
Design, Synthesis and Biological Investigation of 2-Anilino Triazolopyrimidines as Tubulin Polymerization Inhibitors with Anticancer Activities

 ,
,  , , , , ,
, , , , ,  and
and 
Abstract
:1. Introduction
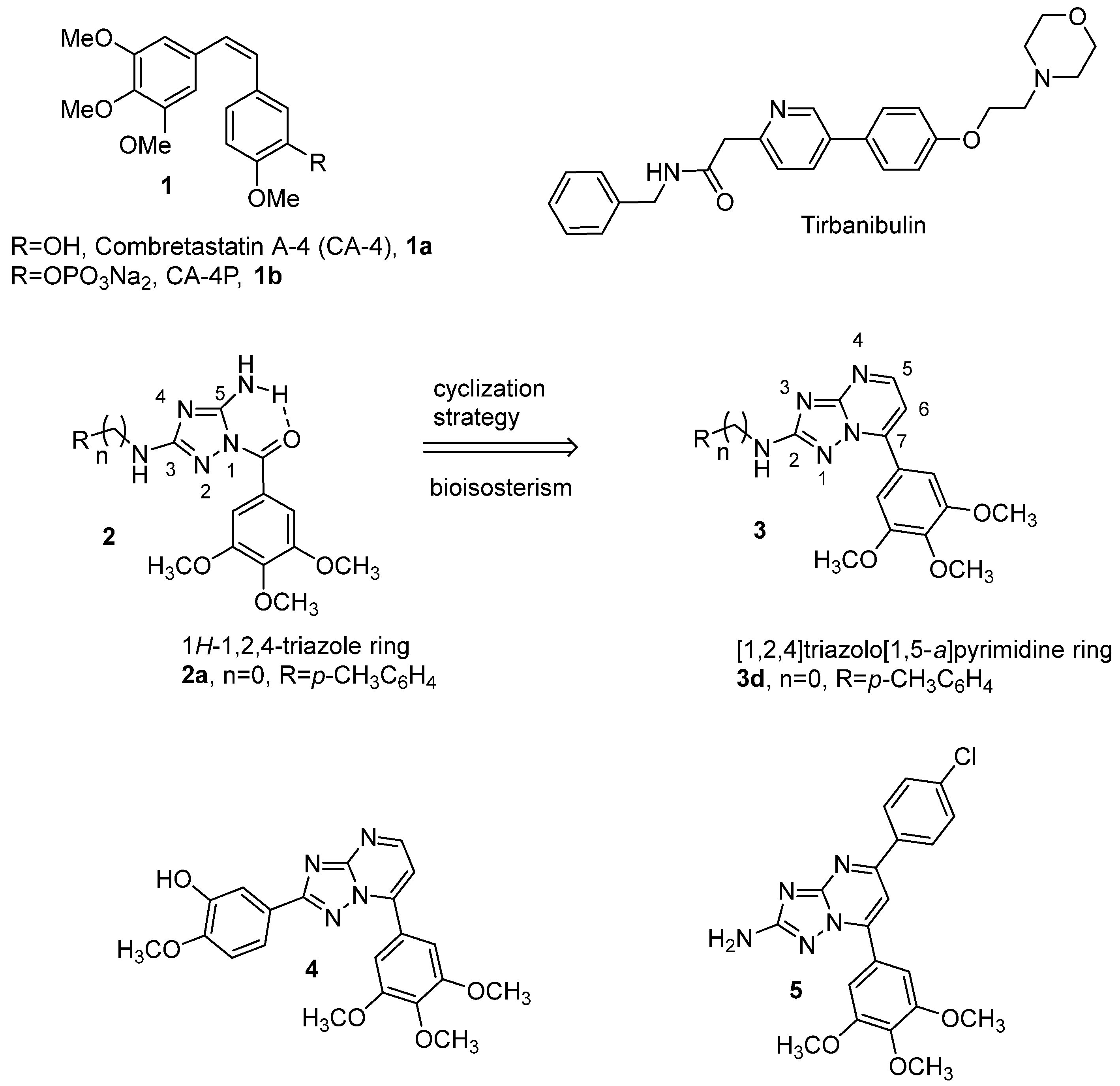
2. Results and Discussion
2.1. Chemistry
2.2. Biological Activity and Molecular Docking Studies
2.2.1. In Vitro Antiproliferative Activities
2.2.2. In Vitro Inhibition of Tubulin Polymerization and Colchicine Binding
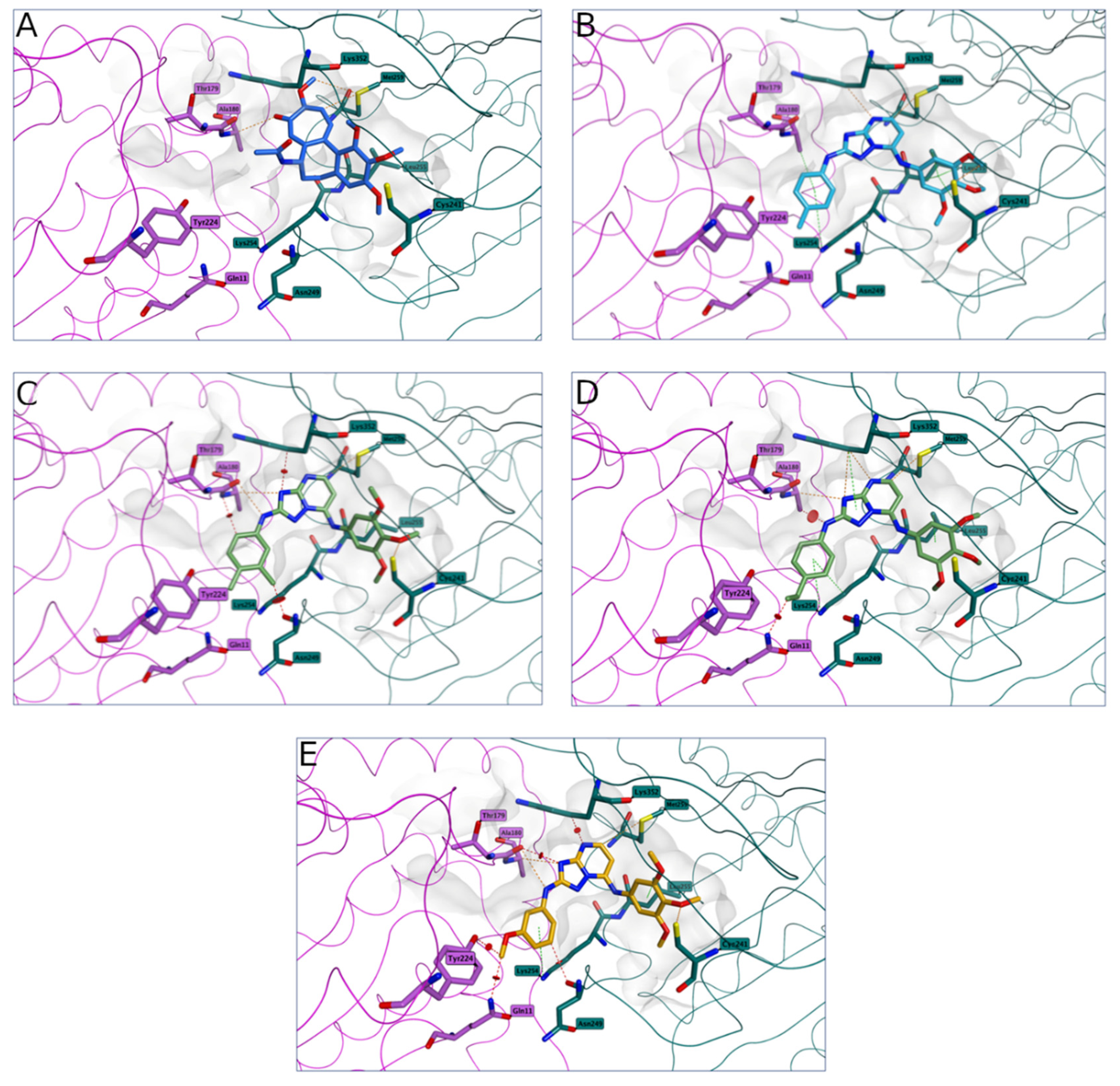
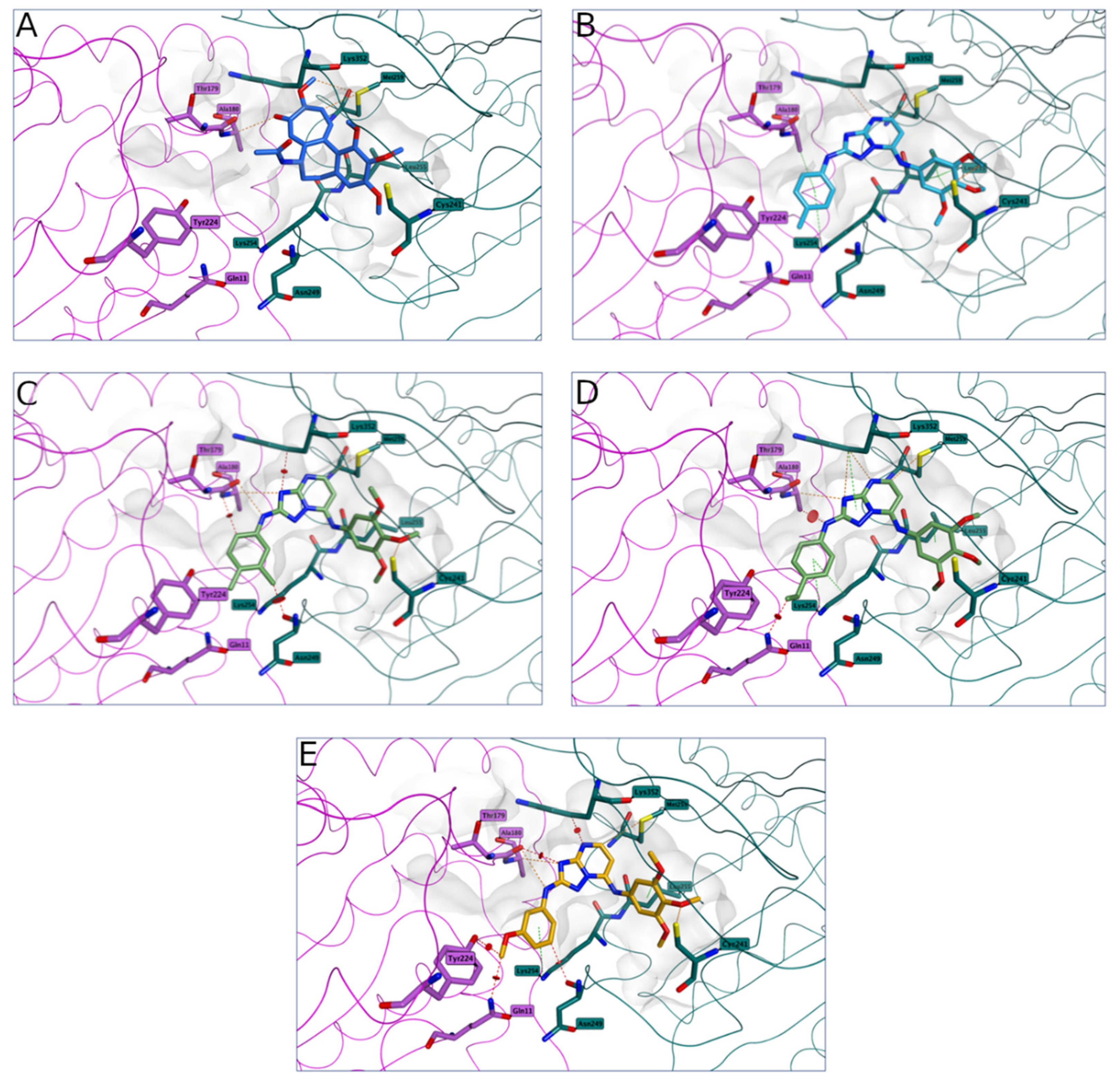
2.2.3. Molecular Modeling Studies
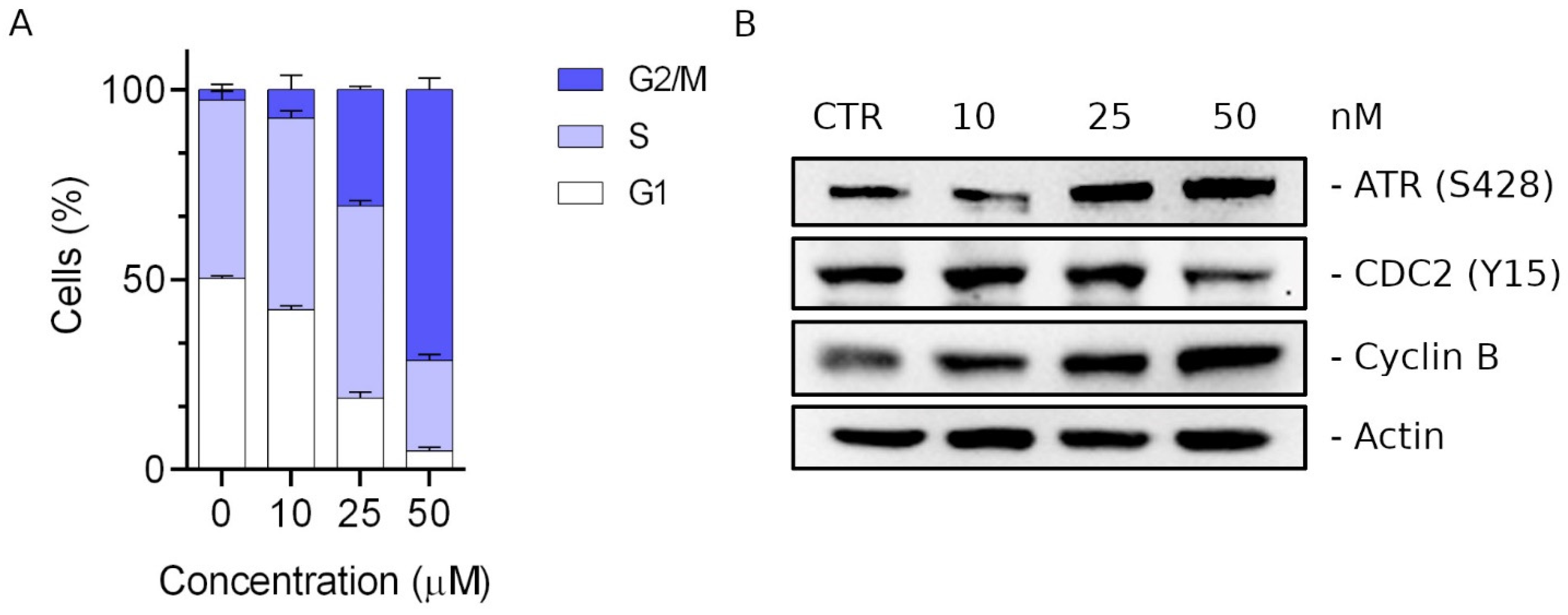
2.2.4. Compound 3d Induced G2/M Arrest of the Cell Cycle
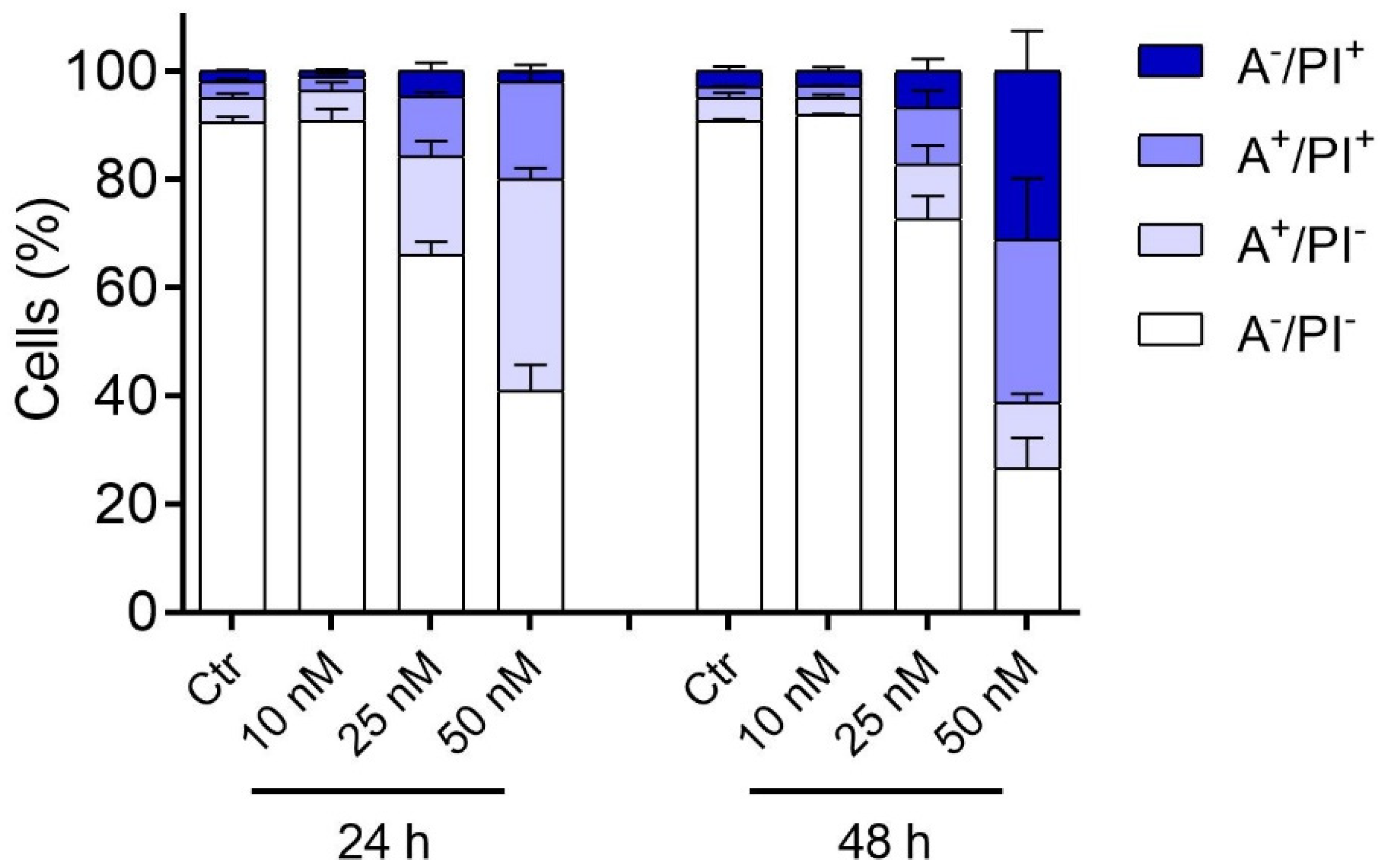
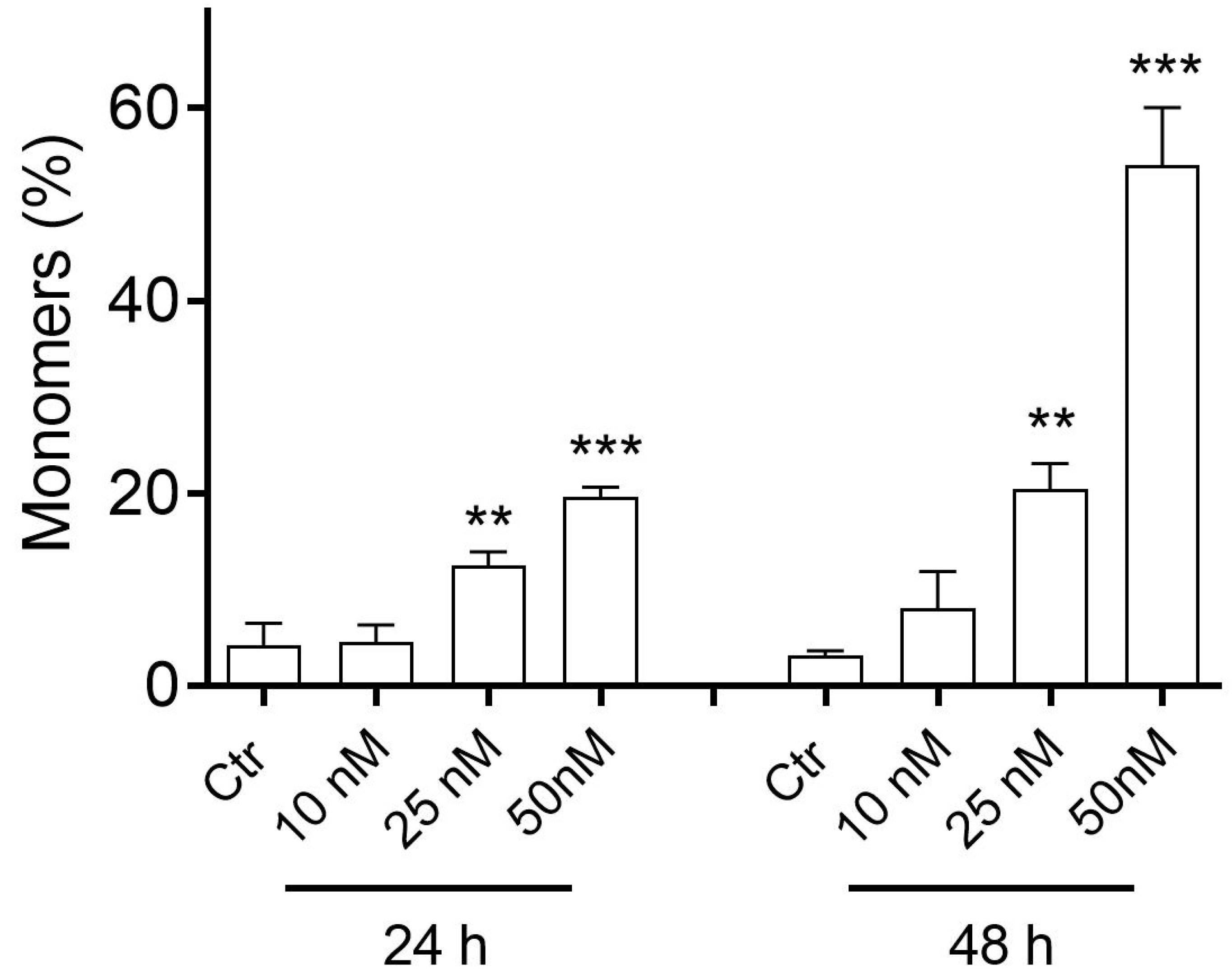
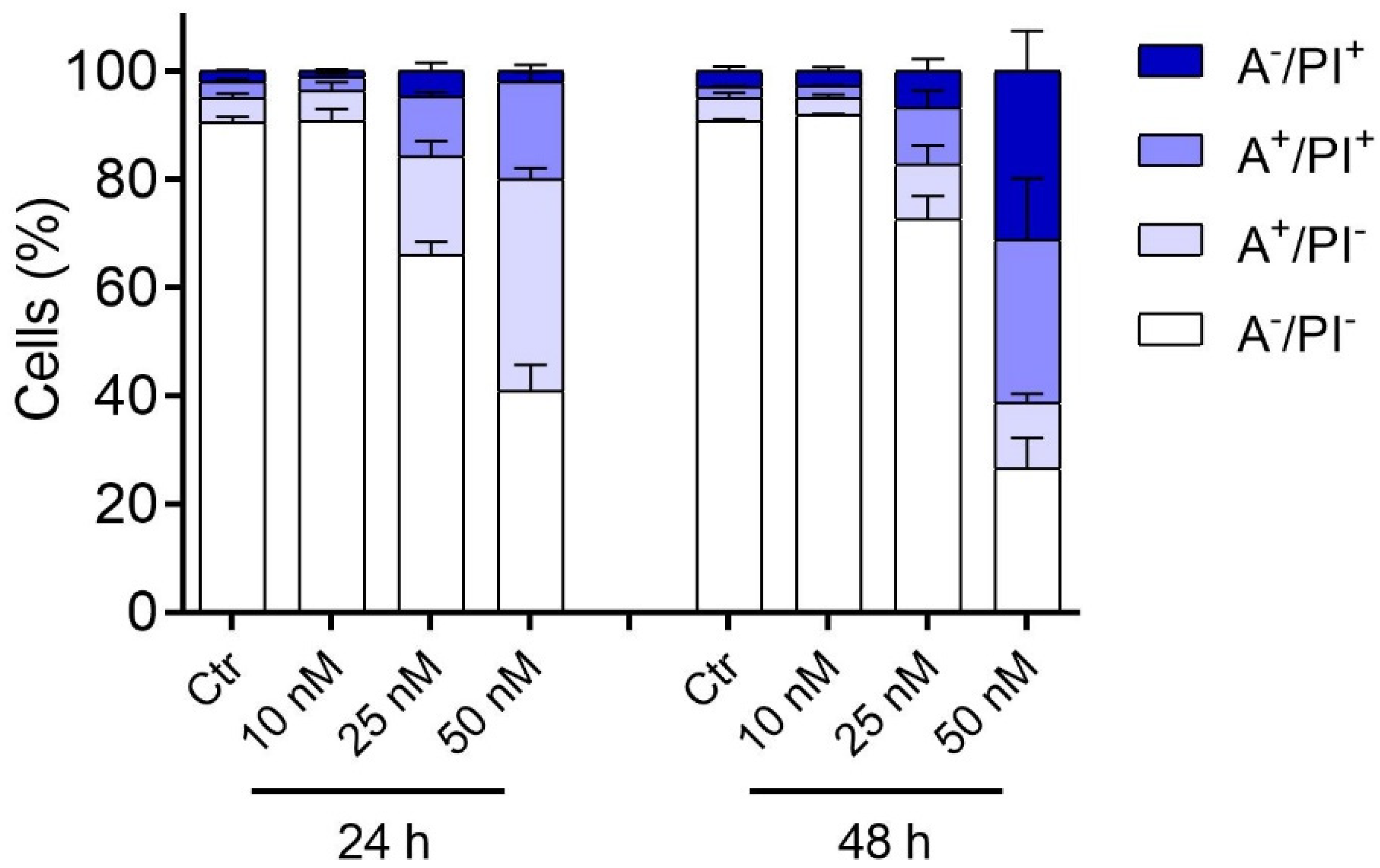
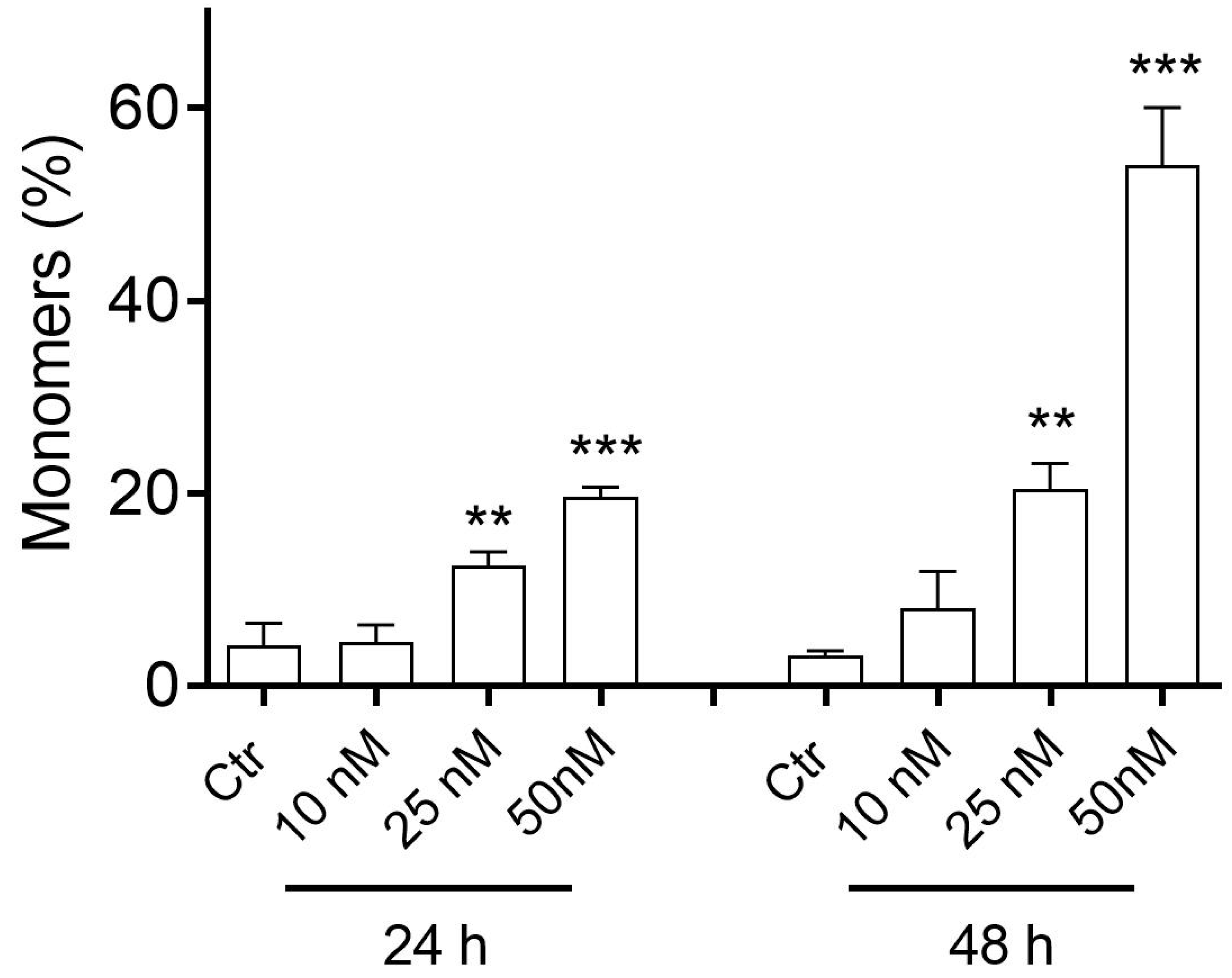
2.2.5. Compound 3d Induced Apoptosis in HeLa Cells through Mitochondrial Depolarization
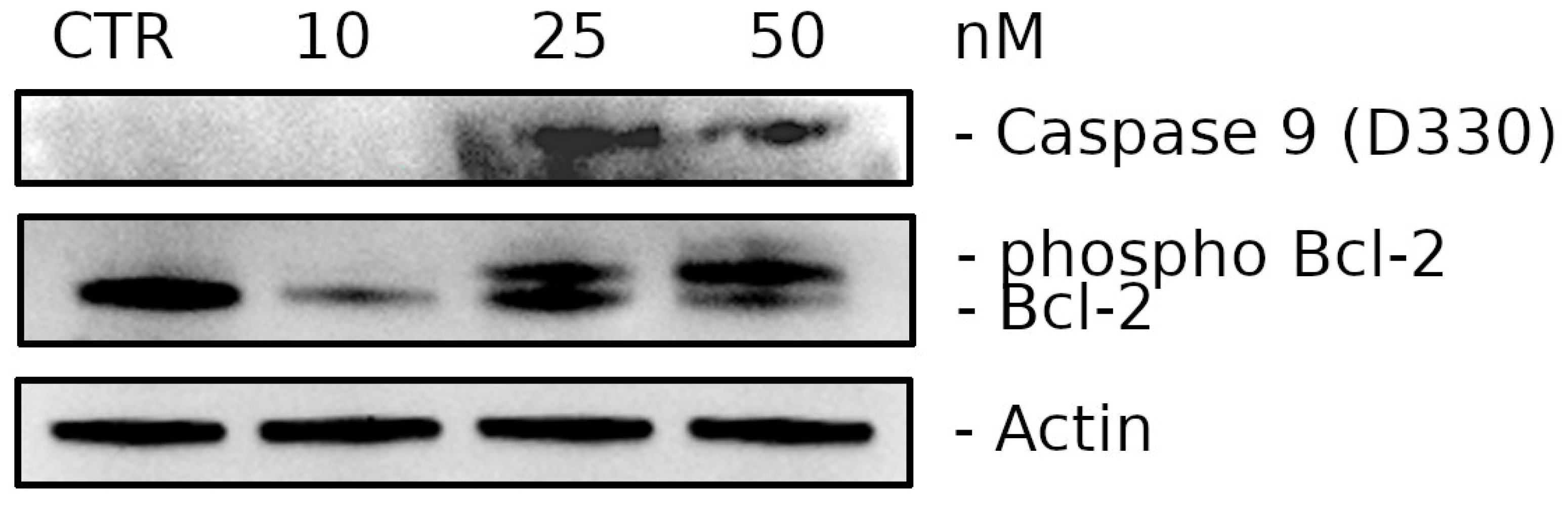
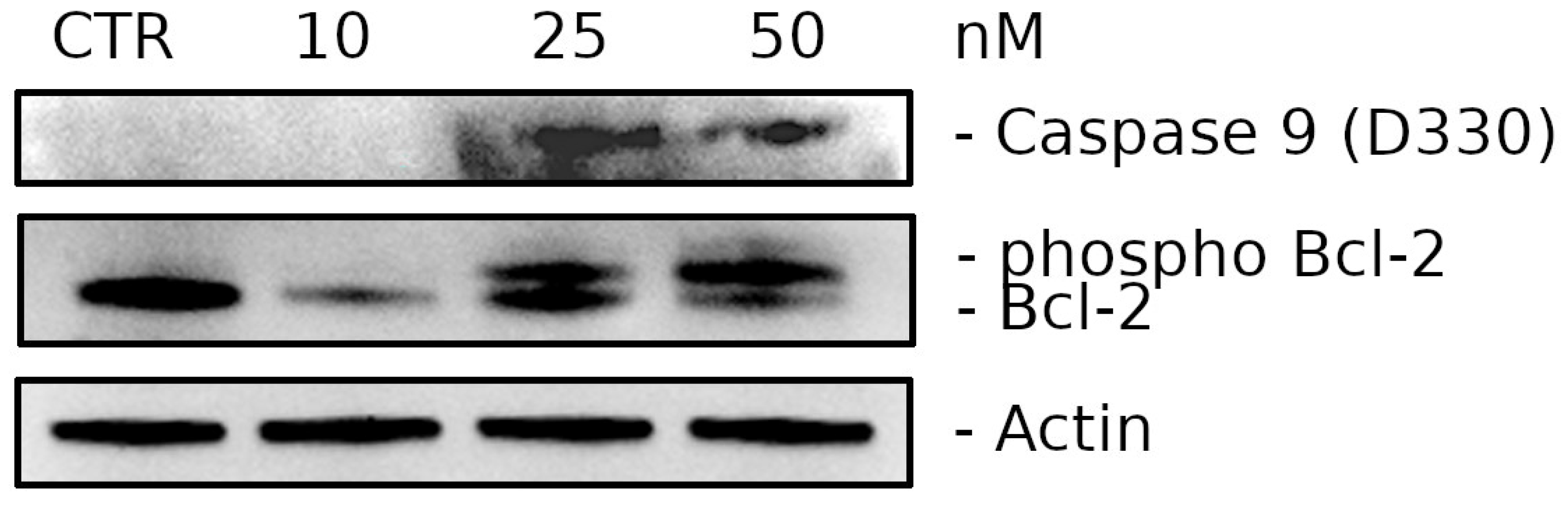
2.2.6. Compound 3d Induces Caspase-9 Activation and Causes a Decrease in the Expression of Bcl-2 Protein
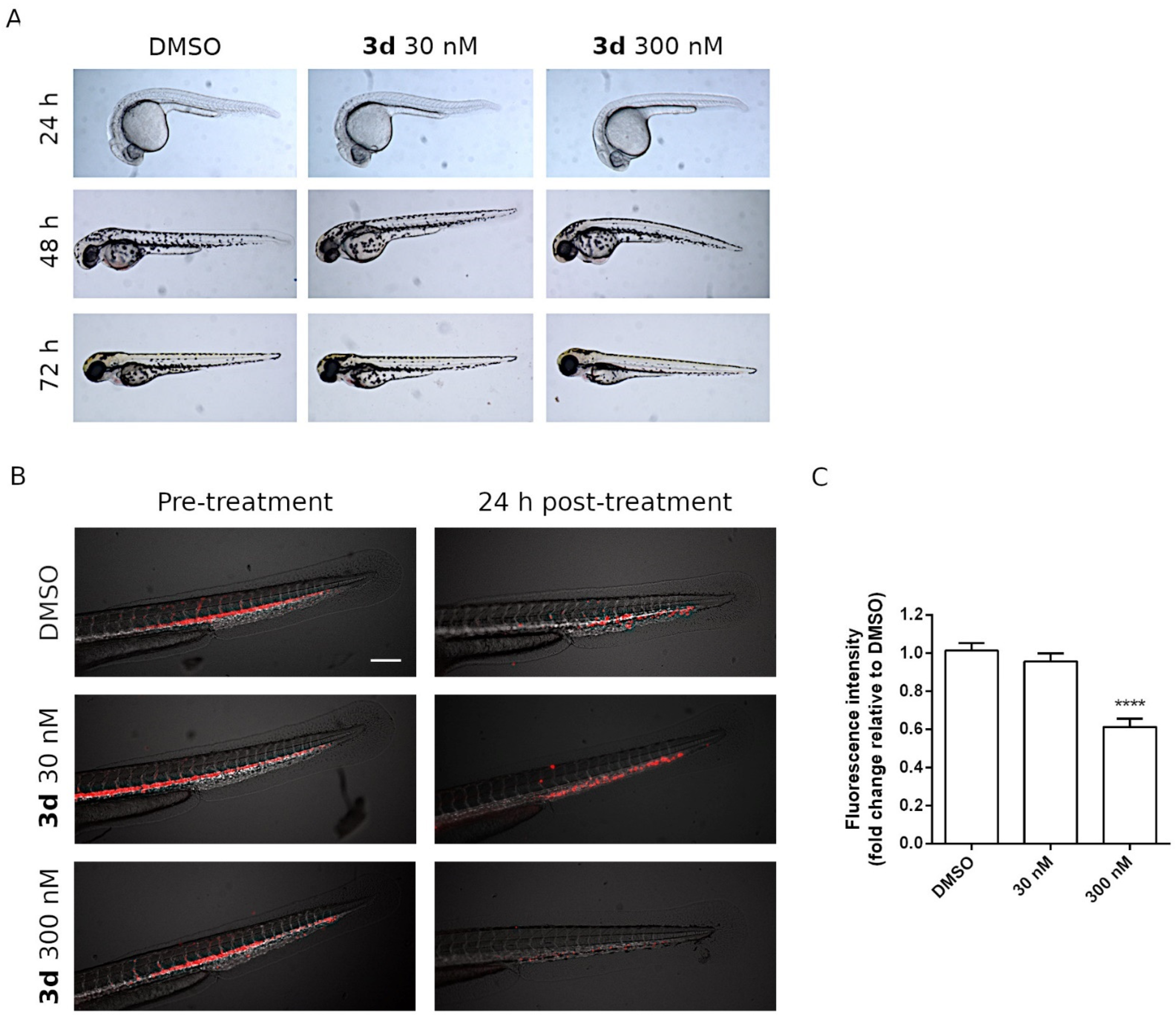
2.2.7. Effects of 3d Treatments on Zebrafish Embryos
2.2.8. In Vivo Antitumor Activity of Compound 3d in a Zebrafish Xenograft Model
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedure A for the Synthesis of Compounds 8a–v
(Z)-Methyl N′-Cyano-N-(pyridin-3-yl)carbamimidothioate (8b)
(Z)-Methyl N′-Cyano-N-(3,5-dimethylphenyl)carbamimidothioate (8g)
(Z)-Methyl N-(4-n-Propylphenyl)-N′-cyanocarbamimidothioate (8i)
(Z)-Methyl N′-Cyano-N-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)carbamimidothioate (8o)
3.1.2. General Procedure B for the Synthesis of Compounds 9a–v
N3-(Pyridin-3-yl)-1H-1,2,4-triazole-3,5-diamine (9b)
N3-(3,5-Dimethylphenyl)-1H-1,2,4-triazole-3,5-diamine (9g)
N3-(4-n-Propylphenyl)-1H-1,2,4-triazole-3,5-diamine (9i)
N3-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-1H-1,2,4-triazole-3,5-diamine (9o)
N3-Benzyl-1H-1,2,4-triazole-3,5-diamine (9p)
N3-(4-Chlorobenzyl)-1H-1,2,4-triazole-3,5-diamine (9q)
N3-(4-Methylbenzyl)-1H-1,2,4-triazole-3,5-diamine (9r)
N3-(4-Methoxybenzyl)-1H-1,2,4-triazole-3,5-diamine (9s)
N3-(Benzo[d][1,3]dioxol-5-ylmethyl)-1H-1,2,4-triazole-3,5-diamine (9t)
N3-(2-Phenylethyl)-1H-1,2,4-triazole-3,5-diamine (9u)
N3-(3-Phenylpropyl)-1H-1,2,4-triazole-3,5-diamine (9v)
3.1.3. General Procedure C for the Synthesis of Compounds 3a–v
N-Phenyl-7-(3,4,5-trimethoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-amine (3a)
N-(Pyridin-3-yl)-7-(3,4,5-trimethoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-amine (3b)
N-(4-Fluorophenyl)-7-(3,4,5-trimethoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-amine (3c)
N-(p-Tolyl)-7-(3,4,5-trimethoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-amine (3d)
N-(m-Tolyl)-7-(3,4,5-trimethoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-amine (3e)
N-(3,4-Dimethylphenyl)-7-(3,4,5-trimethoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-amine (3f)
N-(3,5-Dimethylphenyl)-7-(3,4,5-trimethoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-amine (3g)
N-(4-Ethylphenyl)-7-(3,4,5-trimethoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-amine (3h)
N-(4-Propylphenyl)-7-(3,4,5-trimethoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-amine (3i)
N-(4-Isopropylphenyl)-7-(3,4,5-trimethoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-amine (3j)
N-(4-Methoxyphenyl)-7-(3,4,5-trimethoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-amine (3k)
N-(3-Methoxyphenyl)-7-(3,4,5-trimethoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-amine (3l)
N-(4-Ethoxyphenyl)-7-(3,4,5-trimethoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-amine (3m)
N-(Benzo[d][1,3]dioxol-5-yl)-7-(3,4,5-trimethoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-amine (3n)
N-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-7-(3,4,5-trimethoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-amine (3o)
N-Benzyl-7-(3,4,5-trimethoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-amine (3p)
N-(4-Chlorobenzyl)-7-(3,4,5-trimethoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-amine (3q)
N-(4-Methylbenzyl)-7-(3,4,5-trimethoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-amine (3r)
N-(4-Methoxybenzyl)-7-(3,4,5-trimethoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-amine (3s)
N-(Benzo[d][1,3]dioxol-5-ylmethyl)-7-(3,4,5-trimethoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-amine (3t)
N-Phenethyl-7-(3,4,5-trimethoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-amine (3u)
N-(3-Phenylpropyl)-7-(3,4,5-trimethoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-amine (3v)
3.2. Biological Assays and Computational Studies
3.2.1. Cell Cultures and Viability Assay
3.2.2. Effects on Tubulin Polymerization and on Colchicine Binding to Tubulin
3.2.3. Molecular Modeling
3.2.4. Analysis of Cell Cycle by Flow Cytometry
3.2.5. Apoptosis Assay
3.2.6. Analysis of Mitochondrial Potential
3.2.7. Western Blot Analysis
3.2.8. In Vivo Experiments on Zebrafish Model
Husbandry and Maintenance
Drug Toxicity Assessment on Zebrafish Embryos
Xenograft Model: Injection and Treatment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Akhmanova, A.; Steinmetz, M.O. Control of microtubule organization and dynamics: Two ends in the limelight. Nat. Rev. Mol. Cell. Biol. 2015, 16, 711–726. [Google Scholar] [CrossRef] [PubMed]
- Ludueña, R.F. A hypothesis on the origin and evolution of tubulin. In International Review of Cell and Molecular Biology; Jeon, K.W., Ed.; Academic Press: San Diego, CA, USA, 2013; pp. 41–185. [Google Scholar]
- Knossow, M.; Campanacci, V.; Khodja, L.A.; Gigant, B. The mechanism of tubulin assembly into microtubules: Insights from structural studies. iScience 2020, 23, 101511. [Google Scholar] [CrossRef] [PubMed]
- Mukhtar, E.; Adhami, V.M.; Mukhtar, H. Targeting microtubules by natural agents for cancer therapy. Mol. Cancer Ther. 2014, 13, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Karahalil, B.; Yardım-Akaydin, S.; Nacak Baytas, S. An overview of microtubule targeting agents for cancer therapy. Arh. Hig. Rada Toksikol. 2019, 70, 160–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Čermák, V.; Dostál, V.; Jelínek, M.; Libusová, L.; Kovář, J.; Rösel, D.; Brábek, J. Microtubule-targeting agents and their impact on cancer treatment. Eur. J. Cell Biol. 2020, 99, 151075. [Google Scholar] [CrossRef]
- Field, J.J.; Kanakkanthara, A.; Miller, J.H. Microtubule-targeting agents are clinically successful due to both mitotic and interphase impairment of microtubule function. Bioorg. Med. Chem. 2014, 22, 5050–5059. [Google Scholar] [CrossRef]
- Cao, Y.N.; Zheng, L.L.; Wang, D.; Liang, X.X.; Gao, F.; Zhou, X.L. Recent advances in microtubule-stabilizing agents. Eur. J. Med. Chem. 2018, 143, 806–828. [Google Scholar] [CrossRef] [Green Version]
- Seligmann, J.; Twelves, C. Tubulin: An example of targeted chemotherapy. Future Med. Chem. 2013, 5, 339–352. [Google Scholar] [CrossRef]
- Gigant, B.; Wang, C.; Ravelli, R.B.G.; Roussi, F.; Steinmetz, M.O.; Curmi, P.A.; Sobel, A.; Knossow, M. Structural basis for the regulation of tubulin by vinblastine. Nature 2005, 435, 519–522. [Google Scholar] [CrossRef]
- Ranaivoson, F.M.; Gigant, B.; Berritt, S.; Joullié, M.; Knossow, M. Structural plasticity of tubulin assembly probed by vinca-domain ligands. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 927–934. [Google Scholar] [CrossRef]
- Coderch, C.; Morreale, A.; Gago, F. Tubulin-based structure-affinity relationships for antimitotic vinca alkaloids. Anticancer Agents Med. Chem. 2012, 12, 219–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parness, J.; Horwitz, S. Taxol binds to polymerized tubulin in vitro. J. Cell Biol. 1981, 91, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Prota, A.E.; Bargsten, K.; Zurwerra, D.; Field, J.J.; Diaz, J.F.; Altmann, K.H.; Steinmetz, M.O. Molecular mechanism of action of microtubule-stabilizing anticancer agents. Science 2013, 339, 587–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.H.; Horwitz, S.B. Taxol(®): The First Microtubule Stabilizing Agent. Int. J. Mol. Sci. 2017, 18, 1733. [Google Scholar] [CrossRef] [Green Version]
- Katsetos, C.D.; Dráber, P. Tubulins as therapeutic targets in cancer: From bench to bedside. Curr. Pharm. Des. 2012, 18, 2778–2792. [Google Scholar] [CrossRef]
- Steinmetz, M.O.; Prota, A.E. Microtubule-targeting agents: Strategies to hijack the cytoskeleton. Trends Cell Biol. 2018, 28, 776–792. [Google Scholar] [CrossRef]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef] [Green Version]
- Stanton, R.A.; Gernert, K.M.; Nettles, J.H.; Aneja, R. Drugs that target dynamic microtubules: A new molecular perspective. Med. Res. Rev. 2011, 31, 443–481. [Google Scholar] [CrossRef] [Green Version]
- Arnst, K.E.; Banerjee, S.; Chen, H.; Deng, S.; Hwang, D.J.; Li, W.; Miller, D.D. Current advances of tubulin inhibitors as dual acting small molecules for cancer therapy. Med. Res. Rev. 2019, 39, 1398–1426. [Google Scholar] [CrossRef]
- Florian, S.; Mitchison, T.J. Anti-microtubule drugs. Methods Mol. Biol. 2016, 1413, 403–421. [Google Scholar]
- Vindya, N.G.; Sharma, N.; Yadav, M.; Ethiraj, K.R. Tubulins-the target for anticancer therapy. Curr. Top. Med. Chem. 2015, 15, 73–82. [Google Scholar] [CrossRef]
- van Vuuren, R.J.; Visagie, M.H.; Theron, A.E.; Joubert, A.M. Antimitotic drugs in the treatment of cancer. Cancer Chemother. Pharmacol. 2015, 76, 1101–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukhari, S.N.A.; Kumar, G.B.; Revankar, H.M.; Qin, H.-L. Development of combretastatins as potent tubulin polymerization inhibitors. Bioorg. Chem. 2017, 72, 130–147. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sun, H.; Xu, S.; Zhu, Z.; Xu, J. Tubulin inhibitors targeting the colchicine binding site: A perspective of privileged structures. Future Med. Chem. 2017, 9, 1765–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Chen, J.J.; Xiao, M.; Li, W.; Miller, D.D. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm. Res. 2012, 29, 2943–2971. [Google Scholar] [CrossRef] [Green Version]
- Kaur, R.; Kaur, G.; Gill, R.K.; Soni, R.; Bariwal, J. Recent developments in tubulin polymerization inhibitors: An overview. Eur. J. Med. Chem. 2014, 87, 89–124. [Google Scholar] [CrossRef]
- Pettit, G.R.; Cragg, G.M.; Singh, S.B. Antineoplastic agents, 122. Constituents of Combretum caffrum. J. Nat. Prod. 1987, 50, 386–391. [Google Scholar] [CrossRef]
- Lin, C.M.; Ho, H.H.; Pettit, G.R.; Hamel, E. Antimitotic natural products combretastatin A-4 and combretastatin A-2: Studies on the mechanism of their inhibition of the binding of colchicine to tubulin. Biochemistry 1989, 28, 6984–6991. [Google Scholar] [CrossRef]
- Griggs, J.; Metcalfe, J.C.; Hesketh, R. Targeting tumour vasculature: The development of combretastatin A4. Lancet Oncol. 2001, 2, 82–87. [Google Scholar] [CrossRef]
- Nagaiah, G.; Remick, S.C. Combretastatin A4 phosphate: A novel vascular disrupting agent. Future Oncol. 2010, 6, 1219–1228. [Google Scholar] [CrossRef]
- Tewari, K.S.; Sill, M.W.; Coleman, R.L.; Aghajanian, C.; Mannel, R.; DiSilvestro, P.A.; Powell, M.; Randall, L.M.; Farley, J.; Rubin, S.C.; et al. Bevacizumab plus fosbretabulin in recurrent ovarian cancer: Overall survival and exploratory analyses of a randomized phase II NRG oncology/gynecologic oncology group study. Gynecol. Oncol. 2020, 159, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.; Arnold, S.; Slone, S.A.; Flynn, H.; Weiss, H.; Anthony, L.B. A phase I/II study of fosbretabulin in combination with everolimus in neuroendocrine tumors that have progressed after at least one prior regimen for metastatic disease. J. Clin. Oncol. 2018, 36, TPS4148. [Google Scholar] [CrossRef]
- Patil, P.O.; Patil, A.G.; Rane, R.A.; Patil, P.C.; Deshmukh, P.K.; Bari, S.B.; Patil, D.A.; Naphade, S.S. Recent advancement in discovery and development of natural product combretastatin-inspired anticancer agents. Anticancer Agents Med. Chem. 2015, 15, 955–969. [Google Scholar] [CrossRef] [PubMed]
- Greene, L.M.; Meegan, M.J.; Zisterer, D.M. Combretastatins: More than just vascular targeting agents? J. Pharmacol. Exp. Ther. 2015, 355, 212–227. [Google Scholar] [CrossRef]
- Mikstacka, R.; Stefański, T.; Różański, J. Tubulin-interactive stilbene derivatives as anticancer agents. Cell Mol. Biol. Lett. 2013, 18, 368–397. [Google Scholar] [CrossRef]
- Niu, L.; Yang, J.; Yan, W.; Yu, Y.; Zheng, Y.; Ye, H.; Chen, X.Q.; Chen, L. Reversible binding of the anticancer drug KXO1(tirbanibulin) to the colchicine-binding site of tubulin explains KXO1’s low clinical toxicity. J. Biol. Chem. 2019, 294, 18099–18108. [Google Scholar] [CrossRef]
- Amirall. Almirall Announces FDA Approval of Klisyri® (Tirbanibulin), A New Innovative Topical Treatment for Actinic Keratosis. Available online: https://www.almirall.com/newsroom/news/almirall-announces-fda-approval-of-klisyri®-tirbanibulin-a-new-innovative-topical-treatment-for-actinic-keratosis (accessed on 10 December 2020).
- Mosca, L.; Ilari, A.; Fazi, F.; Assaraf, Y.G.; Colotti, G. Taxanes in cancer treatment: Activity, chemoresistance and its overcoming. Drug Resist. Updates 2021, 54, 100742. [Google Scholar] [CrossRef]
- Martino, E.; Casamassima, G.; Castiglione, S.; Cellupica, E.; Pantalone, S.; Papagni, F.; Rui, M.; Siciliano, A.M.; Collina, S. Vinca alkaloids and analogues as anti-cancer agents: Looking back, peering ahead. Bioorg. Med. Chem. Lett. 2018, 28, 2816–2826. [Google Scholar] [CrossRef]
- Kavallaris, M.; Tait, A.S.; Walsh, B.J.; He, L.F.; Horwitz, S.B.; Norris, M.D.; Haber, M. Multiple microtubule alterations are associated with Vinca alkaloid resistance in human leukemia cells. Cancer Res. 2001, 61, 5803–5809. [Google Scholar]
- Verrills, N.M.; Kavallaris, M. Improving the targeting of tubulin-binding agents: Lessons from drug resistance studies. Curr. Pharm. Des. 2005, 11, 1719–1733. [Google Scholar] [CrossRef]
- Fojo, A.T.; Menefee, M. Microtubule targeting agents: Basic mechanisms of multidrug resistance (MDR). Semin. Oncol. 2005, 32, S3–S8. [Google Scholar] [CrossRef] [PubMed]
- Maloney, S.M.; Hoover, C.A.; Morejon-Lasso, L.V.; Prosperi, J.R. Mechanisms of taxane resistance. Cancers 2020, 12, 3323. [Google Scholar] [CrossRef] [PubMed]
- Kavallaris, M. Microtubules and resistance to tubulin-binding agents. Nat. Rev. Cancer 2010, 10, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Stengel, C.; Newman, S.P.; Leese, M.P.; Potter, B.V.; Reed, M.J.; Purohit, A. Class III beta-tubulin expression and in vitro resistance to microtubule targeting agents. Br. J. Cancer 2010, 102, 316–324. [Google Scholar] [CrossRef]
- Sève, P.; Dumontet, C. Is class III β-tubulin a predictive factor in patients receiving tubulin-binding agents? Lancet Oncol. 2008, 9, 168–175. [Google Scholar] [CrossRef]
- Haider, K.; Rahaman, S.; Yar, M.S.; Kamal, A. Tubulin inhibitors as novel anticancer agents: An overview on patents (2013–2018). Expert Opin. Ther. Pat. 2019, 29, 623–641. [Google Scholar] [CrossRef]
- Bates, D.; Eastman, A. Microtubule destabilising agents: Far more than just antimitotic anticancer drugs. Br. J. Clin. Pharmacol. 2017, 83, 255–268. [Google Scholar] [CrossRef] [Green Version]
- McLoughlin, E.C.; O’Boyle, N.M. Colchicine-binding site inhibitors from chemistry to clinic: A review. Pharmaceuticals 2020, 13, 8. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Chen, J.; Wang, J.; Ahn, S.; Li, C.M.; Lu, Y.; Loveless, V.S.; Dalton, J.T.; Miller, D.D.; Li, W. Novel tubulin polymerization inhibitors overcome multidrug resistance and reduce melanoma lung metastasis. Pharm. Res. 2012, 29, 3040–3052. [Google Scholar] [CrossRef] [Green Version]
- Nainwal, L.M.; Alam, M.M.; Shaquiquzzaman, M.; Marella, A.; Kamal, A. Combretastatin-based compounds with therapeutic characteristics: A patent review. Expert Opin. Ther. Pat. 2019, 29, 703–731. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Kimatrai Salvador, M.; Prencipe, F.; Bertolasi, V.; Cancellieri, M.; Brancale, A.; Hamel, E.; Castagliuolo, I.; Consolaro, F.; et al. Synthesis, antimitotic and antivascular activity of 1-(3’,4’,5’-trimethoxybenzoyl)-3-arylamino-5-amino-1,2,4-triazoles. J. Med. Chem. 2014, 57, 6795–6808. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Tawa, G.; Wallqvist, A. Classification of scaffold-hopping approaches. Drug Discov. Today 2012, 17, 310–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazzara, P.R.; Moore, T.W. Scaffold-hopping as a strategy to address metabolic liabilities of aromatic compounds. RSC Med. Chem. 2020, 11, 18–29. [Google Scholar] [CrossRef]
- Li, C.M.; Lu, Y.; Narayanan, R.; Miller, D.D.; Dalton, J.T. Drug metabolism and pharmacokinetics of 4-substituted methoxybenzoyl-aryl-thiazoles. Drug Metab. Dispos. 2010, 38, 2032–2039. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J.; Wang, J.; Li, C.M.; Ahn, S.; Barrett, C.M.; Dalton, J.T.; Li, W.; Miller, D.D. Design, synthesis, and biological evaluation of stable colchicine binding site tubulin inhibitors as potential anticancer agents. J. Med. Chem. 2014, 57, 7355–7366. [Google Scholar] [CrossRef] [Green Version]
- Hwang, D.-J.; Wang, J.; Li, W.; Miller, D.D. Structural optimization of indole derivatives acting at colchicine binding site as potential anticancer agents. ACS Med. Chem. Lett. 2015, 6, 993–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Y.; Wang, Y.; Li, G.; Zhang, Z.; Ma, L.; Cheng, B.; Chen, J. Discovery of novel benzimidazole and indazole analogues as tubulin polymerization inhibitors with potent anticancer activities. J. Med. Chem. 2021, 64, 4498–4515. [Google Scholar] [CrossRef]
- Chen, H.; Deng, S.; Albadari, N.; Yun, M.-K.; Zhang, S.; Li, Y.; Ma, D.; Parke, D.N.; Yang, L.; Seagroves, T.N.; et al. Design, synthesis, and biological evaluation of stable colchicine-binding site tubulin inhibitors 6-aryl-2-benzoyl-pyridines as potential anticancer agents. J. Med. Chem. 2021, 64, 12049–12074. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Ayral-Kaloustian, S.; Nguyen, T.; Afragola, J.; Hernandez, R.; Lucas, J.; Gibbons, J.; Beyer, C. Synthesis and SAR of [1,2,4]triazolo[1,5-a]pyrimidines, a class of anticancer agents with a unique mechanism of tubulin inhibition. J. Med. Chem. 2007, 50, 319–327. [Google Scholar] [CrossRef]
- Yang, F.; Yu, L.Z.; Diao, P.C.; Jian, X.E.; Zhou, M.F.; Jiang, C.S.; You, W.W.; Ma, V.; Zhao, P.L. Novel[1,2,4]triazolo[1,5-a]pyrimidine derivatives as potent antitubulin agents: Design, multicomponent synthesis and antiproliferative activities. Bioorg. Chem. 2019, 92, 103260. [Google Scholar] [CrossRef]
- Huo, X.-S.; Jian, X.-E.; Ou-Yang, J.; Chen, L.; Yang, F.; Lv, D.-X.; You, W.-W.; Rao, J.-J.; Zhao, P.-L. Discovery of highly potent tubulin polymerization inhibitors: Design, synthesis, and structure-activity relationships of novel 2,7-diaryl-[1,2,4]triazolo[1,5-a]pyrimidines. Eur. J. Med. Chem. 2021, 220, 113449. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, H.S.; Amin, N.H.; El-Saadi, M.T.; Abdel-Rahman, H.M. Design, synthesis, biological assessment, and in-silico studies of 1,2,4-triazolo[1,5-a]pyrimidine derivatives as tubulin polymerization inhibitors. Bioorg. Chem. 2022, 121, 105687. [Google Scholar] [CrossRef] [PubMed]
- Oliva, P.; Romagnoli, R.; Cacciari, B.; Manfredini, S.; Padroni, C.; Brancale, A.; Ferla, S.; Hamel, E.; Corallo, D.; Aveic, S.; et al. Synthesis and biological evaluation of highly active 7-anilino triazolopyrimidines as potent antimicrotubule agents. Pharmaceutics 2022, 14, 1191. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Dastagiri, D.; Ramaiah, M.J.; Reddy, J.S.; Bharathi, E.V.; Reddy, M.K.; Sagar, M.V.P.; Reddy, T.L.; Pushpavalli, S.N.C.V.L.; Pal-Bhadra, M. Synthesis and apoptosis inducing ability of new anilino substituted pyrimidine sulfonamides as potential anticancer agents. Eur. J. Med. Chem. 2011, 46, 5817–5824. [Google Scholar] [CrossRef] [PubMed]
- Donzelli, M.; Draetta, G.F. Regulating mammalian checkpoints through cdc25 inactivation. EMBO Rep. 2003, 4, 671–677. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Zheng, M.; Lu, R.; Du, J.; Zhao, Q.; Li, Z.; Li, Y.; Zhang, S. The role of CDC25C in cell cycle regulation and clinical cancer therapy: A systematic review. Cancer Cell Int. 2020, 20, 213. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C. Microtubules, microtubule-interfering agents and apoptosis. Apoptosis 2003, 8, 413–450. [Google Scholar] [CrossRef] [Green Version]
- Rovini, A.; Savry, A.; Braguer, D.; Carré, M. Microtubule-targeted agents: When mitochondria become essential to chemotherapy. Biochim. Biophys. Acta-Bioenerg. 2011, 1807, 679–688. [Google Scholar] [CrossRef] [Green Version]
- Romagnoli, R.; Baraldi, P.G.; Lopez Cara, C.; Hamel, E.; Basso, G.; Bortolozzi, R.; Viola, G. Synthesis and biological evaluation of 2-(3’,4’,5’-trimethoxybenzoyl)-3-aryl/arylaminobenzo[b]thiophene derivatives as a novel class of antiproliferative agents. Eur. J. Med. Chem. 2010, 45, 5781–5791. [Google Scholar] [CrossRef] [Green Version]
- Romagnoli, R.; Baraldi, P.G.; Kimatrai Salvador, M.; Preti, D.; Tabrizi, M.A.; Brancale, A.; Fu, X.-H.; Li, J.; Zhang, S.-Z.; Hamel, E.; et al. Discovery and optimization of a series of 2-aryl-4-amino-5-(3’,4’,5’-trimethoxybenzoyl)thiazoles as novel anticancer agents. J. Med. Chem. 2012, 55, 5433–5445. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Lopez-Cara, C.; Preti, D.; Aghazadeh Tabrizi, M.; Balzarini, J.; Bassetto, M.; Brancale, A.; Fu, X.-H.; Gao, Y.; et al. Concise synthesis and biological evaluation of 2-aroyl-5-amino benzo[b]thiophene derivatives as a novel class of potent antimitotic agents. J. Med. Chem. 2013, 56, 9296–9309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romagnoli, R.; Baraldi, P.G.; Lopez-Cara, C.; Kimatrai Salvador, M.; Preti, D.; Aghazadeh Tabrizi, M.; Balzarini, J.; Nussbaumer, P.; Brancale, A.; Fu, X.-H.; et al. Design, synthesis and biological evaluation of 3,5-disubstituted 2-amino thiophene derivatives as a novel class of antitumor agents. Bioorg. Med. Chem. 2014, 22, 5097–5109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, J.M.; Sakurikar, N.; Alford, S.E.; Chu, R.; Chambers, T.C. Critical role of anti-apoptotic Bcl-2 protein phosphorylation in mitotic death. Cell Death Dis. 2013, 4, e834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitaker, R.H.; Placzek, W.J. Regulating the BCL2 family to improve sensitivity to microtubule targeting agents. Cells 2019, 8, 346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliva, P.; Onnis, V.; Balboni, E.; Hamel, E.; Estévez-Sarmiento, F.; Quintana, J.; Estévez, F.; Brancale, A.; Ferla, S.; Manfredini, S.; et al. Synthesis and biological evaluation of 2-substituted benzyl/phenylethylamino-4-amino-5-aroyl thiazoles as apoptosis inducing anticancer agents. Molecules 2020, 25, 2177. [Google Scholar] [CrossRef]
- Hamel, E.; Lin, C.M. Separation of active tubulin and microtubule-associated proteins by ultracentrifugation and isolation of a component causing the formation of microtubule bundles. Biochemistry 1984, 23, 4173–4184. [Google Scholar] [CrossRef]
- Hamel, E. Evaluation of antimitotic agents by quantitative comparisons of their effects on the polymerization of purified tubulin. Cell Biochem. Biophys. 2003, 38, 1–21. [Google Scholar] [CrossRef]
- Verdier-Pinard, P.; Lai, J.-Y.; Yoo, H.-D.; Yu, J.; Marquez, B.; Nagle, D.G.; Nambu, M.; White, J.D.; Falck, J.R.; Gerwick, W.H.; et al. Structure-activity analysis of the interaction of curacin A, the potent colchicine site antimitotic agent, with tubulin and effects of analogs on the growth of MCF-7 breast cancer cells. Mol. Pharmacol. 1998, 53, 62–67. [Google Scholar] [CrossRef] [Green Version]
- Molecular Operating Environment (MOE), 2022.02. Chemical Computing Group ULC: Montreal, QC, Canada. Available online: http://www.chemcomp.com (accessed on 1 July 2022).
- Schrödinger Release 2022-2; Maestro, Schrödinger, LLC: New York, NY, USA, 2022.
- Lawson, N.D.; Weinstein, B.M. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev. Biol. 2002, 248, 307–318. [Google Scholar] [CrossRef] [Green Version]

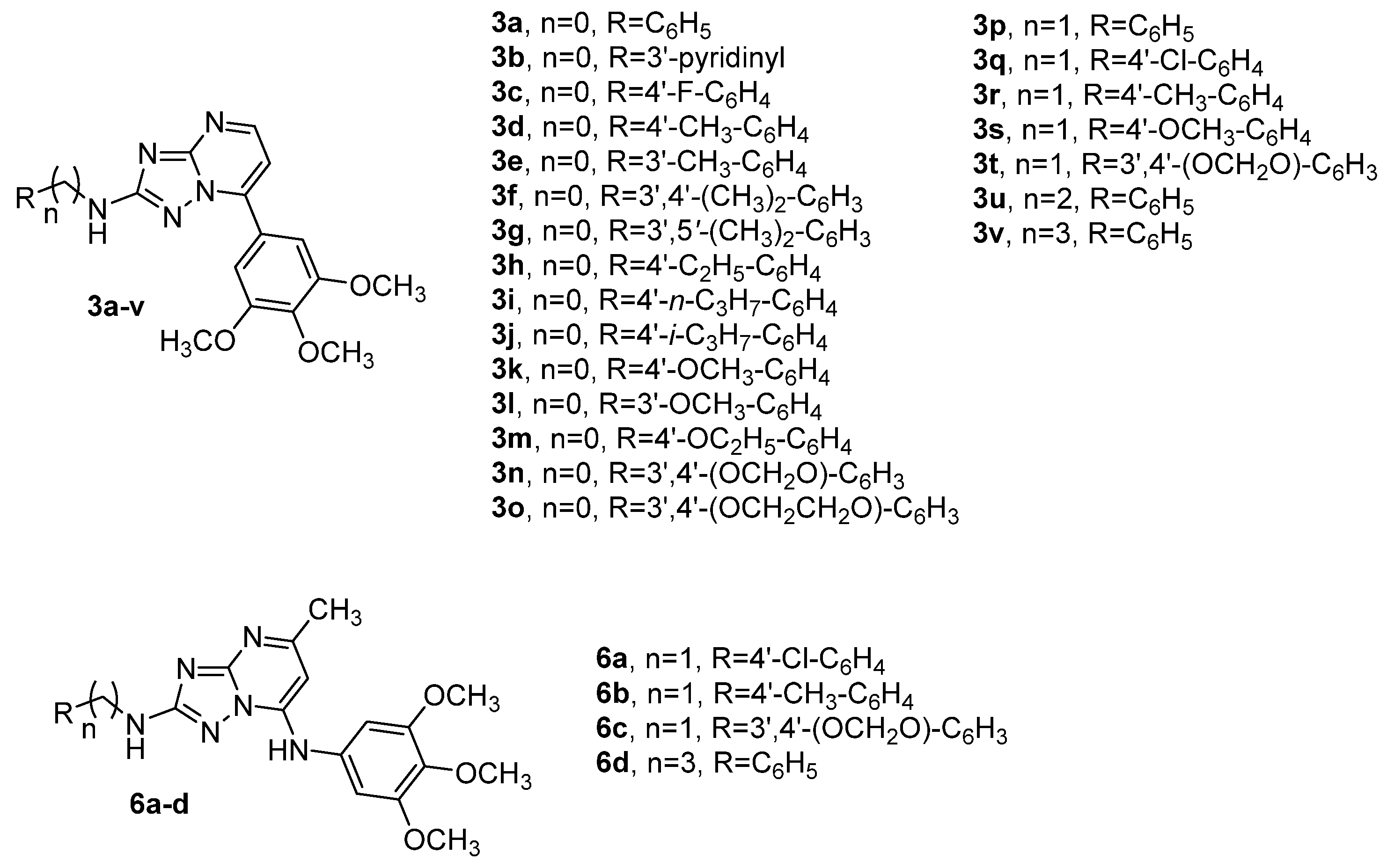


{kind=link}
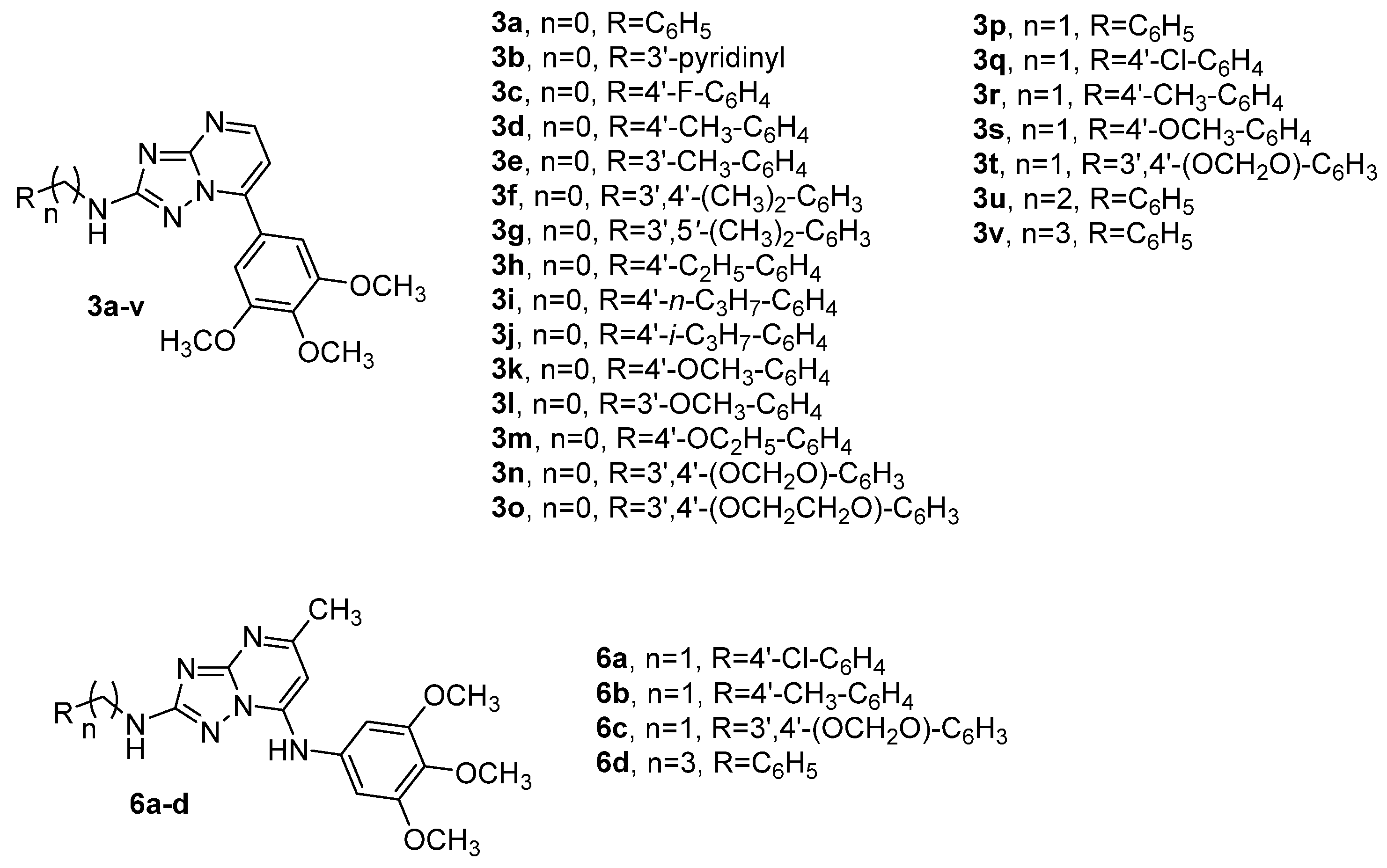{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
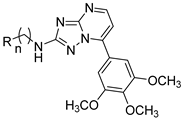
| Compound | R | n | IC50 (μM) a | |||
|---|---|---|---|---|---|---|
| MDA-MB-231 | HeLa | A549 | HT29 | |||
| 3a | C6H5 | 0 | 2.27 ± 0.31 | 0.80 ± 0.09 | 1.56 ± 0.06 | 1.02 ± 0.67 |
| 3b | 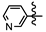 | 0 | >10 | >10 | >10 | 3.42 ± 0.41 |
| 3c | 4′-F-C6H4 | 0 | >10 | 5.44 ± 1.05 | >10 | >10 |
| 3d | 4′-CH3-C6H4 | 0 | 0.43 ± 0.11 | 0.038 ± 0.009 | 0.043 ± 0.024 | 0.030 ± 0.003 |
| 3e | 3′-CH3-C6H4 | 0 | >10 | 0.39 ± 0.02 | 1.87 ± 0.17 | 1.38 ± 0.09 |
| 3f | 3′,4′-(CH3)2-C6H3 | 0 | >10 | 0.067 ± 0.0023 | 0.16 ± 0.045 | 0.16 ± 0.041 |
| 3g | 3′,5′-(CH3)2-C6H3 | 0 | >10 | >10 | >10 | >10 |
| 3h | 4′-C2H5-C6H4 | 0 | >10 | 0.24 ± 0.074 | 0.2 ± 0.044 | 0.16 ± 0.033 |
| 3i | 4′-n-C3H7-C6H4 | 0 | >10 | >10 | >10 | >10 |
| 3j | 4′-i-C3H7-C6H4 | 0 | >10 | 4.7 ± 0.59 | >10 | >10 |
| 3k | 4′-OCH3-C6H4 | 0 | 5.2 ± 1.8 | 1.27 ± 0.08 | 5.32 ± 1.43 | 2.8 ± 0.20 |
| 3l | 3′-OCH3-C6H4 | 0 | 1.76 ± 0.9 | 0.44 ± 0.08 | 0.76 ± 0.18 | 0.76 ± 0.18 |
| 3m | 4′-OC2H5-C6H4 | 0 | >10 | >10 | >10 | 6.16 ± 0.97 |
| 3n |  | 0 | 6.1 ± 1.72 | 0.84 ± 0,19 | 1.44 ± 0.26 | 1.39 ± 0.06 |
| 3o | 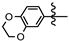 | 0 | >10 | 0.29 ± 0.031 | 2.42 ± 0.12 | 0.67 ± 0.12 |
| 3p | C6H5 | 1 | 9.3 ± 0.22 | >10 | >10 | >10 |
| 3q | 4′-Cl-C6H4 | 1 | >10 | >10 | >10 | >10 |
| 3r | 4′-CH3-C6H4 | 1 | >10 | >10 | >10 | >10 |
| 3s | 4′-OCH3-C6H4 | 1 | >10 | >10 | >10 | >10 |
| 3t | | 1 | >10 | >10 | >10 | >10 |
| 3u | C6H5 | 2 | 8.7 ± 1.1 | >10 | >10 | 6.74 ± 0.50 |
| 3v | C6H5 | 3 | >10 | >10 | >10 | >10 |
| CA-4 (1a) | - | - | 0.005 ± 0.002 | 0.004 ± 0.001 | 0.18 ± 0.05 | 3.10 ± 0.03 |
| Compounds | Tubulin Assembly a IC50 ± SD (µM) | Colchicine Binding b % Inhibition ± SD |
|---|---|---|
| 3d | 0.45 ± 0.1 | 72 ± 5 |
| 3f | 0.80 ± 0.1 | 18 ± 5 |
| 3h | 1.9 ± 0.2 | 21 ± 0.9 |
| 3l | 2.2 ± 0.2 | 39 ± 5 |
| CA-4 (1a) | 0.75 ± 0.06 | 98 ± 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romagnoli, R.; Oliva, P.; Prencipe, F.; Manfredini, S.; Budassi, F.; Brancale, A.; Ferla, S.; Hamel, E.; Corallo, D.; Aveic, S.; et al. Design, Synthesis and Biological Investigation of 2-Anilino Triazolopyrimidines as Tubulin Polymerization Inhibitors with Anticancer Activities. Pharmaceuticals 2022, 15, 1031. https://doi.org/10.3390/ph15081031
Romagnoli R, Oliva P, Prencipe F, Manfredini S, Budassi F, Brancale A, Ferla S, Hamel E, Corallo D, Aveic S, et al. Design, Synthesis and Biological Investigation of 2-Anilino Triazolopyrimidines as Tubulin Polymerization Inhibitors with Anticancer Activities. Pharmaceuticals. 2022; 15(8):1031. https://doi.org/10.3390/ph15081031
Chicago/Turabian StyleRomagnoli, Romeo, Paola Oliva, Filippo Prencipe, Stefano Manfredini, Federica Budassi, Andrea Brancale, Salvatore Ferla, Ernest Hamel, Diana Corallo, Sanja Aveic, and et al. 2022. "Design, Synthesis and Biological Investigation of 2-Anilino Triazolopyrimidines as Tubulin Polymerization Inhibitors with Anticancer Activities" Pharmaceuticals 15, no. 8: 1031. https://doi.org/10.3390/ph15081031
APA StyleRomagnoli, R., Oliva, P., Prencipe, F., Manfredini, S., Budassi, F., Brancale, A., Ferla, S., Hamel, E., Corallo, D., Aveic, S., Manfreda, L., Mariotto, E., Bortolozzi, R., & Viola, G. (2022). Design, Synthesis and Biological Investigation of 2-Anilino Triazolopyrimidines as Tubulin Polymerization Inhibitors with Anticancer Activities. Pharmaceuticals, 15(8), 1031. https://doi.org/10.3390/ph15081031