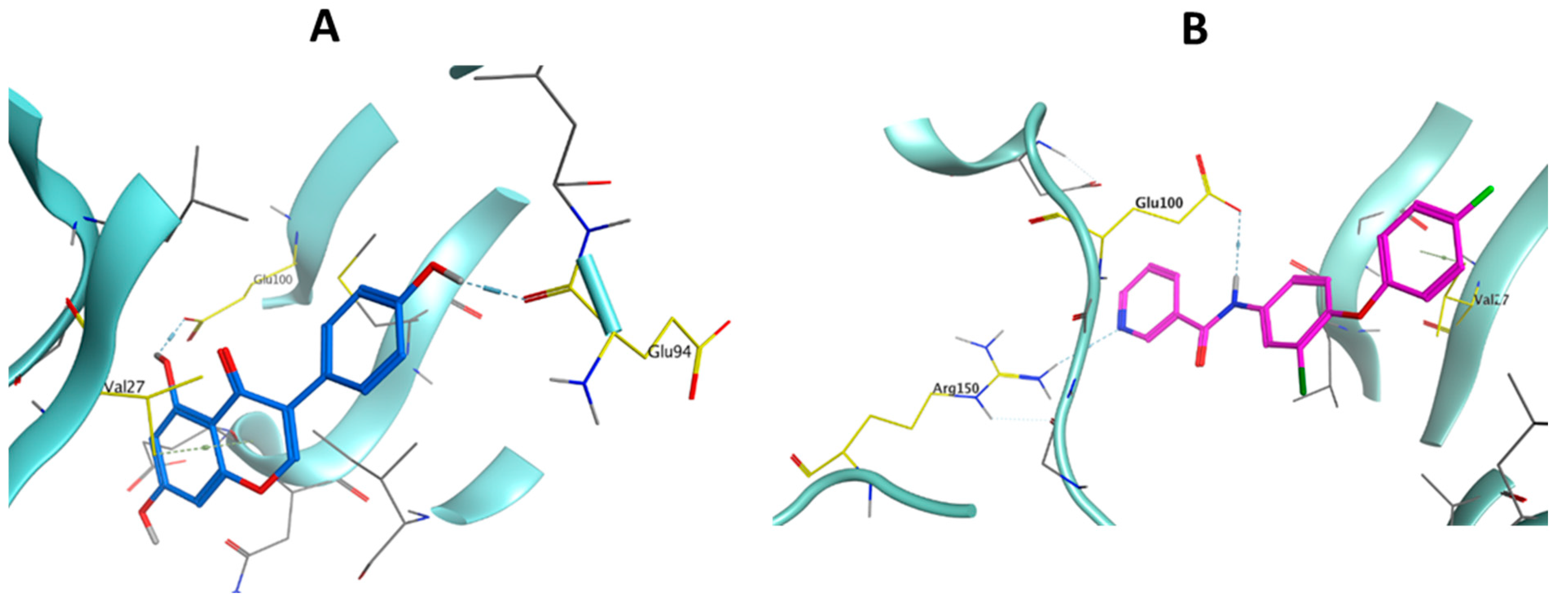
2.2.5. Docking Study
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedure of 2-Chloro-4-nitrophenoxybenzene Derivatives (2a–c)
- 2-Chloro-1-(4-fluorophenoxy)-4-nitrobenzene (2a)
- 2-Chloro-1-(3-fluorophenoxy)-4-nitrobenzene (2b)
- 2-Chloro-1-(3-chlorophenoxy)-4-nitrobenzene (2c)
3.1.2. General Procedure of 3-Choro-4-phenoxyaniline Derivatives (3a–c)
- 3-Chloro-4-(4-fluorophenoxy) aniline (3a)
- 3-Chloro-4-(3-fluorophenoxy) aniline (3b)
- 3-Chloro-4-(3-chlorophenoxy) aniline (3c)
3.1.3. General Procedure of Target Compounds 4a–r
- N-(3-chloro-4-(4-fluorophenoxy) phenyl) picolinamide (4a)
- N-(3-chloro-4-(4-fluorophenoxy) phenyl) pyridazine-3-carboxamide (4b)
- N-(3-chloro-4-(4-fluorophenoxy) phenyl) pyrazine-2-carboxamide (4c)
- N-(3-chloro-4-(4-fluorophenoxy) phenyl) nicotinamide (4d)
- N-(3-chloro-4-(4-fluorophenoxy) phenyl) isonicotinamide (4e)
- N-(3-chloro-4-(4-fluorophenoxy) phenyl) pyridazine-4-carboxamide (4f)
- N-(3-chloro-4-(3-fluorophenoxy) phenyl) picolinamide (4g)
- N-(3-chloro-4-(3-fluorophenoxy) phenyl) pyridazine-3-carboxamide (4h)
- N-(3-chloro-4-(3-fluorophenoxy) phenyl) pyrazine-2-carboxamide (4i)
- N-(3-chloro-4-(3-fluorophenoxy) phenyl) nicotinamide (4j)
- N-(3-chloro-4-(3-fluorophenoxy) phenyl) isonicotinamide (4k)
- N-(3-chloro-4-(3-fluorophenoxy) phenyl) pyridazine-4-carboxamide (4l)
- N-(3-chloro-4-(3-chlorophenoxy) phenyl) picolinamide (4m)
- N-(3-chloro-4-(3-chlorophenoxy) phenyl) pyridazine-3-carboxamide (4n)
- N-(3-chloro-4-(3-chlorophenoxy) phenyl) pyrazine-2-carboxamide (4o)
- N-(3-chloro-4-(3-chlorophenoxy) phenyl) nicotinamide (4p)
- N-(3-chloro-4-(3-chlorophenoxy) phenyl) isonicotinamide (4q)
- N-(3-chloro-4-(3-chlorophenoxy) phenyl) pyridazine-4-carboxamide (4r)
3.2. Biological Evaluation
3.2.1. In Vitro Kinase Screening
3.2.2. NCI Cell Line Screening
3.2.3. Docking Methodology
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Deiss, L.P.; Feinstein, E.; Berissi, H.; Cohen, O.; Kimchi, A. Identification of a novel serine/threonine kinase and a novel 15-kD protein as potential mediators of the gamma interferon-induced cell death. Genes Dev. 1995, 9, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Bialik, S.; Kimchi, A. The death-associated protein kinases: Structure, function, and beyond. Annu. Rev. Biochem. 2006, 75, 189–210. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Ravanan, P.; Talwar, P. Death Associated Protein Kinase 1 (DAPK1): A Regulator of Apoptosis and Autophagy. Front. Mol. Neurosci. 2016, 9, 46. [Google Scholar] [CrossRef] [PubMed]
- Farag Ahmed, K.; Roh Eun, J. Death-associated protein kinase (DAPK) family modulators: Current and future therapeutic outcomes. Med. Res. Rev. 2018, 39, 349–385. [Google Scholar] [CrossRef]
- Pelled, D.; Raveh, T.; Riebeling, C.; Fridkin, M.; Berissi, H.; Futerman, A.H.; Kimch, A. Death-associated protein (DAP) kinase plays a central role in ceramide-induced apoptosis in cultured hippocampal neurons. J. Biol. Chem. 2002, 277, 1957–1961. [Google Scholar] [CrossRef]
- Bialik, S.; Kimchi, A. DAP-kinase as a target for drug design in cancer and diseases associated with accelerated cell death. Semin. Cancer Biol. 2004, 14, 283–294. [Google Scholar] [CrossRef]
- Hainsworth, A.H.; Allsopp, R.C.; Jim, A.; Potter, J.F.; Lowe, J.; Talbot, C.J.; Prettyman, R.J. Death-associated protein kinase (DAPK1) in cerebral cortex of late-onset Alzheimer’s disease patients and aged controls. Neuropathol. Appl. Neurobiol. 2010, 36, 17–24. [Google Scholar] [CrossRef]
- Gade, P.; Manjegowda, S.B.; Nallar, S.C.; Maachani, U.B.; Cross, A.S.; Kalvakolanu, D.V. Regulation of the Death-Associated Protein Kinase 1 Expression and Autophagy via ATF6 Requires Apoptosis Signal-Regulating Kinase 1. Mol. Cell. Biol. 2014, 34, 4033–4048. [Google Scholar] [CrossRef]
- Gozuacik, D.; Bialik, S.; Raveh, T.; Mitou, G.; Shohat, G.; Sabanay, H.; Mizushima, N.; Yoshimori, T.; Kimchi, A. DAP-kinase is a mediator of endoplasmic reticulum stress-induced caspase activation and autophagic cell death. Cell Death Differ. 2008, 15, 1875–1886. [Google Scholar] [CrossRef]
- Shohat, G.; Spivak-Kroizman, T.; Cohen, O.; Bialik, S.; Shani, G.; Berrisi, H.; Eisenstein, M.; Kimchi, A. The Pro-apoptotic Function of Death-associated Protein Kinase Is Controlled by a Unique Inhibitory Autophosphorylation-based Mechanism. J. Biol. Chem. 2001, 276, 47460–47467. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Grupe, A.; Rowland, C.; Nowotny, P.; Kauwe, J.S.K.; Smemo, S.; Hinrichs, A.; Tacey, K.; Toombs, T.A.; Kwok, S.; et al. DAPK1 variants are associated with Alzheimer’s disease and allele-specific expression. Hum. Mol. Genet. 2006, 15, 2560–2568. [Google Scholar] [CrossRef] [PubMed]
- Mor, I.; Carlessi, R.; Ast, T.; Feinstein, E.; Kimchi, A. Death-associated protein kinase increases glycolytic rate through binding and activation of pyruvate kinase. Oncogene 2011, 31, 683. [Google Scholar] [CrossRef]
- Velentza, A.V.; Wainwright, M.S.; Zasadzki, M.; Mirzoev, S.; Schumacher, A.M.; Haiech, J.; Focia, P.J.; Egli, M.; Watterson, D.M. An aminopyridazine-based inhibitor of a pro-apoptotic protein kinase attenuates hypoxia-ischemia induced acute brain injury. Bioorganic Med. Chem. Lett. 2003, 13, 3465–3470. [Google Scholar] [CrossRef]
- Herce, H.D.; Deng, W.; Helma, J.; Leonhardt, H.; Cardoso, M.C. Visualization and targeted disruption of protein interactions in living cells. Nat. Commun. 2013, 4, 2660. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, Y.; Zhang, X.; Zhang, W.; Guo, S.; Jin, F. Recent progress of cell-penetrating peptides as new carriers for intracellular cargo delivery. J. Control. Release 2014, 174, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Watterson, D.M.; Velentza, A.V.; Zasadzki, M.; Craft, J.M.; Haiech, J.; van Eldik, L.J. Discovery of a new class of synthetic protein kinase inhibitors that suppress selective aspects of glial activation and protect against β-amyloid induced injury. J. Mol. Neurosci. 2003, 20, 411–423. [Google Scholar] [CrossRef]
- Mirzoeva, S.; Sawkar, A.; Zasadzki, M.; Guo, L.; Velentza, A.V.; Dunlap, V.; Bourguignon, J.-J.; Ramstrom, H.; Haiech, J.; van Eldik, L.J.; et al. Discovery of a 3-Amino-6-phenyl-pyridazine Derivative as a New Synthetic Antineuroinflammatory Compound. J. Med. Chem. 2002, 45, 563–566. [Google Scholar] [CrossRef]
- Wilbek, T.S.; Skovgaard, T.; Sorrell, F.J.; Knapp, S.; Berthelsen, J.; Strømgaard, K. Identification and characterization of a small-molecule inhibitor of death-associated protein kinase 1. ChemBioChem 2015, 16, 59–63. [Google Scholar] [CrossRef]
- Okamoto, M.; Takayama, K.; Shimizu, T.; Ishida, K.; Takahashi, O.; Furuya, T. Identification of Death-Associated Protein Kinases Inhibitors Using Structure-Based Virtual Screening. J. Med. Chem. 2009, 52, 7323–7327. [Google Scholar] [CrossRef]
- Al-Ghabkari, A.; Deng, J.-T.; McDonald, P.C.; Dedhar, S.; Alshehri, M.; Walsh, M.P.; MacDonald, J.A. A novel inhibitory effect of oxazol-5-one compounds on ROCKII signaling in human coronary artery vascular smooth muscle cells. Sci. Rep. 2016, 6, 32118. [Google Scholar] [CrossRef] [PubMed]
- Carlson, D.A.; Franke, A.S.; Weitzel, D.H.; Speer, B.L.; Hughes, P.F.; Hagerty, L.; Fortner, C.N.; Veal, J.M.; Barta, T.E.; Zieba, B.J.; et al. Fluorescence Linked Enzyme Chemoproteomic Strategy for Discovery of a Potent and Selective DAPK1 and ZIPK Inhibitor. ACS Chem. Biol. 2013, 8, 2715–2723. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, J.A.; Sutherland, C.; Carlson, D.A.; Bhaidani, S.; Al-Ghabkari, A.; Swärd, K.; Haystead, T.A.J.; Walsh, M.P. A Small Molecule Pyrazolo[3,4-d]Pyrimidinone Inhibitor of Zipper-Interacting Protein Kinase Suppresses Calcium Sensitization of Vascular Smooth Muscle. Mol. Pharmacol. 2016, 89, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Irie, T.; Sawa, M. 7-Azaindole: A Versatile Scaffold for Developing Kinase Inhibitors. Chem. Pharm. Bull. 2018, 66, 29–36. [Google Scholar] [CrossRef]
- Okamoto, M.; Takayama, K.; Shimizu, T.; Muroya, A.; Furuya, T. Structure–activity relationship of novel DAPK inhibitors identified by structure-based virtual screening. Bioorganic Med. Chem. 2010, 18, 2728–2734. [Google Scholar] [CrossRef]
- Yang, Z.-X.; Chen, H.B.; Zou, Y.-Z.; Hou, Y.; Wang, T.-P.; Liang, Y. Synthesis of 7-chloro-4-hydroxy-6-(phenoxy)-3-quinolinecarboxylic acid ethyl ester derivatives and determination of their activity as coccidiostats. Youji Huaxue 2008, 28, 432–435. [Google Scholar]
- Marafie, J.A.; Moseley, J.D. The application of stop-flow microwave technology to scaling-out SNAr reactions using a soluble organic base. Org. Biomol. Chem. 2010, 8, 2219–2227. [Google Scholar] [CrossRef]
- Hennequin, L.F.A.; Chen, H.; Zou, Y.; Zhu, J.; Wang, Y. Preparation of Quinazolines as Antitumor Agents; AstraZeneca AB: Södertälje, Sweden; AstraZeneca UK Limited: Cambridge, UK, 2003; p. 218. [Google Scholar]
- Bolea, C. Preparation of Amido Derivatives and Their Use as Positive Allosteric Modulators of Metabotropic Glutamate Receptors; Addex Pharma S.A.: Geneva, Switzerland, 2009; p. 68. [Google Scholar]
- Yang, Z.; Chen, H.; Zou, Y.; Zhu, J.; Wang, Y. Method for Synthesizing 6-aryloxy-7-chloro-4-hydroxyl-3-quinolinecarboxylate. CN 200610022419, 11 July 2007. [Google Scholar]
- Ishikawa, T.; Seto, M.; Banno, H.; Kawakita, Y.; Oorui, M.; Taniguchi, T.; Ohta, Y.; Tamura, T.; Nakayama, A.; Miki, H. Design and Synthesis of Novel Human Epidermal Growth Factor Receptor 2 (HER2)/Epidermal Growth Factor Receptor (EGFR) Dual Inhibitors Bearing a Pyrrolo [3,2-d] pyrimidine Scaffold. J. Med. Chem. 2011, 54, 8030–8050. [Google Scholar] [CrossRef] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
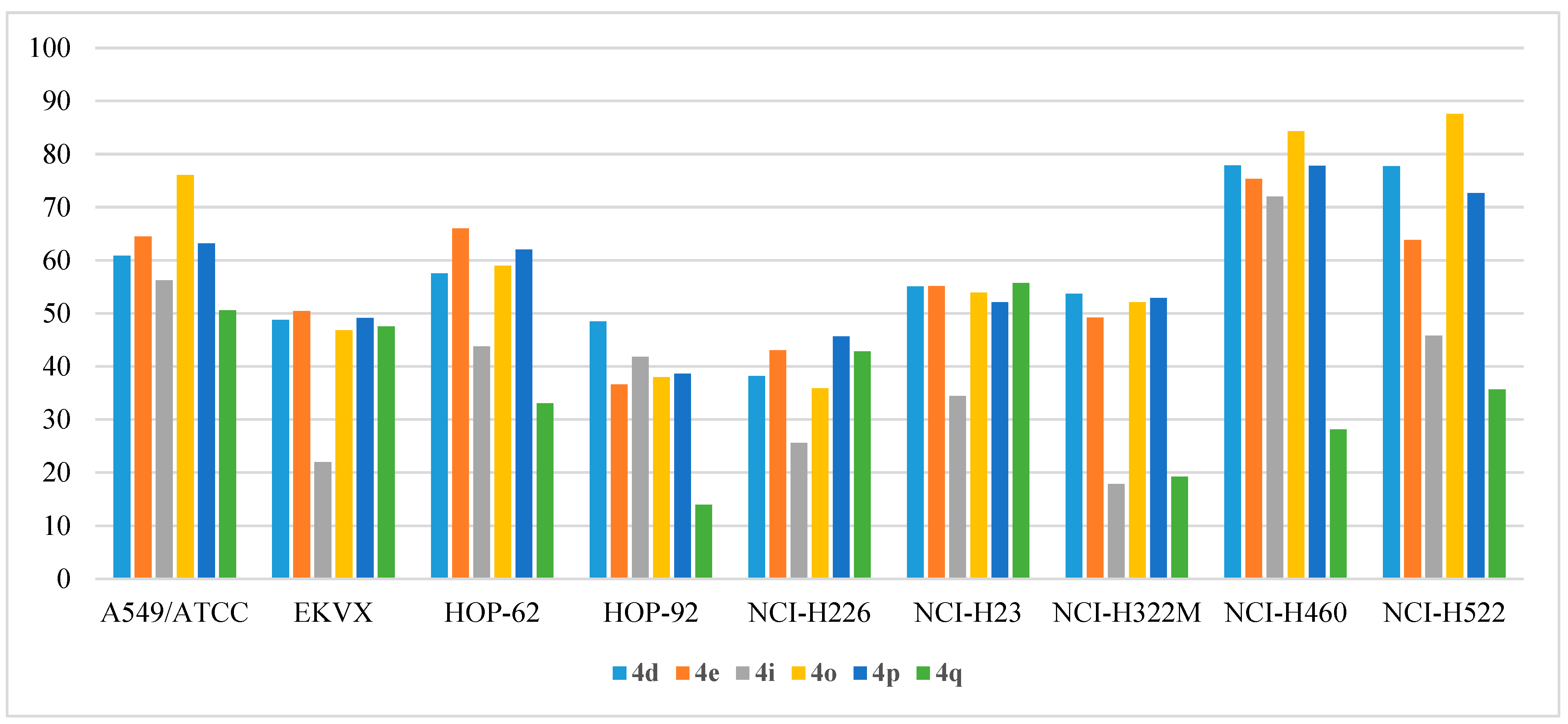{kind=link}
{kind=link}
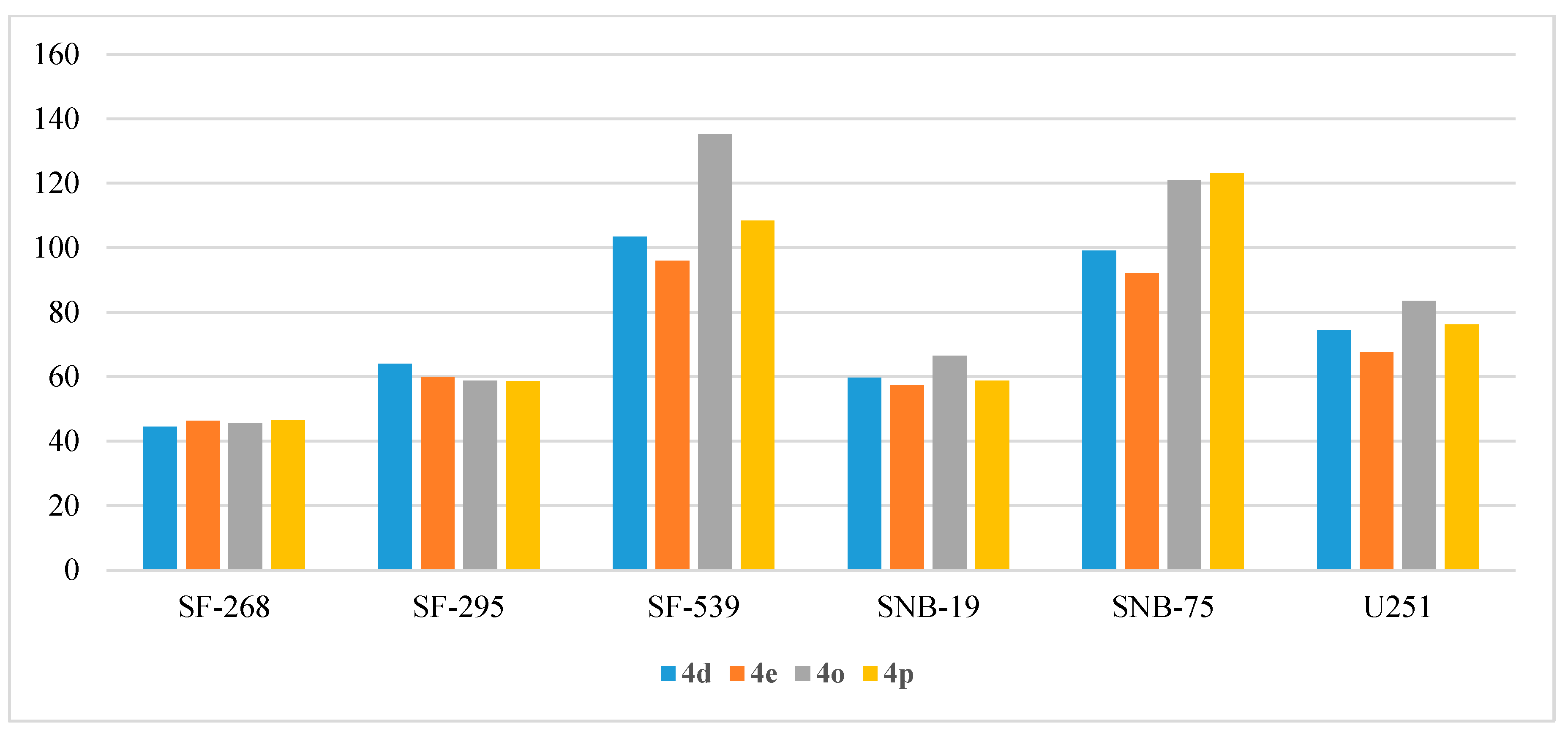{kind=link}
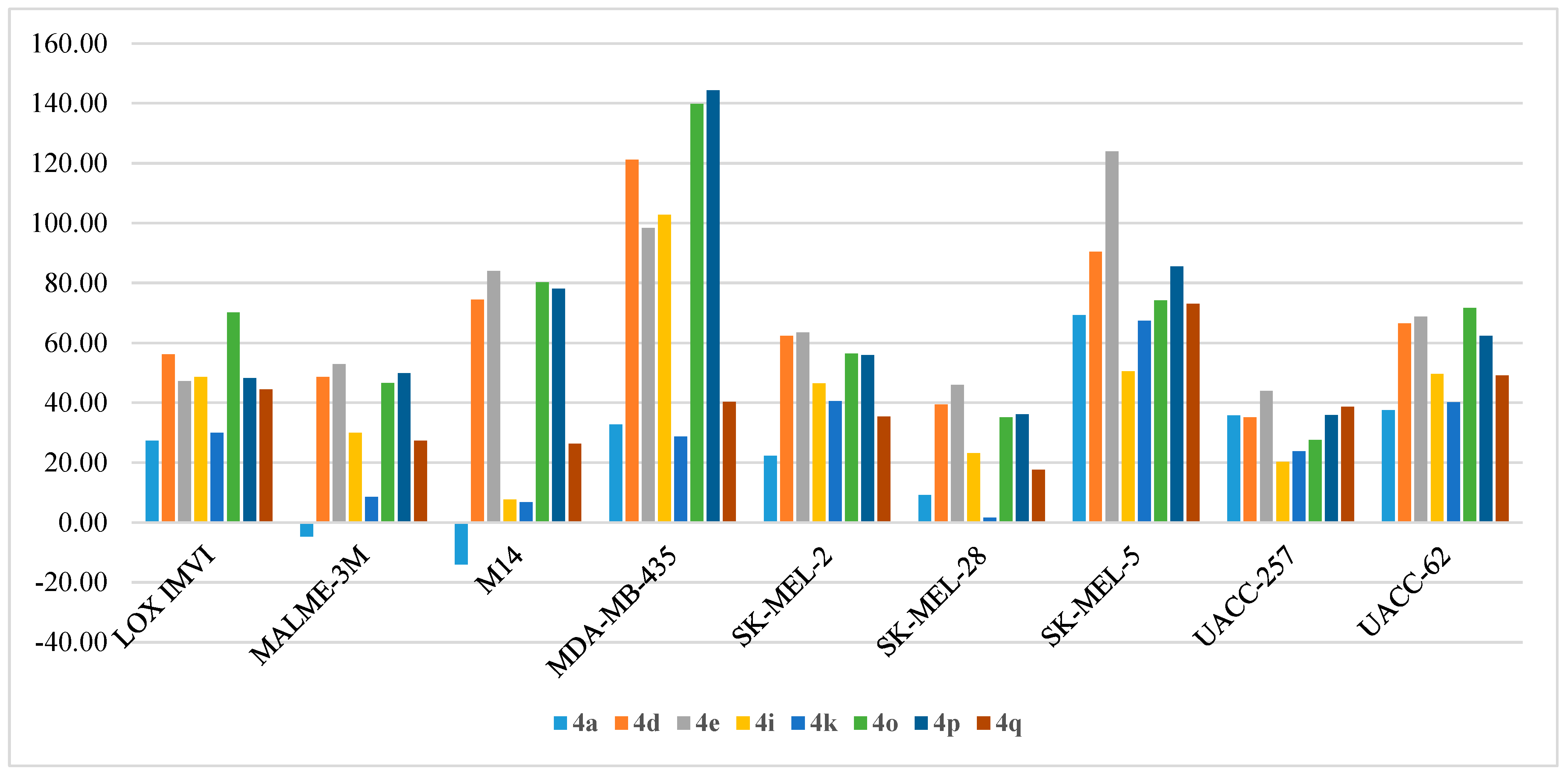{kind=link}
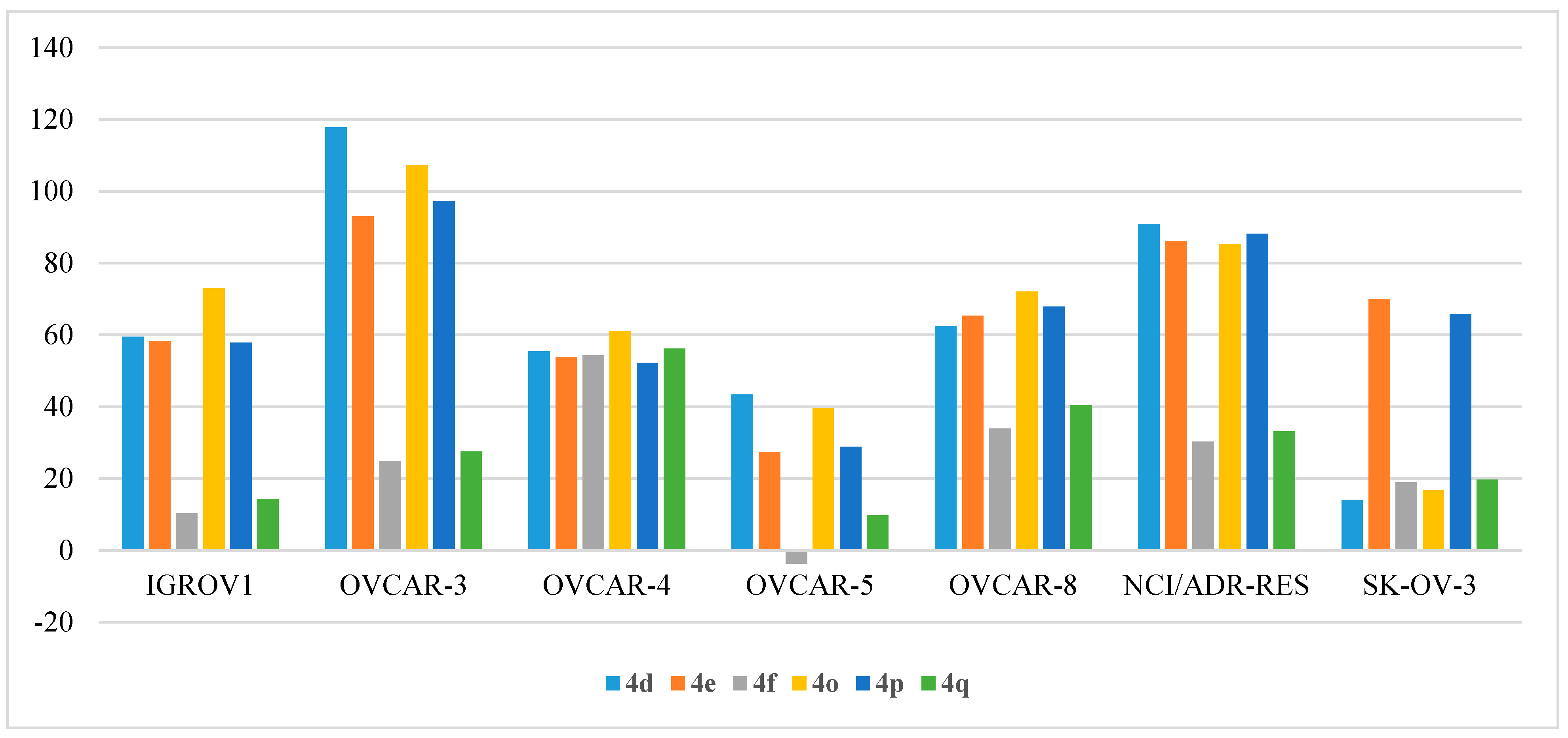{kind=link}
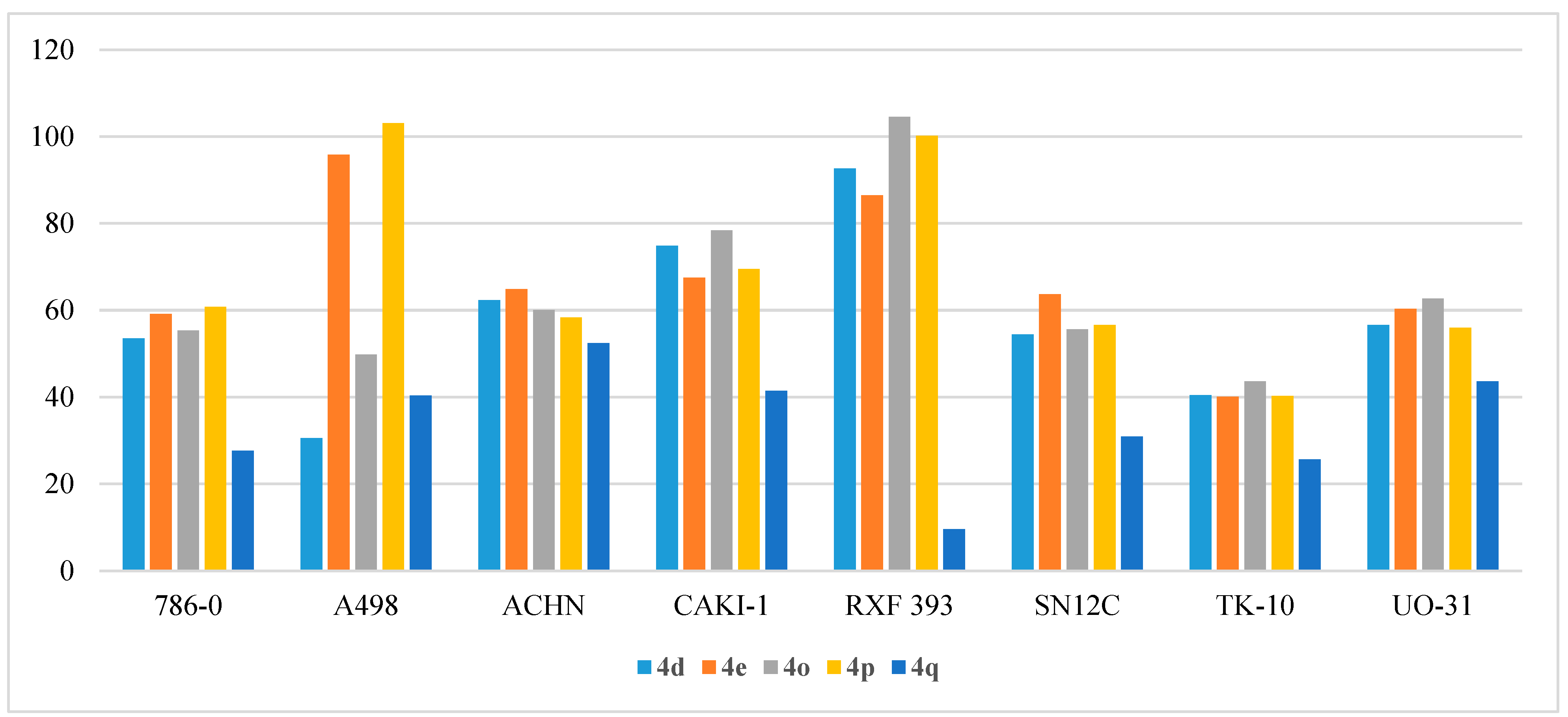{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd | R | Ar | Yield% | Cpd | R | Ar | Yield% |
|---|---|---|---|---|---|---|---|
| 4a | 4-F |  | 73 | 4j | 4-F |  | 70 |
| 4b | 3-F |  | 50 | 4k | 4-F |  | 56 |
| 4c | 3-F |  | 63 | 4l | 4-F |  | 46 |
| 4d | 3-F |  | 59 | 4m | 3-Cl |  | 62 |
| 4e | 3-F |  | 23 | 4n | 3-Cl |  | 72 |
| 4f | 3-F |  | 29 | 4o | 3-Cl |  | 65 |
| 4g | 3-F |  | 62 | 4p | 3-Cl |  | 49 |
| 4h | 4-F |  | 82 | 4q | 3-Cl |  | 12 |
| 4i | 4-F |  | 86 | 4r | 3-Cl |  | 53 |
| Kinase | DAPK1% Inhibition at 10 μM Dose a | Kinase | DAPK1% Inhibition at 10 μM Dose a |
|---|---|---|---|
| ABL1 | 12.29 ± 0.11 | JAK1 | 8.11 ± 1.85 |
| ACK1 | 0.53 ± 3.32 | JNK1 | −9.39 ± 0.22 |
| AKT1 | −6.56 ± 1.56 | KDR/VEGFR2 | −4.28 ± 0.11 |
| AMPK(A1/B1/G1) | 2.10 ± 2.96 | LCK | −15.69 ± 0.45 |
| Aurora A | −5.10 ± 0.78 | LRRK2 | 23.80 ± 1.19 |
| BRAF | −4.67 ± 0.03 | MARK1 | −3.12 ± 0.15 |
| BRAF (V599E) | −9.72 ± 0.04 | MEK1 | −16.18 ± 0.42 |
| BTK | 1.97 ± 0.21 | OSR1/OXSR1 | −6.76 ± 1.93 |
| CAMK1a | −49.53 ± 0.28 | P38a/MAPK14 | −22.83 ± 0.63 |
| CDK1/cyclin A | −8.34 ± 0.93 | PAK1 | 1.12 ± 2.73 |
| CHK1 | −7.21 ± 0.77 | PDGFRa | 19.18 ± 0.68 |
| c-Kit | 15.53 ± 3.14 | RAF1 | 4.51 ± 1.32 |
| CLK1 | −0.14 ± 0.13 | RET | −12.78 ± 0.99 |
| c-MET | 7.03 ± 0.34 | ROCK1 | −6.35 ± 1.35 |
| c-Src | 10.63 ± 1.61 | ROCK2 | 2.96 ± 1.06 |
| DAPK1 | 44.19 ± 1.95 | ROS/ROS1 | −7.04 ± 1.71 |
| DDR1 | 5.76 ± 1.69 | STK39/STLK3 | −2.47 ± 1.50 |
| ERBB2/HER2 | −7.42 ± 1.74 | SYK | −13.26 ± 0.10 |
| FGFR1 | −14.24 ± 0.24 | TAK1 | −4.08 ± 1.16 |
| FLT3 | 0.13 ± 1.08 | TIE2/TEK | −13.18 ± 2.15 |
| FMS | 3.36 ± 0.31 | TLK1 | −2.23 ± 3.33 |
| IGF1R | −12.67 ± 0.72 | TRKA | −8.71 ± 0.17 |
| IKKb/IKBKB | −15.92 ± 0.74 |
| Cpd | % Inhibition against DAPK1 a | Cpd | % Inhibition against DAPK1 a |
|---|---|---|---|
| 4a | 44.19 | 4j | 79.06 |
| 4b | 58.95 | 4k | 80.61 |
| 4c | 58.83 | 4l | 61.83 |
| 4d | 50.91 | 4m | 61.73 |
| 4e | 59.98 | 4n | 61.54 |
| 4f | 64.06 | 4o | 37.99 |
| 4g | 50.18 | 4p | 61.48 |
| 4h | 81.00 | 4q | 72.10 |
| 4i | 63.69 | 4r | 59.98 |
| Cpd | DAPK1 a, IC50 (μM) |
|---|---|
| 4h | 6.81 |
| 4j | 1.70 |
| 4k | 7.26 |
| 4q | 1.09 |
| Cpd. | CCRF-CEM | HL-60(TB) | K-562 | MOLT-4 | RPMI-8226 | SR |
|---|---|---|---|---|---|---|
| 4a | 24.87 | 43.74 | 44.47 | 40.16 | 37.38 | 2.35 |
| 4b | 24.49 | 26.45 | 38.23 | 19.72 | 42.72 | −6.10 |
| 4c | 6.71 | 11.44 | 10.98 | 6.93 | 21.08 | −4.05 |
| 4d | 81.59 | 96.88 | 86.57 | 58.79 | 82.05 | 79.23 |
| 4e | 76.04 | 87.94 | 83.79 | 59.83 | 84.21 | 76.25 |
| 4f | 41.83 | 32.93 | 67.22 | 27.56 | 54.45 | 36.39 |
| 4g | 40.77 | 30.04 | 44.68 | 24.07 | 55.39 | 8.58 |
| 4h | −5.45 | −5.22 | 4.80 | −24.34 | −1.85 | −26.54 |
| 4i | 35.87 | 78.57 | 78.88 | 54.90 | 19.59 | 50.71 |
| 4j | −0.75 | 16.79 | 49.09 | -5.20 | 12.69 | 24.85 |
| 4k | 21.90 | 16.39 | 53.90 | 5.43 | 34.21 | 10.68 |
| 4l | 21.39 | 15.06 | 19.57 | 12.50 | 33.55 | −2.96 |
| 4m | −0.92 | 12.35 | −7.67 | −12.52 | 0.39 | −26.02 |
| 4n | 16.03 | 10.46 | 23.01 | 19.89 | 28.89 | 13.01 |
| 4o | 83.35 | 98.50 | 86.78 | 73.85 | 81.94 | 73.68 |
| 4p | 82.54 | 93.94 | 88.94 | 62.59 | 88.07 | 80.15 |
| 4q | 43.23 | 40.60 | 71.93 | 32.16 | 59.20 | 18.60 |
| 4r | 34.26 | 18.01 | 41.54 | 14.16 | 46.55 | −11.83 |
| Cpd | A549/ ATCC | EKVX | HOP-62 | HOP-92 | NCI-H226 | NCI-H23 | NCI-H322M | NCI-H460 | NCI-H522 |
|---|---|---|---|---|---|---|---|---|---|
| 4a | 34.51 | 25.04 | 15.76 | 33.54 | 45.00 | 37.68 | 12.42 | 23.21 | 38.20 |
| 4b | 32.62 | 25.29 | 0.90 | 17.89 | 30.57 | 23.57 | 6.18 | 3.78 | 27.10 |
| 4c | 18.95 | 10.63 | −7.22 | 15.05 | 1.09 | 6.75 | 2.94 | 0.03 | 17.87 |
| 4d | 60.88 | 48.75 | 57.56 | 48.48 | 38.23 | 55.06 | 53.70 | 77.90 | 77.71 |
| 4e | 64.48 | 50.41 | 65.97 | 36.59 | 43.08 | 55.14 | 49.18 | 75.35 | 63.79 |
| 4f | 44.90 | 46.82 | 31.88 | 12.60 | 37.44 | 49.29 | 10.81 | 22.18 | 24.20 |
| 4g | 36.08 | 35.47 | 10.18 | NT | 30.80 | 33.47 | −0.14 | 11.06 | 22.76 |
| 4h | 1.70 | 13.06 | −8.34 | 7.68 | 5.54 | 14.36 | 1.66 | 0.15 | 20.06 |
| 4i | 56.25 | 21.95 | 43.80 | 41.84 | 25.60 | 34.41 | 17.83 | 71.99 | 45.79 |
| 4j | 14.04 | 18.08 | −6.37 | 8.10 | 0.24 | 20.04 | −0.70 | 3.01 | 27.70 |
| 4k | 12.58 | 60.37 | 29.52 | 20.18 | 17.62 | 51.60 | 1.00 | 18.70 | 37.11 |
| 4l | 24.02 | 19.22 | 1.57 | 16.91 | 26.86 | 25.71 | 7.09 | 5.29 | 20.81 |
| 4m | 8.08 | −0.64 | −6.77 | 3.98 | −8.86 | −5.18 | −5.68 | −0.87 | 5.22 |
| 4n | 24.70 | 12.67 | −0.67 | 15.62 | 21.16 | 15.91 | 6.13 | 0.21 | 26.35 |
| 4o | 76.05 | 46.82 | 59.01 | 37.99 | 35.91 | 53.91 | 52.07 | 84.28 | 87.52 |
| 4p | 63.21 | 49.14 | 62.02 | 38.66 | 45.68 | 52.09 | 52.92 | 77.80 | 72.66 |
| 4q | 50.61 | 47.56 | 33.07 | 13.96 | 42.84 | 55.74 | 19.25 | 28.11 | 35.65 |
| 4r | 32.61 | 28.53 | 6.68 | 5.58 | 29.92 | 32.81 | 1.73 | 7.58 | 27.65 |
| Cpd | COLO 205 | HCC-2998 | HCT-116 | HCT-15 | HT29 | KM12 | SW-620 |
|---|---|---|---|---|---|---|---|
| 4a | −11.74 | 10.88 | 3.23 | 22.43 | 23.27 | 20.85 | 5.86 |
| 4b | −0.78 | 3.23 | 44.58 | 48.54 | 13.03 | 17.64 | −3.69 |
| 4c | −10.09 | 7.89 | 14.49 | 24.71 | 15.35 | 4.65 | −2.86 |
| 4d | 84.52 | 43.81 | 67.97 | 68.78 | 90.87 | 69.66 | 77.31 |
| 4e | 77.29 | 42.26 | 66.25 | 70.11 | 87.31 | 70.73 | 74.22 |
| 4f | 9.29 | −6.85 | 52.29 | 19.16 | 3.92 | 29.08 | 15.56 |
| 4g | 1.75 | −5.80 | 21.38 | 20.12 | 0.43 | 25.11 | 1.33 |
| 4h | −24.87 | 6.42 | 8.49 | 3.76 | −3.35 | 1.13 | 5.68 |
| 4i | 30.70 | 23.68 | 41.31 | 52.26 | 76.34 | 72.49 | 63.04 |
| 4j | −20.33 | 8.79 | 11.85 | 15.67 | 11.48 | 23.60 | 7.30 |
| 4k | 4.83 | 27.39 | 56.60 | 13.33 | 1.72 | 22.04 | 14.96 |
| 4l | −2.63 | 3.36 | 6.44 | 9.12 | 12.54 | 10.52 | −5.05 |
| 4m | −12.61 | −17.45 | −3.79 | −0.91 | −2.08 | −1.10 | −0.09 |
| 4n | −5.90 | 4.76 | 33.28 | 43.26 | 10.11 | 14.71 | −4.84 |
| 4o | 87.71 | 49.22 | 79.08 | 76.69 | 97.86 | 79.27 | 77.28 |
| 4p | 84.61 | 42.04 | 70.09 | 63.16 | 91.43 | 71.89 | 74.59 |
| 4q | 17.91 | −0.82 | 55.73 | 24.37 | 15.41 | 32.88 | 23.45 |
| 4r | −1.57 | −10.77 | 15.72 | 17.28 | 3.02 | 21.25 | −0.48 |
| Cpd | SF-268 | SF-295 | SF-539 | SNB-19 | SNB-75 | U251 |
|---|---|---|---|---|---|---|
| 4a | 19.64 | 26.63 | 14.50 | 33.85 | 12.24 | 37.09 |
| 4b | 15.24 | 19.22 | 3.77 | 13.63 | 19.94 | 30.61 |
| 4c | 3.67 | 6.83 | −4.47 | 8.03 | 20.26 | 21.79 |
| 4d | 44.50 | 63.96 | 103.46 | 59.62 | 99.15 | 74.34 |
| 4e | 46.35 | 59.88 | 95.94 | 57.35 | 92.15 | 67.54 |
| 4f | 15.83 | 59.58 | 17.49 | 32.89 | 10.07 | 27.52 |
| 4g | 18.94 | 22.54 | 7.93 | 18.58 | 14.60 | 21.98 |
| 4h | −1.32 | 8.92 | 4.25 | 8.73 | NT | 12.52 |
| 4i | 27.80 | 27.12 | 32.90 | 36.26 | 52.28 | 61.85 |
| 4j | 5.00 | 14.00 | 5.58 | 6.79 | NT | 10.97 |
| 4k | 16.97 | 58.92 | 18.58 | 26.78 | NT | 14.64 |
| 4l | 18.14 | 14.27 | 4.69 | 11.91 | 28.81 | 15.77 |
| 4m | −1.72 | 5.99 | −3.28 | 4.33 | 12.89 | 9.88 |
| 4n | 16.12 | 17.78 | 4.69 | 10.47 | 22.28 | 23.31 |
| 4o | 45.68 | 58.77 | 135.24 | 66.47 | 120.94 | 83.53 |
| 4p | 46.58 | 58.61 | 108.37 | 58.71 | 123.19 | 76.10 |
| 4q | 20.45 | 67.42 | 24.18 | 41.32 | 18.27 | 37.68 |
| 4r | 17.40 | 27.73 | 4.39 | 18.63 | 12.21 | 19.46 |
| Cpd | LOX IMVI | MALME-3M | M14 | MDA-MB-435 | SK-MEL-2 | SK-MEL-28 | SK-MEL-5 | UACC-257 | UACC-62 |
|---|---|---|---|---|---|---|---|---|---|
| 4a | 27.24 | −4.65 | −14.00 | 32.75 | 22.30 | 9.15 | 69.25 | 35.74 | 37.47 |
| 4b | 25.22 | 7.97 | 7.10 | 19.00 | 8.32 | 1.43 | 32.67 | 7.10 | 31.60 |
| 4c | 2.62 | 2.27 | −1.30 | 8.65 | 5.50 | −0.68 | 9.49 | −9.00 | 16.27 |
| 4d | 56.13 | 48.64 | 74.39 | 121.17 | 62.33 | 39.39 | 90.39 | 35.06 | 66.46 |
| 4e | 47.19 | 52.84 | 84.06 | 98.36 | 63.46 | 45.96 | 123.95 | 43.92 | 68.80 |
| 4f | 38.68 | 16.33 | 15.91 | 33.96 | 30.20 | 13.40 | 58.84 | 41.05 | 48.32 |
| 4g | 18.38 | −3.55 | 11.13 | 23.66 | 14.50 | 2.85 | 41.79 | 19.72 | 36.67 |
| 4h | 3.90 | −1.10 | −5.94 | 5.26 | −4.51 | −14.37 | 2.21 | −12.41 | 12.42 |
| 4i | 48.55 | 29.89 | 7.62 | 102.80 | 46.40 | 23.13 | 50.48 | 20.25 | 49.67 |
| 4j | 8.76 | −2.94 | 2.72 | 58.51 | 8.69 | −2.54 | 20.48 | 8.59 | 18.73 |
| 4k | 29.89 | 8.49 | 6.80 | 28.75 | 40.51 | 1.54 | 67.41 | 23.72 | 40.20 |
| 4l | 10.38 | −2.67 | 5.36 | 10.47 | 2.21 | −0.94 | 21.76 | 6.33 | 25.61 |
| 4m | -2.57 | −9.79 | −4.28 | 6.16 | −7.38 | 0.87 | −3.94 | −4.90 | 6.68 |
| 4n | 11.48 | −3.78 | 7.23 | 22.44 | −2.18 | 0.55 | 16.20 | −3.76 | 25.33 |
| 4o | 70.14 | 46.63 | 80.21 | 139.73 | 56.37 | 35.07 | 74.19 | 27.57 | 71.64 |
| 4p | 48.29 | 49.87 | 78.15 | 144.28 | 55.91 | 36.16 | 85.47 | 35.81 | 62.37 |
| 4q | 44.43 | 27.35 | 26.33 | 40.27 | 35.39 | 17.55 | 73.04 | 38.68 | 49.14 |
| 4r | 14.41 | 2.90 | 15.11 | 21.71 | 14.60 | 5.10 | 29.20 | 16.23 | 33.19 |
| Cpd | IGROV1 | OVCAR-3 | OVCAR-4 | OVCAR-5 | OVCAR-8 | NCI/ADR-RES | SK-OV-3 |
|---|---|---|---|---|---|---|---|
| 4a | 2.39 | 26.40 | NT | 15.95 | 28.75 | 30.57 | 11.16 |
| 4b | 7.95 | 21.21 | 38.86 | 3.48 | 6.93 | 7.85 | −3.63 |
| 4c | 5.29 | 5.30 | 21.99 | 2.65 | −2.78 | −3.02 | −7.73 |
| 4d | 59.49 | 117.85 | 55.46 | 43.39 | 62.49 | 90.94 | 14.03 |
| 4e | 58.24 | 93.03 | 53.83 | 27.40 | 65.37 | 86.14 | 69.98 |
| 4f | 10.32 | 24.90 | 54.30 | −3.67 | 33.96 | 30.30 | 18.97 |
| 4g | −0.70 | 18.15 | 26.00 | −8.04 | 11.94 | 8.76 | −1.53 |
| 4h | 1.79 | −2.65 | 11.49 | −5.31 | −5.64 | 11.70 | −14.73 |
| 4i | 37.38 | 56.36 | NT | 18.94 | 28.56 | 59.16 | −0.47 |
| 4j | 4.59 | 14.21 | 14.85 | 0.07 | 1.40 | 14.87 | −10.36 |
| 4k | 7.73 | 31.85 | 22.92 | 7.00 | 27.11 | 36.38 | 19.08 |
| 4l | 3.35 | 12.54 | 24.55 | −2.71 | 9.85 | 7.17 | 3.83 |
| 4m | −4.42 | −2.87 | 1.25 | 6.70 | −3.01 | −14.69 | −3.50 |
| 4n | 8.57 | 14.44 | 31.35 | 8.86 | 8.33 | 15.03 | −2.30 |
| 4o | 72.96 | 107.23 | 61.05 | 39.64 | 72.05 | 85.16 | 16.73 |
| 4p | 57.85 | 97.34 | 52.16 | 28.80 | 67.92 | 88.11 | 65.75 |
| 4q | 14.33 | 27.51 | 56.18 | 9.72 | 40.42 | 33.17 | 19.67 |
| 4r | 7.86 | 14.64 | 31.04 | −5.97 | 10.63 | 4.76 | 0.32 |
| Cpd | 786-0 | A498 | ACHN | CAKI-1 | RXF 393 | SN12C | TK-10 | UO-31 |
|---|---|---|---|---|---|---|---|---|
| 4a | −5.04 | 38.95 | 26.30 | 45.20 | 17.36 | 36.18 | 35.56 | 45.01 |
| 4b | 23.45 | 21.77 | 14.60 | 24.59 | 20.89 | 15.74 | 25.76 | 33.84 |
| 4c | 21.26 | 10.29 | 2.04 | 14.58 | −4.88 | 7.62 | 22.97 | 17.68 |
| 4d | 53.47 | 30.57 | 62.30 | 74.84 | 92.63 | 54.42 | 40.46 | 56.60 |
| 4e | 59.13 | 95.84 | 64.82 | 67.45 | 86.46 | 63.63 | 40.07 | 60.28 |
| 4f | 23.38 | 41.23 | 44.98 | 33.92 | −6.80 | 18.18 | 22.50 | 31.83 |
| 4g | 15.44 | 24.78 | 31.68 | 37.07 | 0.38 | 18.44 | 18.94 | 31.99 |
| 4h | 5.85 | 22.23 | −2.59 | 15.45 | −0.44 | −4.70 | 1.08 | 22.52 |
| 4i | 18.95 | 32.33 | 5.89 | 50.35 | 27.74 | 30.22 | 14.22 | 34.97 |
| 4j | 7.73 | 29.16 | 0.47 | 13.15 | 9.82 | 4.16 | 2.30 | 19.09 |
| 4k | 16.26 | 56.91 | 25.12 | 30.56 | 8.16 | 16.37 | 15.85 | 28.68 |
| 4l | 13.93 | 19.59 | 12.22 | 22.93 | −11.93 | 14.96 | 18.03 | 40.38 |
| 4m | 9.93 | 5.35 | −1.09 | 7.21 | −11.85 | 2.65 | 7.95 | 12.70 |
| 4n | 22.57 | 12.65 | 2.11 | 19.24 | 9.70 | 16.79 | 24.95 | 35.06 |
| 4o | 55.36 | 49.81 | 60.04 | 78.37 | 104.58 | 55.60 | 43.63 | 62.66 |
| 4p | 60.79 | 103.10 | 58.34 | 69.44 | 100.14 | 56.56 | 40.23 | 55.92 |
| 4q | 27.67 | 40.37 | 52.45 | 41.45 | 9.56 | 30.90 | 25.70 | 43.59 |
| 4r | 14.13 | 27.83 | 26.38 | 33.64 | −20.50 | 16.91 | 17.18 | 35.82 |
| Cpd | PC-3 | DU-145 |
|---|---|---|
| 4a | 35.59 | 23.29 |
| 4b | 47.62 | 17.49 |
| 4c | 33.62 | 8.58 |
| 4d | 83.31 | 80.87 |
| 4e | 75.41 | 78.17 |
| 4f | 20.74 | 9.42 |
| 4g | 28.39 | 16.44 |
| 4h | 2.71 | 3.23 |
| 4i | 23.96 | 20.47 |
| 4j | 9.30 | 12.96 |
| 4k | 24.42 | 22.70 |
| 4l | 13.07 | 5.67 |
| 4m | 9.03 | −3.53 |
| 4n | 42.83 | 13.68 |
| 4o | 88.90 | 77.35 |
| 4p | 85.15 | 83.88 |
| 4q | 25.02 | 22.55 |
| 4r | 19.59 | 15.68 |
| Cpd | MCF7 | MDA-MB-231/ATCC | HS 578T | BT-549 | T-47D | MDA-MB-468 |
|---|---|---|---|---|---|---|
| 4a | 34.71 | 11.88 | 4.36 | −3.11 | 62.39 | 60.73 |
| 4b | 12.80 | 6.93 | −4.53 | 15.34 | 42.13 | 41.58 |
| 4c | 11.41 | −0.05 | 1.61 | 7.71 | 23.18 | 14.77 |
| 4d | 82.54 | 46.50 | 72.17 | 40.15 | 61.71 | 99.92 |
| 4e | 80.44 | 39.78 | 68.75 | 51.44 | 66.51 | 109.22 |
| 4f | 44.11 | 14.71 | 7.30 | 40.61 | 46.75 | 76.00 |
| 4g | 18.46 | 1.84 | 8.39 | 8.12 | 39.23 | 55.16 |
| 4h | 13.86 | 0.09 | −0.49 | 2.31 | 27.84 | 20.32 |
| 4i | 70.32 | 41.29 | 37.62 | −0.40 | 57.08 | 68.17 |
| 4j | 23.32 | 6.34 | 8.51 | 7.52 | 29.40 | 42.77 |
| 4k | 39.38 | 19.70 | 6.71 | 36.00 | 56.60 | 78.17 |
| 4l | 9.28 | 9.19 | −7.24 | 5.75 | 30.08 | 36.63 |
| 4m | 3.69 | −2.42 | 3.47 | 1.02 | −4.39 | −2.35 |
| 4n | 8.35 | 4.49 | −6.22 | 1.47 | 27.64 | 36.69 |
| 4o | 79.69 | 68.29 | 81.27 | 42.55 | 59.27 | 99.57 |
| 4p | 78.44 | 41.02 | 68.95 | 46.72 | 64.41 | 109.21 |
| 4q | 56.26 | 20.80 | 10.84 | 40.15 | 51.56 | 74.71 |
| 4r | 14.03 | 5.90 | 2.82 | 1.87 | 38.96 | 52.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elkamhawy, A.; Paik, S.; Ali, E.M.H.; Hassan, A.H.E.; Kang, S.J.; Lee, K.; Roh, E.J. Identification of Novel Aryl Carboxamide Derivatives as Death-Associated Protein Kinase 1 (DAPK1) Inhibitors with Anti-Proliferative Activities: Design, Synthesis, In Vitro, and In Silico Biological Studies. Pharmaceuticals 2022, 15, 1050. https://doi.org/10.3390/ph15091050
Elkamhawy A, Paik S, Ali EMH, Hassan AHE, Kang SJ, Lee K, Roh EJ. Identification of Novel Aryl Carboxamide Derivatives as Death-Associated Protein Kinase 1 (DAPK1) Inhibitors with Anti-Proliferative Activities: Design, Synthesis, In Vitro, and In Silico Biological Studies. Pharmaceuticals. 2022; 15(9):1050. https://doi.org/10.3390/ph15091050
Chicago/Turabian StyleElkamhawy, Ahmed, Sora Paik, Eslam M. H. Ali, Ahmed H. E. Hassan, So Jin Kang, Kyeong Lee, and Eun Joo Roh. 2022. "Identification of Novel Aryl Carboxamide Derivatives as Death-Associated Protein Kinase 1 (DAPK1) Inhibitors with Anti-Proliferative Activities: Design, Synthesis, In Vitro, and In Silico Biological Studies" Pharmaceuticals 15, no. 9: 1050. https://doi.org/10.3390/ph15091050
APA StyleElkamhawy, A., Paik, S., Ali, E. M. H., Hassan, A. H. E., Kang, S. J., Lee, K., & Roh, E. J. (2022). Identification of Novel Aryl Carboxamide Derivatives as Death-Associated Protein Kinase 1 (DAPK1) Inhibitors with Anti-Proliferative Activities: Design, Synthesis, In Vitro, and In Silico Biological Studies. Pharmaceuticals, 15(9), 1050. https://doi.org/10.3390/ph15091050