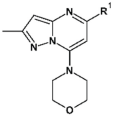
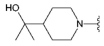
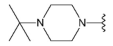
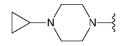
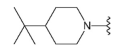
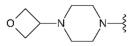
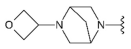
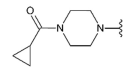
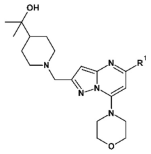
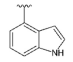
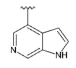
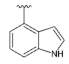
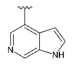
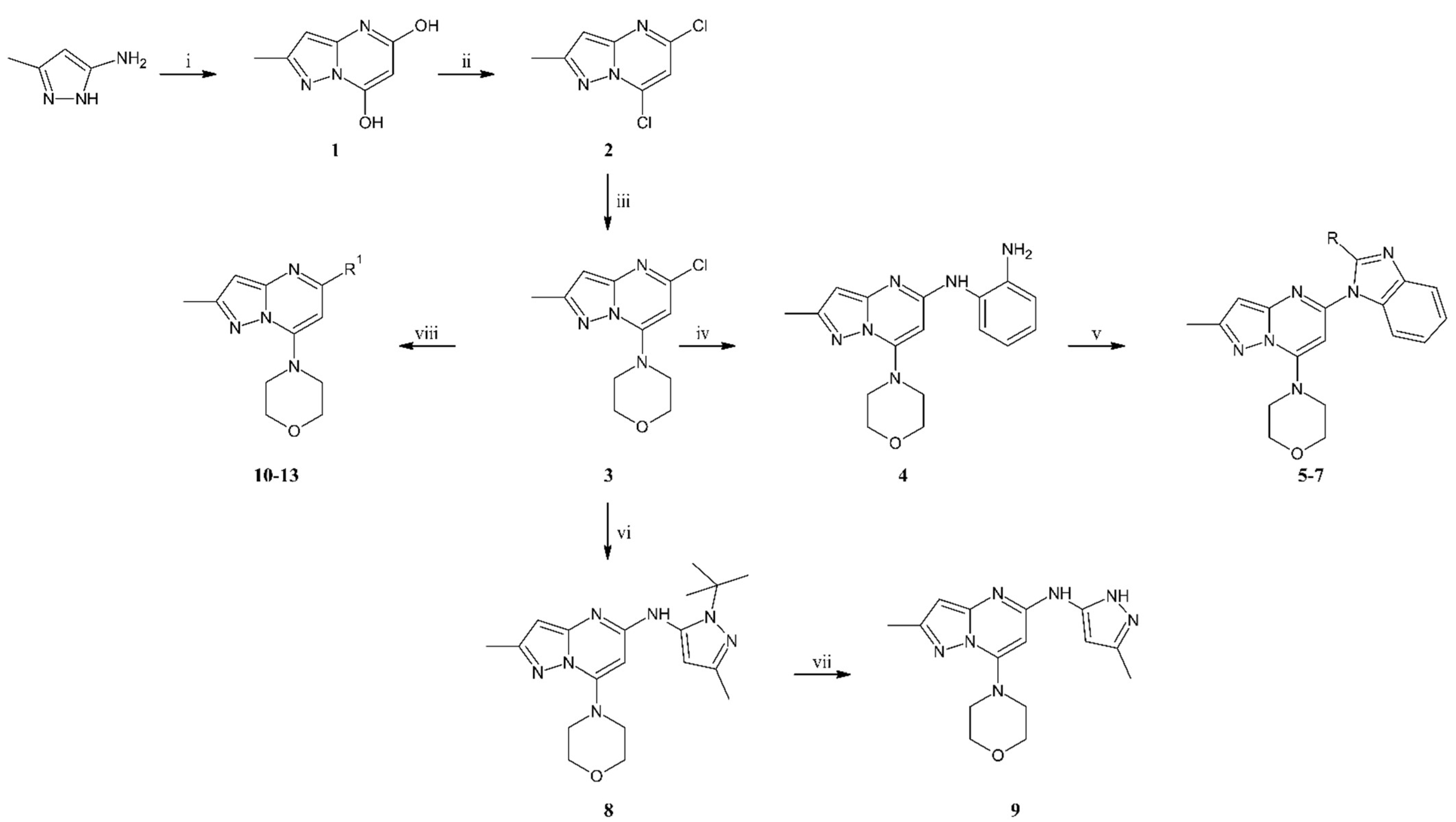
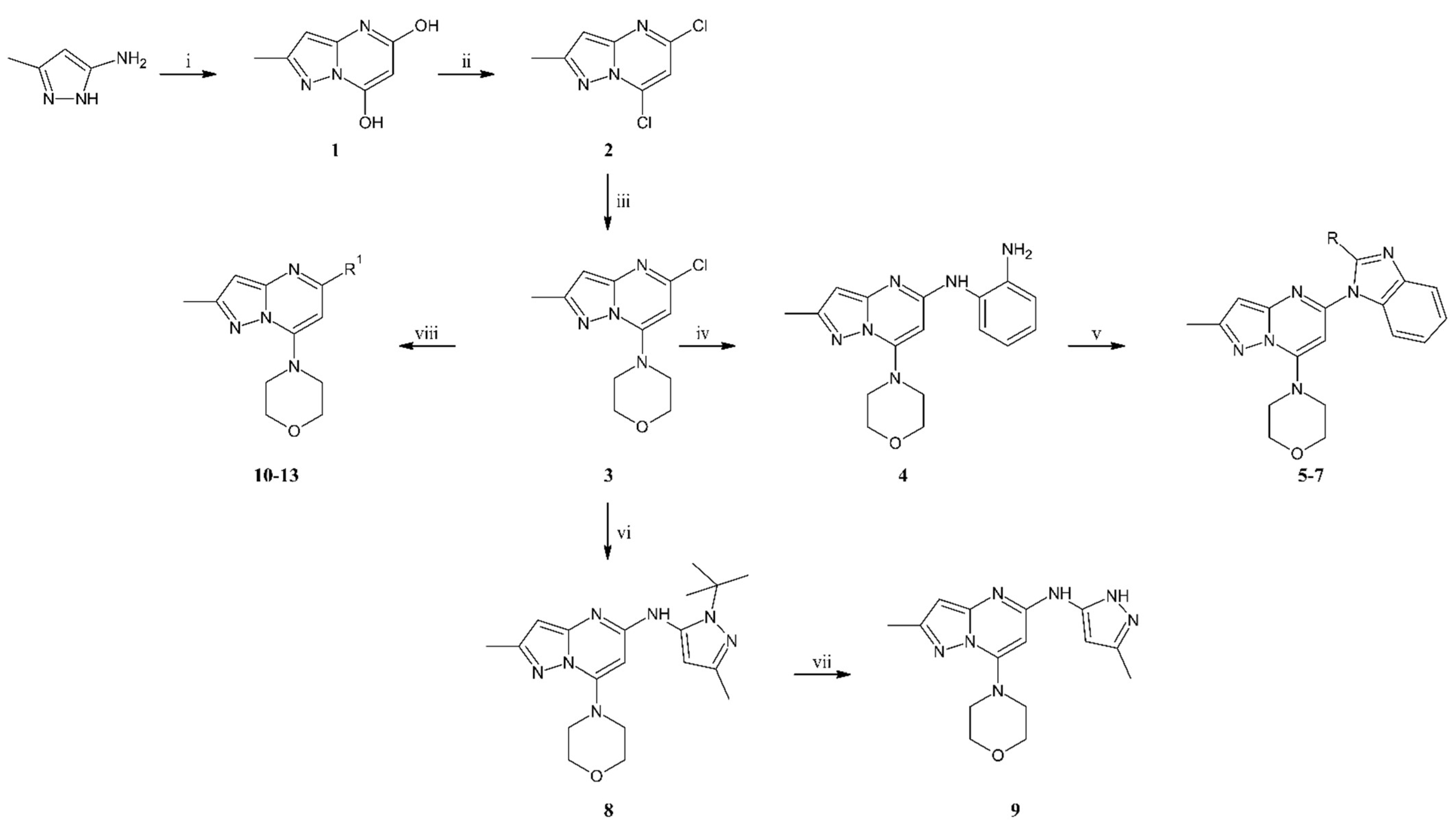
3.1.2. Synthesis
To the flask with sodium ethoxide solution (obtained from sodium (4.73 g, 0.21 mol) and ethanol (175 mL) a solution of 3-amino-5-methylpyrazole (10.0 g, 0.10 mol) in ethanol (100 mL) and diethyl malonate (23.5 mL, 0.15 mol) were added. The reaction was carried out at reflux for 24 h. The reaction mixture was cooled to room temperature, and then the solvent was evaporated under reduced pressure. The residue was dissolved in 1200 mL of water and acidified with concentrated hydrochloric acid to a pH of about 2. Creamy solid precipitated from the solution was filtered off, washed, and dried. The title compound 1 (15.2 g, 0.08 mol) was obtained as an off-white solid with 89% yield. MS-ESI: m/z calcd for C7H7N3O2 [M+Na]+: 188.04; found 187.9.
To the cooled to 0 °C POCl3 (90 mL, 0.963 mol), compound 1 (15.2 g, 0.092 mol) was added. The reaction was carried out at reflux for 24 h. The reaction mixture was cooled to room temperature and poured into the water with ice. The mixture was quenched with a 6 M sodium hydroxide solution to pH 6. The aqueous phase was extracted with ethyl acetate, and after separation, the organic phase was dried with anhydrous sodium sulfate. After filtration of the drying agent and evaporation of the solvent, the residue was purified by column chromatography (0–40% ethyl acetate gradient in heptane) to give compound 2 (11.4 g, 0.056 mol) obtained as an off-white solid with 61% yield. 1H NMR (300 MHz, CDCl3) δ: 6.90 (s, 1H, Ar-H), 6.53 (s, 1H, Ar-H), 2.56 (s, 3H, CH3). MS-ESI: m/z calcd for C7H5Cl2N3 [M+H]+: 201.99; found 201.9.
To the solution of compound 2 (2.0 g, 9.9 mmol) in acetone (50 mL), potassium carbonate (1.64 g, 11.9 mmol), and morpholine (1.35 mL, 15.5 mmol) were added. The reaction was carried out at room temperature for 1.5 h. Then water (100 mL) was added to the reaction mixture, and the precipitated white solid was filtered off. The obtained solid was washed with water (50 mL) and water/acetone mixture (2/1, v/v) (50 mL), then dried. Compound 3 (2.36 g, 0.09 mol) was obtained as a white solid with 94% yield. 1H NMR (300 MHz, CDCl3) δ: 6.29 (s, 1H, Ar-H), 6.01 (s, 1H, Ar-H), 4.00–3.92 (m, 4H, morph.), 3.81–3.72 (m, 4H, morph.), 2.46 (s, 3H, CH3). MS-ESI: m/z calcd for C11H13ClN4O [M+H]+: 253.09; found 253.0.
The mixture of compound 3 (1.0 g, 3.96 mmol), benzene-1,2-diamine (1.31 g, 11.9 mmol), cesium carbonate (3.87 g, 11.9 mmol), tris(dibenzylideneacetone)dipalladium(0) (0.181 g, 0.20 mmol), 9,9-dimethyl-4,5-bis(diphenylphosphine)xanthene (0.229 g, 0.40 mmol) and dry toluene (40 mL) were introduced to the reaction Schlenk flask. The mixture was flushed with argon and stirred at 110 °C for 24 h. After cooling to room temperature, the reaction mixture was filtered through Celite®, and the solid was washed with ethyl acetate. The filtrate was concentrated under reduced pressure using an evaporator. The residue was resolved and purified by column chromatography (50–100% ethyl acetate gradient in heptane) to give the title compound 4 (0.78 g, 2.4 mmol) with 61% yield. 1H NMR (300 MHz, CDCl3) δ 7.23–7.17 (m, 1H, Ar-H), 7.16–7.09 (m, 1H, Ar-H), 6.88–6.76 (m, 2H, Ar-H), 6.37 (s, 1H, Ar-H), 5.92–5.86 (m, 1H), 5.30 (s, 1H), 4.01–3.81 (m, 4H, morph.), 3.58–3.45 (m, 4H, morph.), 2.39 (s, 3H, CH3). MS-ESI: m/z calcd for C17H20N6O [M+H]+: 325.18; found 325.1.
In the solution of compound 4 (1.0 eq) dissolved in dry DCM (10 mL/1g of compound 4), the carboxylic acid (2.0 eq), HOBt × H2O (1.2 eq), EDCI × HCl (2.4 eq), and TEA (3.0 eq) were added. The whole reaction mixture was stirred at room temperature for 48 h. To the reaction, mixture water was added, and organic and water phases were separated. The aqueous phase was washed three times with DCM. Combined organic phases were dried over anhydrous sodium sulfate. After the drying agent was filtered off and the solvent evaporated, the reaction mixture was dissolved in glacial acetic acid. The reaction mixture was refluxed for 24 h. Then the reaction mixture was cooled and concentrated under reduced pressure. The residue was diluted with water and neutralized with a saturated sodium bicarbonate solution. The aqueous phase was extracted three times with ethyl acetate. Combined organic phases were dried over sodium sulfate. Once the drying agent was filtered off, the solvent was evaporated under reduced pressure using an evaporator. The reaction mixture was purified by column chromatography.
Compound 5 was prepared from compound 4 (0.20 g, 0.62 mmol), acetic acid (70 μL, 74 mg, 1.23 mmol), HOBt (0.10 g, 0.74 mmol), EDCI (0.28 g, 1.48 mmol), TEA (0.26 mL, 0.19 g, 1.85 mmol) and DCM (6.0 mL). The crude product was purified by flash chromatography to give 5 (0.16 g, 0.46 mmol) as a light yellow solid with 73% yield. 1H NMR (300 MHz, CDCl3) δ 7.78–7.72 (m, 1H, Ar-H), 7.50–7.45 (m, 1H, Ar-H), 7.34–7.22 (m, 2H, Ar-H), 6.42 (s, 1H, Ar-H), 6.16 (s, 1H, Ar-H), 4.03–3.97 (m, 4H, morph.), 3.88–3.82 (m, 4H, morph.), 2.76 (s, 3H, CH3), 2.53 (s, 3H, CH3). 13C NMR (75 MHz, CDCl3) δ 155.0, 151.6, 151.2, 150.4, 148.5, 142.7, 134.5, 123.0, 122.9, 119.4, 110.4, 96.1, 87.5, 66.2, 48.4, 15.6, 14.8. MS-ESI: m/z calcd for C19H20N6O [M+H]+: 349.18; found 349.1.
Compound 6 was prepared from compound 4 (0.20 g, 0.62 mmol), propionic acid (92 μL, 91 mg, 1.23 mmol), HOBt (0.10 g, 0.74 mmol), EDCI (0.28 g, 1.48 mmol), TEA (0.26 mL, 0.19 g, 1.85 mmol) and DCM (6.0 mL). The crude product was purified by flash chromatography to give 6 (0.17 g, 0.47 mmol) as a white solid with 75% yield. 1H NMR (300 MHz, CDCl3) δ 7.83–7.76 (m, 1H, Ar-H), 7.47–7.41 (m, 1H, Ar-H), 7.34–7.21 (m, 2H, Ar-H), 6.42 (s, 1H, Ar-H), 6.15 (s, 1H, Ar-H), 4.03–3.97 (m, 4H, morph.), 3.88–3.82 (m, 4H, morph.), 3.11 (q, J = 7.5 Hz, 2H, CH2), 2.53 (s, 3H, CH3), 1.41 (t, J = 7.5 Hz, 3H, CH3). 13C NMR (75 MHz, CDCl3) δ 156.2, 155.0, 151.2, 150.4, 148.4, 142.7, 134.6, 123.0, 122.7, 119.5, 110.2, 96.2, 87.7, 66.2, 48.4, 22.1, 14.8, 11.9. MS-ESI: m/z calcd for C20H22N6O [M+H]+: 363.19; found 363.1.
Compound 7 was prepared from compound 4 (0.20 g, 0.62 mmol), difluoroacetic acid (77 μL, 0.12 g, 1.23 mmol), HOBt (0.10 g, 0.74 mmol), EDCI (0.28 g, 1.48 mmol), TEA (0.26 mL, 0.19 g, 1.85 mmol) and DCM (6.0 mL). The crude product was purified by flash chromatography to give 7 (0.18 g, 0.47 mmol) as a white solid with 76% yield. 1H NMR (300 MHz, CDCl3) δ 7.92 (d, J = 7.1 Hz, 1H, Ar-H), 7.65 (d, J = 7.4 Hz, 1H, Ar-H), 7.47–7.38 (m, 2H, Ar-H), 7.24 (t, J = 26.8 Hz, 1H, CHF2), 6.42 (s, 1H, Ar-H), 6.28 (s, 1H, Ar-H), 4.02–3.97 (m, 4H, morph.), 3.92–3.87 (m, 4H, morph.), 2.53 (s, 3H, CH3). MS-ESI: m/z calcd for C19H18F2N6O [M+H]+: 385.16; found 385.0.
The mixture of compound 3 (0.64 g, 2.53 mmol), 1-tert-butyl-3-methyl-1H-pyrazol-5-amine (0.59 g, 3.86 mmol), cesium carbonate (1.70 g, 5.16 mmol), tris(dibenzylideneacetone)dipalladium(0) (0.13 g, 0.12 mmol), 9,9-dimethyl-4,5-bis(diphenylphosphine)xanthene (0.15 g, 0.25 mmol) and dry toluene (30 mL) were introduced to the reaction Schlenk flask. The whole mixture was flushed with argon and stirred at 100 °C for 18 h. After cooling to room temperature, the reaction mixture was filtered through the Celite®, and the solid was washed with CHCl3 (50 mL). The filtrate was concentrated under reduced pressure. The residue was resolved on a chromatographic column (amine-functionalized silica gel) (0–10% ethyl acetate gradient in heptane) to give compound 8 (0.51g, 1.38 mmol) with 54% yield. 1H NMR (300 MHz, CDCl3) δ 7.02 (s, 1H, Ar-H), 6.01 (s, 1H, Ar-H), 5.77 (s, 1H), 3.96–3.86 (m, 4H, morph.), 3.59–3.49 (m, 4H, morph.), 2.38 (s, 3H, CH3), 2.29 (s, 3H, CH3), 1.60 (s, 9H, t-Bu.). 13C NMR (75 MHz, CDCl3) δ 156.9, 154.3, 151.9, 151.0, 146.1, 137.3, 104.6, 92.1, 79.2, 66.5, 59.8, 48.8, 30.3, 15.0, 14.6. MS-ESI: m/z calcd for C19H27N7O [M+H]+: 370.24; found 370.1.
Compound 8 (0.20 g, 0.545 mmol), trifluoroacetic acid (1.0 mL), and water (4.0 mL) were refluxed for 20 h. Then, the reaction mixture was cooled to room temperature, water (10 mL) was added and the whole mixture was alkalized with saturated sodium carbonate solution (12 mL). Precipitation was observed and obtained solid was filtered off, washed with water (5 mL), and dried. The title compound 9 (0.15 g, 0.48 mmol) was isolated as a white solid with 89% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.86 (s, 1H, NH), 9.41 (s, 1H, NH), 6.30 (s, 1H, Ar-H), 6.06 (s, 1H, Ar-H), 5.85 (s, 1H, Ar-H), 3.80–3.78 (m, 4H, morph.), 3.52–3.50 (m, 4H, morph.), 2.27 (s, 3H, CH3), 2.20 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ 153.6, 151.6, 150.7, 150.1, 95.1, 91.4, 81.8, 65.6, 48.0, 14.4, 10.9. MS-ESI: m/z calcd for C15H19N7O [M+H]+: 314.17; found 314.1.
To the solution of compound 3 (1.0 eq) dissolved in 1,2-dimethoxyethane (DME) (10 mL/1 g of compound 3), boronic acid pinacol ester or boronic acid (1.5 eq), tetrakis(triphenylphosphino)palladium (0) (0.2 eq) and 2M aqueous sodium carbonate solution (2.0 eq) were added. The reaction mixture was refluxed overnight. Then, the reaction mixture was cooled to room temperature, filtered through the pad of Celite®, and obtained solid washed with ethyl acetate. The filtrate was concentrated under reduced pressure using an evaporator and the residue was purified by column chromatography.
Synthesized from compound 3 (0.15 g, 0.594 mmol), 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole-1-carboxylic acid tert-butyl ester (0.26 g, 0.890 mmol), tetrakis(triphenylphosphine)palladium(0) (0.14 g, 0.119 mmol), 2M aqueous sodium carbonate solution (0.59 mL, 1.19 mmol) and DME (6 mL). The crude product was purified by flash chromatography (0–100% ethyl acetate gradient in heptane) to give 10 (0.095 g, 0.33 mmol) with 56% yield. 1H NMR (300 MHz, DMSO-d6) δ 13.18 (s, 1H, NH), 8.49 (s, 1H, Ar-H), 8.13 (s, 1H, Ar-H), 6.60 (s, 1H, Ar-H), 6.24 (s, 1H, Ar-H), 3.90–3.78 (m, 4H, morph.), 3.78–3.63 (m, 4H, morph.), 2.37 (s, 3H, CH3). 13C NMR (75 MHz, DMSO-d6) δ 152.8, 151.7, 151.4, 149.8, 138.1, 128.7, 121.7, 93.9, 89.3, 65.9, 48.3, 14.7. MS-ESI: m/z calcd for C14H16N6O [M+H]+: 285.15; found 284.9.
Synthesized from compound 3 (0.10 g, 0.396 mmol), 2-aminopyridine-5-boronic acid pinacol ester (0.14 g, 0.594 mmol), tetrakis(triphenylphosphine)palladium(0) (91 mg, 0.079 mmol), 2M aqueous sodium carbonate solution (0.40 mL, 0.791 mmol) and DME (4 mL). the crude product was purified by flash chromatography (0–100% ethyl acetate gradient in heptane) to give 11 (0.075 g, 0.032 mol) with 61% yield. 1H NMR (300 MHz, CDCl3+CD3OD) δ 8.58 (d, J = 1.8 Hz, 1H), 8.11 (dd, J = 8.8, 1.8 Hz, 1H), 6.68 (d, J = 8.8 Hz, 1H, Ar-H), 6.47 (s, 1H, Ar-H), 6.34 (s, 1H), 4.04–3.98 (m, 4H, morph.), 3.80–3.75 (m, 4H, morph.), 2.49 (s, 3H, CH3). 13C NMR (75 MHz, CDCl3+CD3OD) δ 146.6, 136.8, 108.8, 100.2, 94.7, 88.7, 74.8, 70.2, 66.1, 29.5, 16.55, 14.0. MS-ESI: m/z calcd for C16H18N6O [M+H]+: 311.16; found 311.0.
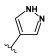
Synthesized from compound 3 (0.10 g, 0.396 mmol), 1H-indazole-4-boronic acid (0.10 g, 0.594 mmol), tetrakis(triphenylphosphine)palladium(0) (91 mg, 0.079 mmol), 2M aqueous sodium carbonate solution (0.40 mL, 0.791 mmol) and DME (4 mL). The crude product was purified by flash chromatography (0–100% ethyl acetate gradient in heptane) to give 12 (0.077 g, 0.23 mmol) with 58% yield. 1H NMR (300 MHz, CDCl3) δ 8.76–8.74 (m, 1H, NH), 7.69–7.65 (m, 1H), 7.61–7.57 (m, 1H, Ar-H), 7.53–7.46 (m, 1H), 6.59 (s, 1H, Ar-H), 6.50 (s, 1H, Ar-H), 4.06–3.98 (m, 4H, morph.), 3.85–3.76 (m, 4H, morph.), 2.54 (s, 3H, CH3). 13C NMR (75 MHz, CDCl3) δ 156.8, 154.8, 151.9, 150.6, 141.2, 135.8, 132.3, 128.9, 126.9, 121.0, 111.7, 96.3, 91.2, 66.6, 48.7, 15.2. MS-ESI: m/z calcd for C18H18N6O [M+H]+: 335.16; found 335.1.
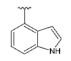
Synthesized from compound 3 (0.10 g, 0.404 mmol), indole-4-boronic acid pinacol ester (0.15 g, 0.606 mmol), tetrakis(triphenylphosphine)palladium(0) (93 mg, 0.081 mmol), 2M aqueous sodium carbonate solution (0.40 mL, 0.80 mmol) and DME (5 mL). The crude product was purified by flash chromatography (0–50% ethyl acetate gradient in heptane) to give 13 (0.074 g, 0.22 mmol) with 55% yield. 1H NMR (300 MHz, CDCl3) δ 8.68 (s, 1H, NH), 7.63–7.55 (m, 1H, Ar-H), 7.48–7.39 (m, 1H, Ar-H), 7.33–7.21 (m, 2H, Ar-H), 7.10–7.04 (m, 1H, Ar-H), 6.61 (s, 1H, Ar-H), 6.45 (s, 1H, Ar-H), 4.05–3.93 (m, 4H, morph.), 3.82–3.70 (m, 4H, morph.), 2.52 (s, 3H, CH3). 13C NMR (75 MHz, CDCl3) δ 158.4, 154.1, 151.7, 150.1, 136.6, 131.3, 125.9, 125.4, 121.9, 120.2, 112.6, 102.6, 95.5, 92.1, 66.3, 48.4, 14.8. MS-ESI: m/z calcd for C19H19N5O [M+H]+: 334.17; found 334.0.
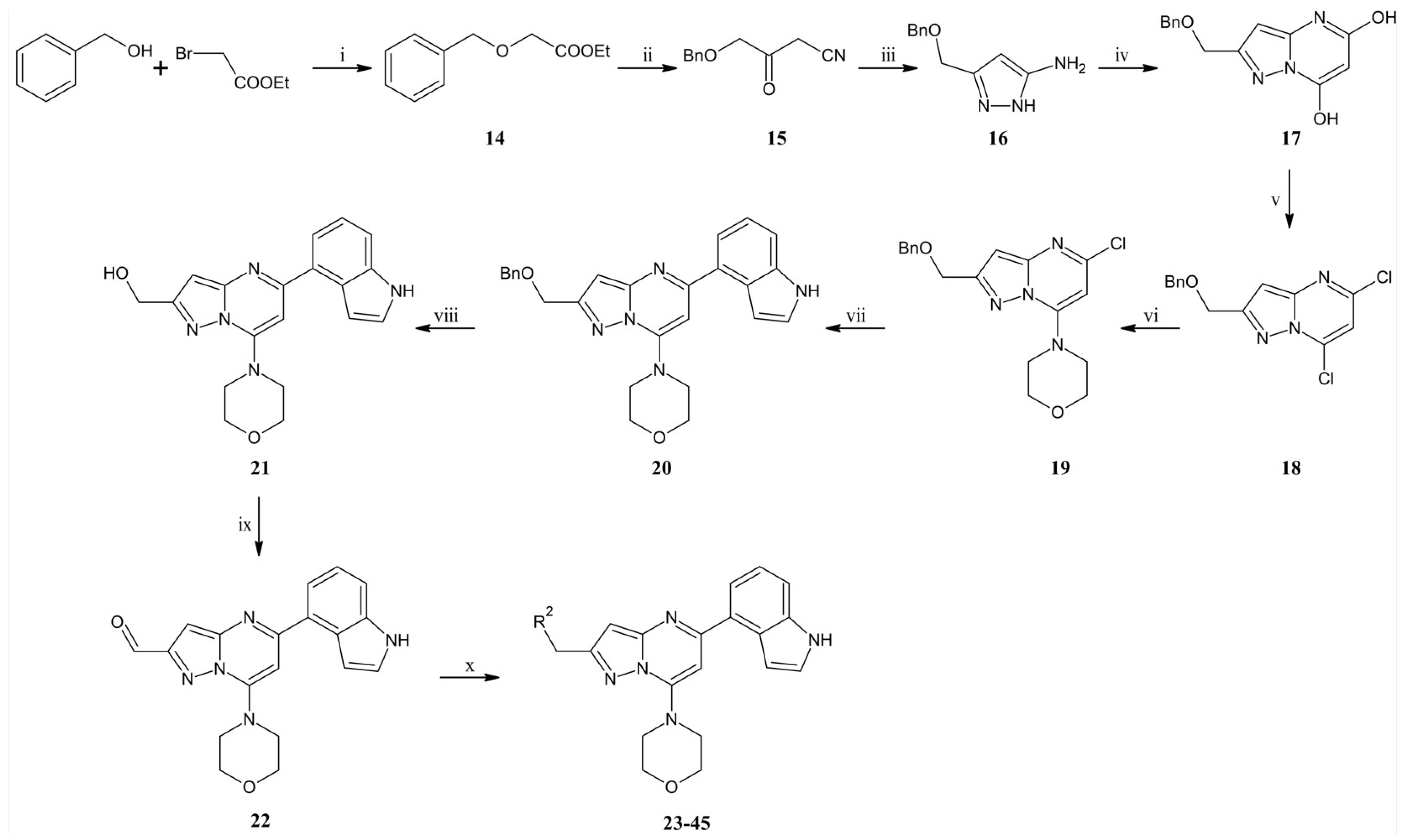
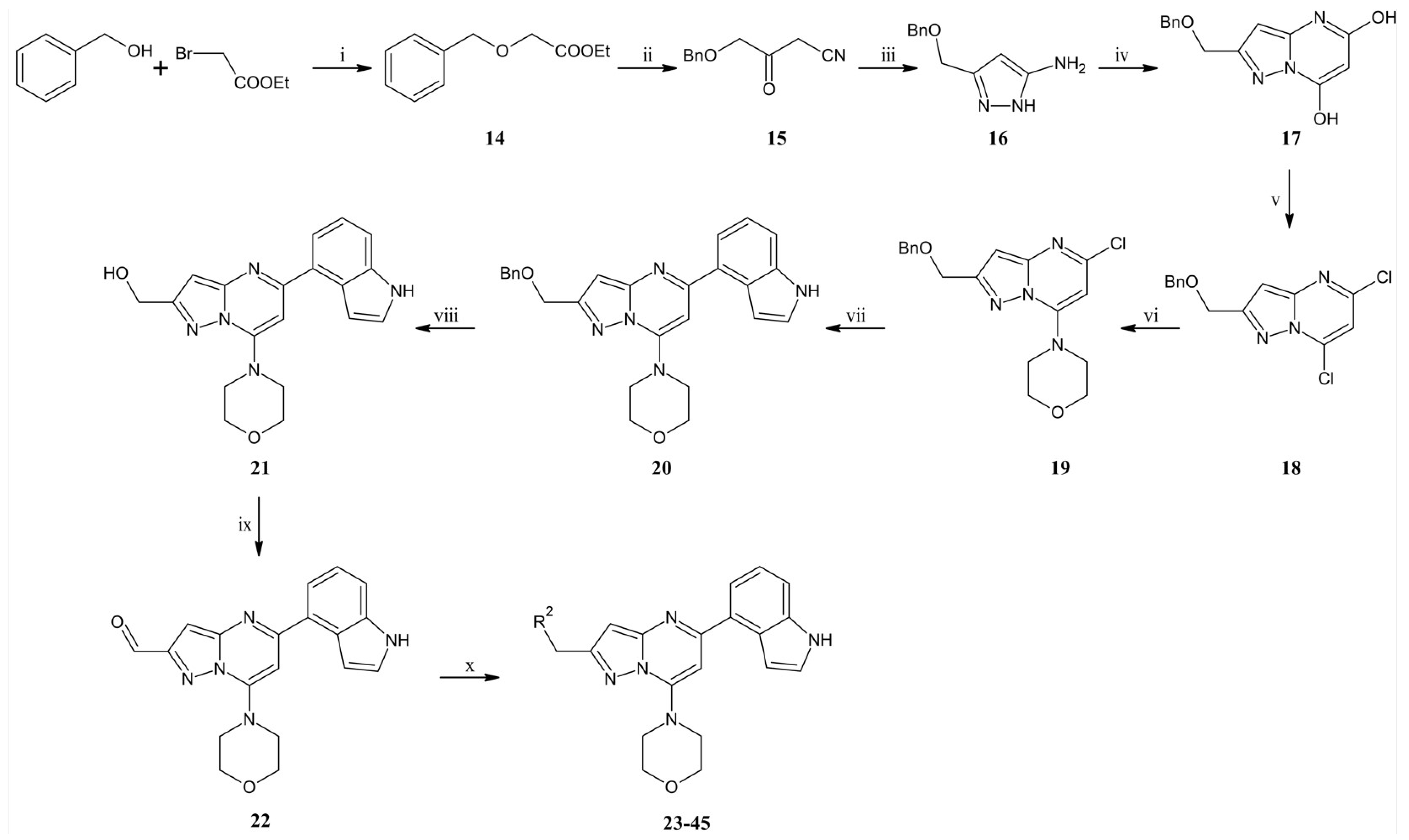
To the suspension of 60% NaH (21.8 g, 0.545 mol) in dry toluene (1000 mL), benzyl alcohol (47 mL, 0.454 mol) was added dropwise over 30 min. The whole mixture was stirred at room temperature for 4 h. The suspension was cooled in a water-ice bath and ethyl bromoacetate (66 mL, 0.595 mol) was added dropwise for 45 min. The reaction mixture was heated to room temperature and stirred for one h. The whole mixture was poured onto ice water (1200 mL) acidified with concentrated hydrochloric acid (10 mL) to pH 4. Phases were separated and the aqueous phase was extracted three times with diethyl ether (200 mL). Combined organic phases were washed with brine and dried over anhydrous magnesium sulfate. After filtration of the drying agent, organic solvents were evaporated under reduced pressure. The residue was separated by distillation under reduced pressure to give (66.7 g, 0.34 mol) ethyl 2-benzyloxyacetate (14) with 76% yield as a colorless liquid (Tb = 104–106°C / 0.7 tor). 1H NMR (500 MHz, CDCl3) δ: 7.39–7.28 (m; 5H, Ar-H), 4.63 (s; 2H, CH2), 4.23 (q; J = 7.1 Hz; 2H, CH2), 4.09 (s; 2H, CH2), 1.28 (t; J = 7.1 Hz; 3H, CH3). MS-ESI: m/z calcd for C11H14O3 [M+H]+: 195.23; found 195.1.
A flask filled with dry THF (750 mL) under an argon atmosphere was cooled to −78 °C, then 2.5 M n-BuLi hexane solution (200 mL, 0.5 mol) was added, and after that acetonitrile (28 mL, 0.533 mol) was added dropwise. The whole mixture was stirred at −78 °C for 2 h. The mixture was transferred dropwise to the suspension of ethyl 2-benzyloxyacetate (77.7 g, 0.4 mol) dropwise, and stirring was continued at −78 °C for one h. The reaction was quenched with ammonium chloride solution (500 mL). The reaction mixture was poured onto ice water and acidified with 6 M hydrochloric acid (250 mL) to pH 3. The aqueous phase was extracted twice with diethyl ether (400 mL). Combined organic phases were washed with brine and dried over anhydrous magnesium sulfate. The drying agent was filtered off, and the solvent was evaporated under reduced pressure. Compound 15 was used in the next step without additional purification. MS-ESI: m/z calcd for C11H11NO2 [M+H]+: 190.22; found 190.1.
To compound 15 (75.7 g, 0.4 mol, obtained above), ethanol (500 mL) and hydrazine monohydrate (100 mL, 2.1 mol) were added. The mixture was refluxed for 16 h. After concentration, the residue was dissolved with chloroform and dried over anhydrous sodium sulfate. Then, the drying agent was filtered off, and the solvent was evaporated. The crude product was purified by column chromatography (0–5% methanol gradient in ethyl acetate) to give 16 (70.4 g, 0.34 mol) with 87% yield after two steps as a brown oil. 1H NMR (300 MHz; CDCl3) δ: 7.39–7.28 (m; 5H, Ar-H); 5.59 (s; 1H); 4.53 (s; 2H, CH2); 4.50 (s; 2H, CH2). MS-ESI: m/z calcd for C11H13N3O [M+H]+: 204.25; found 204.1.
To the flask containing sodium ethanolate solution (obtained from sodium ethanolate (53 g, 0.74 mol) and ethanol (700 mL)), compound 16 (70.4 g, 0.35 mol) dissolved in ethanol (200 mL) and diethyl malonate (80 mL, 0.53 mol) was added. The reaction was refluxed for 24 h. Then the reaction mixture was cooled to room temperature, and the solvent was evaporated under reduced pressure. The residue was dissolved in water (1200 mL) and acidified with concentrated hydrochloric acid (250 mL). Creamy solid precipitated from the solution was filtered off, washed, and dried to give 17 (79.0 g, 0.27 mol) with 84% yield as a creamy solid. MS-ESI: m/z calcd for C14H13N3O3 [M+Na]+: 294.26; found 294.1.
The suspension of compound 17 (30 g, 0.11 mol) in acetonitrile (270 mL) was cooled to 0 °C in a water-ice bath, and POCl3 (206 mL, 2.2 mol) was added. The reaction was heated at 80 °C for five h. The reaction mixture was concentrated under reduced pressure to remove acetonitrile and POCl3. The residue was poured onto the water with ice and alkalized to pH 5 with saturated sodium hydrogen carbonate solution (350 mL). The aqueous phase was extracted with ethyl acetate, and after separation, the organic phase was dried over anhydrous sodium sulfate. After filtration of the drying agent and evaporation of the solvent, the residue was purified by column chromatography (0–20% ethyl acetate gradient in heptane to give 18 (13 g, 42.3 mmol) with 38% yield as a slightly yellow oil. 1H NMR (300 MHz, CDCl3) δ: 7.41–7.27 (m; 5H, Ar-H); 6.96 (s; 1H, Ar-H); 6.80 (s; 1H, Ar-H); 4.81 (s; 2H, CH2); 4.65 (s; 2H, CH2). MS-ESI: m/z calcd for C14H11Cl2N3O [M+H]+: 309.17; found 308.0.
To the solution of compound 18 (13 g, 42.3 mmol) dissolved in acetone (450 mL), sodium carbonate (5.38 g, 50.8 mmol), and morpholine (6.65 mL, 76.2 mmol) were added. The reaction was carried out at room temperature for 1.5 h. 500 mL of water were added to the reaction mixture, and the precipitated white solid was filtered off. The solid was washed with water (300 mL) and water/acetone mixture (2/1, v/v) (200 mL), then dried to give 19 (14 g, 39.01 mmol) with 92% yield as a white solid. 1H NMR (300 MHz, CDCl3) δ: 7.41–7.27 (m; 5H, Ar-H); 6.56 (s; 1H, Ar-H); 6.06 (s; 1H, Ar-H); 4.73 (s; 2H, CH2); 4.62 (s; 2H, CH2); 3.98–3.90 (m; 4H, morph.); 3.82–3.74 (m; 4H, morph.). MS-ESI: m/z calcd for C18H19ClN4O2 [M+H]+: 359.83; found 359.2.
To the solution of compound 19 (1.88 g, 5.24 mmol) dissolved in 1,2-dimethoxyethane (DME) (52 mL), indole-4-boronic acid pinacol ester (1.97 g, 7.87 mmol), tetrakis(triphenylphosphino)palladium (0) (0.61 g, 0.52 mmol) and 2M aqueous sodium carbonate solution (5.2 mL) were added. The reaction was refluxed for 16 h. Then, the reaction mixture was cooled to room temperature, filtered through the Celite®, and the solid was washed with ethyl acetate (50 mL). The filtrate was concentrated under reduced pressure using an evaporator. The crude product was purified by column chromatography (0–70% ethyl acetate gradient in heptane) to obtain compound 20 (1.91 g, 4.34 mmol) with an 83% yield. 1H NMR (300 MHz, CDCl3) δ: 8.61 (s; 1H); 7.61 (dd; J = 7.4; 0.8 Hz; 1H, Ar-H); 7.50–7.23 (m; 8H); 7.13–7.07 (m; 1H, Ar-H); 6.74 (s; 1H, Ar-H); 6.66 (s; 1H, Ar-H); 4.81 (s; 2H, CH2); 4.67 (s; 2H, CH2); 4.02–3.95 (m; 4H, morph.); 3.81–3.73 (m; 4H, morph.). MS-ESI: m/z calcd for C26H25N5O2 [M+H]+: 440.21; found 440.1.
To the solution of compound 20 (5.0 g, 9.1 mmol) in DMF (120 mL) and EtOH (60 mL), 10% Pd/C (11.3 g) and formic acid (100 µL) were added. The reaction was heated to 60 °C under hydrogen pressure for 24 h. After cooling the reaction mixture to room temperature, the catalyst was filtered-off on a Celite®, washed with EtOH (50 mL), and the filtrate was then concentrated under reduced pressure using an evaporator. The crude product was purified by column chromatography (0–100% ethyl acetate gradient in heptane) to give 21 (2.08 g, 5.95 mmol) with 66% yield. 1H NMR (300 MHz, DMSO-d6) δ 11.36 (s; 1H, NH); 7.70–7.63 (m; 1H, Ar-H); 7.59–7.52 (m; 1H, Ar-H); 7.52–7.46 (m; 1H, Ar-H); 7.28–7.20 (m; 1H, Ar-H); 7.14–7.09 (m; 1H, Ar-H); 6.78 (s; 1H, Ar-H); 6.55 (s; 1H, Ar-H); 5.36 (t; J = 6.0 Hz; 1H, OH); 4.66 (d; J = 6.0 Hz; 2H, CH2); 3.90–3.83 (m; 4H, morph.); 3.83–3.75 (m; 4H, morph.). MS-ESI: m/z calcd for C19H19N5O2 [M+H]+: 350.39; found 350.2.
To the solution of compound 21 (0.90 g, 2.58 mmol) in dry DMF(26 mL), Dess–Martin reagent (1.31 g, 3.09 mmol) was added. The whole mixture was stirred at room temperature for one h. The obtained solid was filtered off and then washed with ethyl acetate (35 mL). The obtained solution was concentrated under reduced pressure. The crude product was purified by flash chromatography (0–70% ethyl acetate gradient in heptane) to give 22 (0.70 g, 2.01 mmol) with 78% yield. 1H NMR (300 MHz, CDCl3) δ 10.22 (s; 1H, CHO); 8.47 (s; 1H); 7.66–7.59 (m; 1H, Ar-H); 7.57–7.50 (m; 1H, Ar-H); 7.39–7.29 (m; 2H, Ar-H); 7.18–7.09 (m; 2H, Ar-H); 6.83 (s; 1H, Ar-H); 4.08–4.00 (m; 4H, morph.); 3.86–3.77 (m; 4H, morph.). MS-ESI: m/z calcd for C19H17N5O2 [M+H]+: 348.38; found 348.1.
To the solution of compound 22 (1.0 eq) in dry DCM (10 mL/1 g of compound 22), amine derivative (1.2 eq) was added and then stirred at room temperature. After 1 h sodium triacetoxyborohydride (1.5 eq) was added and the mixture was stirred at room temperature for 15 h. To the reaction mixture was added water and phases were separated. The aqueous phase was extracted three times with DCM. Combined organic phases were dried over anhydrous sodium sulfate, filtered, and evaporated under reduced pressure. The residue was purified by flash chromatography.
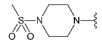
Compound 23 was prepared from aldehyde 22 (0.39 g, 0.65 mmol), 1-methanesulfonylpiperazine (0.13 g, 0.78 mmol), DCM (4.0 mL) and sodium triacetoxyborohydride (0.25 g, 1.18 mmol). The crude product was purified by flash chromatography (0–10% MeOH gradient in AcOEt) to give 23 (0.27 mg, 0.54 mmol) as a light yellow solid with 84% yield. 1HNMR (600 MHz, DMSO-d6) δ 11.31 (s, 1H, NH), 7.64 (dd, J = 7.4, 0.8 Hz, 1H, Ar-H), 7.53 (dt, J = 8.0, 0.8 Hz, 1H, Ar-H), 7.47 (t, J = 2.8 Hz, 1H, Ar-H), 7.22 (t, J = 7.7 Hz, 1H, Ar-H), 7.10–7.09 (m, 1H, Ar-H), 6.77 (s, 1H, Ar-H), 6.51 (s, 1H, Ar-H), 3.86–3.84 (m, 4H, morph.), 3.79–3.77 (m, 4H, morph.), 3.74 (s, 2H, CH2), 3.14–3.13 (m, 4H), 2.86 (s, 3H, CH3), 2.58–2.57 (m, 4H). 13CNMR (151 MHz, DMSO-d6) δ 157.9, 153.5, 151.0, 149.5, 136.7, 129.9, 126.4, 125.6, 120.7, 119.5, 113.2, 101.8, 94.7, 91.5, 65.6, 55.5, 51.8, 47.8, 45.4, 33.7. HRMS (ESI): m/z calcd for C24H29N7O3S [M+H]+: 496.2125; found 496.2134.
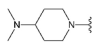
Compound 24 was prepared from aldehyde 22 (0.18 g, 0.52 mmol), 4-(dimethylamino)piperidine dihydrochloride (0.13 g, 0.62 mmol), DCM (3.5 mL), triethylamine (0.17 mL, 1.24 mmol) and sodium triacetoxyborohydride (0.17 g, 0.78 mmol). The crude product was purified by flash chromatography (0–20% MeOH gradient in AcOEt) to give 24 (0.18 g, 0.39 mmol)) as a light yellow solid with 76% yield. 1H NMR (300 MHz, CDCl3) δ: 8.85 (s; 1H, NH); 7.60 (d; J = 7.2 Hz; 1H, Ar-H); 7.51 (d; J = 8.2 Hz; 1H, Ar-H); 7.36–7.28 (m; 2H, Ar-H); 7.12–7.08 (m; 1H, Ar-H); 6.65 (s; 1H, Ar-H); 6.61 (s; 1H, Ar-H); 4.04–3.94 (m; 4H, morph.); 3.80 (s; 2H, CH2); 3.79–3.72 (m; 4H, morph.); 3.19–3.07 (m; 2H, CH2); 2.60–2.49 (m; 1H, CH); 2.44 (s; 6H, 2xCH3); 2.22–2.09 (m; 2H, CH2); 1.98–1.86 (m; 2H); 1.76–1.59 (m; 2H) HRMS (ESI): m/z calcd for C26H33N7O [M+H]+: 460.2819; found 460.2842.
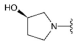
Compound 25 was prepared from aldehyde 22 (0.17 g, 0.48 mmol), (R)-(+)-3-pyrrolidinol (53 mg, 0.58 mmol), DCM (2.0 mL) and sodium triacetoxyborohydride (0.18 mg, 0.86 mmol). The crude product was purified by flash chromatography (0–30% MeOH gradient in AcOEt) to give 25 (50 mg, 0.12 mmol) as a light yellow solid with 25% yield. 1H NMR (600 MHz, DMSO-d6) δ 11.31 (s, 1H, NH), 7.64 (dd, J = 7.4, 0.6 Hz, 1H, Ar-H), 7.53 (d, J = 8.0 Hz, 1H, Ar-H), 7.46 (t, J = 2.8 Hz, 1H, Ar-H), 7.21 (t, J = 7.7 Hz, 1H, Ar-H), 7.09 (t, J = 2.1 Hz, 1H, Ar-H), 6.76 (s, 1H, Ar-H), 6.50 (s, 1H, Ar-H), 4.21–4.19 (m, 1H), 3.86–3.84 (m, 4H, morph.), 3.82–3.72 (m, 6H), 2.80–2.77 (m, 1H), 2.71–2.67 (m, 1H), 2.55–2.52 (m, 1H), 2.44–2.42 (m, 1H), 2.01–1.98 (m, 1H), 1.57–1.54 (m, 1H). 13C NMR (151 MHz, DMSO-d6) δ 157.8, 154.6, 150.9, 149.5, 136.7, 130.0, 126.4, 125.6, 120.7, 119.5, 113.1, 101.8, 94.6, 91.4, 69.4, 65.6, 62.5, 53.3, 52.3, 47.8, 34.5. HRMS (ESI): m/z calcd for C23H26N6O2 [M+H]+: 419.2190; found 419.2191.
Compound 26 was prepared from aldehyde 22 (0.17 mg, 0.48 mmol), (S)-3-pyrrolidinol (52 mg, 0.58 mmol), DCM (2.0 mL) and sodium triacetoxyborohydride (0.18 mg, 0.86 mmol). The crude product was purified by flash chromatography (0–30% MeOH gradient in AcOEt) to give 26 (70 mg, 0.17 mmol) as a light yellow solid with 35% yield. 1H NMR (600 MHz, DMSO-d6) δ 11.33 (s, 1H, NH), 7.64 (dd, J = 7.4, 0.7 Hz, 1H, Ar-H), 7.53 (d, J = 8.0 Hz, 1H, Ar-H), 7.47 (t, J = 2.8 Hz, 1H, Ar-H), 7.22–7.20 (m, 1H, Ar-H), 7.09–7.08 (m, 1H, Ar-H), 6.76 (s, 1H, Ar-H), 6.50 (s, 1H, Ar-H), 4.21–4.19 (m, 1H), 3.86–3.84 (m, 4H, morph.), 3.78–3.72 (m, 6H), 2.78 (m, 1H), 2.68 (m, 1H), 2.54–2.50 (m, 1H), 2.43 (m, 1H), 2.03–1.97 (m, 1H), 1.57–1.54 (m, 1H). 13C NMR (151 MHz, DMSO-d6) δ 157.8, 154.6, 150.9, 149.6, 136.8, 130.0, 126.5, 125.6, 120.8, 119.6, 113.2, 101.8, 94.6, 91.5, 69.5, 65.6, 62.6, 53.4, 52.4, 47.9, 34.5. HRMS (ESI): m/z calcd for C23H26N6O2 [M+H]+: 419.2190; found 419.2200.
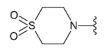
Compound 27 was prepared from aldehyde 22 (85 mg, 0.25 mmol), thiomorpholine-1,1-dioxide (40 mg, 0.29 mmol), DCM (3.0 mL) and sodium triacetoxyborohydride (78 mg, 0.37 mmol). The crude product was purified by flash chromatography (0–10% MeOH gradient in AcOEt) to give 27 (48 mg, 0.10 mmol) as a light yellow solid with 42% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.33 (s, 1H, NH), 7.62 (dd, J = 7.4, 0.8 Hz, 1H, Ar-H), 7.51 (d, J = 8.1 Hz, 1H, Ar-H), 7.44 (t, J = 2.8 Hz, 1H, Ar-H), 7.19 (t, J = 7.7 Hz, 1H, Ar-H), 7.07–7.06 (m, 1H, Ar-H), 6.75 (s, 1H, Ar-H), 6.54 (s, 1H, Ar-H), 3.87 (s, 2H), 3.83–3.81 (m, 3H), 3.75–3.74 (m, 3H), 3.12–3.09 (m, 4H), 2.98–2.95 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 158.0, 153.3, 151.0, 149.6, 136.8, 129.9, 126.5, 125.6, 120.8, 119.6, 113.3, 101.8, 94.8, 91.7, 65.6, 54.2, 50.6, 50.2, 47.9. HRMS (ESI): m/z calcd for C23H26N6O3S [M+H]+: 467.1860; found 467.1866.
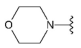
Compound 28 was prepared from aldehyde 22 (0.20 g, 0.23 mmol), morpholine (24 mL, 24 mg, 0.27 mmol), DCM (3.0 mL) and sodium triacetoxyborohydride (95 mg, 0.45 mmol). The crude product was purified by flash chromatography (0–10% MeOH gradient in AcOEt) to give 28 (55 mg, 0.13 mmol) as a light yellow solid with 59% yield. 1H NMR (500 MHz, DMSO-d6) δ 11.32 (s, 1H, NH), 7.65 (d, J = 7.4 Hz, 1H, Ar-H), 7.54 (d, J = 7.4 Hz, 1H, Ar-H), 7.49–7.45 (m, 1H, Ar-H), 7.25–7.19 (m, 1H, Ar-H), 7.12–7.08 (m, 1H, Ar-H), 6.77 (s, 1H, Ar-H), 6.52 (s, 1H, Ar-H), 3.89–3.83 (m, 4H, morph.), 3.81–3.76 (m, 4H, morph.), 3.68 (s, 2H, CH2), 3.63–3.57 (m, 4H, morph.), 2.49–2.45 (m, 4H, morph.). 13C NMR (126 MHz, DMSO-d6) δ: 157.9, 153.6, 151.0, 149.6, 136.8, 129.9, 126.5, 125.6, 120.8, 119.6, 113.2, 101.8, 94.8, 91.5, 66.2, 65.6, 56.4, 53.2, 47.9. HRMS (ESI): m/z calcd for C23H26N6O2 [M+H]+: 419.2190; found 419.2196.
Compound 29 was prepared from aldehyde 22 (0.17 g, 0.50 mmol), 1-methyl-4-(piperidin-4-yl)piperazine (0.11 g, 0.6 mmol), DCM (3.5 mL) and sodium triacetoxyborohydride (0.16 g, 0.75 mmol). The crude product was purified by flash chromatography (0–15% MeOH gradient in AcOEt) to give 29 (0.23 g, 0.45 mmol) as a yellow solid with 89% yield. 1H NMR (300 MHz, CDCl3) δ: 9.56 (s; 1H); 7.62–7.55 (m; 1H, Ar-H); 7.47–7.1 (m; 1H, Ar-H); 7.31–7.21 (m; 2H, Ar-H); 7.11–7.02 (m; 1H, Ar-H); 6.64 (s; 1H, Ar-H); 6.62 (s; 1H, Ar-H); 4.00–3.90 (m; 4H, morph.); 3.86–3.62 (m; 6H); 3.19–3.05 (m; 2H); 2.81–2.45 (m; 8H); 2.34 (s; 3H); 2.39–2.29 (m; 1H); 2.21–2.08 (m; 2H); 1.91–1.79 (m; 2H); 1.75–1.54 (m; 2H). 13C NMR (75 MHz, CDCl3) δ 158.8, 154.2, 151.6, 150.2, 136.8, 131.1, 126.1, 125.8, 121.8, 120.1, 113.0, 102.4, 96.0, 92.5, 66.3, 61.9, 56.5, 54.8, 53.0, 48.5, 48.4, 45.6, 27.9. HRMS (ESI): m/z calcd for C29H38N8O [M+H]+: 515.3241; found 515.3224.
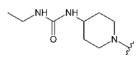
The 3-ethyl-1-(piperidin-4-yl)urea was synthesizedaccording to the van Duzer et al. procedure [
56]. The urea derivative was used in the reductive amination reaction (next step) as is, without additional purification.
Compound 30 was prepared from aldehyde 22 (0.20 g, 0.58 mmol), 3-ethyl-1-(piperidin-4-yl)urea hydrochloride (0.14 g, 0.69 mmol), DCM (4.0 mL), triethylamine (0.194 mL, 1.38 mmol) and sodium triacetoxyborohydride (0.19 g, 0.86 mmol). The crude product was purified by flash chromatography (0–15% MeOH gradient in AcOEt) to give 30 (0.15 g, 0.30 mmol) as a light yellow solid with 52% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.33 (s, 1H), 7.64 (d, J = 7.4 Hz, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.47 (t, J = 2.7 Hz, 1H), 7.21 (t, J = 7.7 Hz, 1H, Ar-H), 7.09–7.09 (m, 1H, Ar-H), 6.76 (s, 1H, Ar-H), 6.48 (s, 1H, Ar-H), 5.71 (d, J = 7.8 Hz, 1H), 5.65 (t, J = 5.5 Hz, 1H), 3.86–3.84 (m, 4H), 3.78–3.77 (m, 4H), 3.64 (s, 2H), 3.36–3.36 (m, 1H), 3.01–2.94 (m, 2H), 2.81–2.78 (m, 2H), 2.15–2.10 (m, 2H), 1.74–1.72 (m, 2H, CH2), 1.38–1.32 (m, 2H, CH2), 0.96 (t, J = 7.1 Hz, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ 157.9, 157.3, 154.3, 151.0, 149.5, 136.8, 130.0, 126.5, 125.6, 120.8, 119.6, 113.2, 101.8, 94.7, 91.5, 65.6, 56.2, 52.0, 47.9, 46.1, 33.9, 32.5, 15.7. HRMS (ESI): m/z calcd for C27H34N8O2 [M+H]+: 503.2877; found 503.2882.
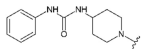
The synthesis of 1-phenyl-3-(piperidin-4-yl)urea was conducted according to the van Duzer et al.procedure [
56]. The urea derivative was used in the reductive amination reaction (next step)as is, without additional purification.
Compound 31 was prepared from aldehyde 22 (0.20 g, 0.58 mmol), 1-phenyl-3-(piperidin-4-yl)urea hydrochloride (0.18 g, 0.69 mmol), DCM (4.0 mL), triethylamine (0.194 mL, 1.38 mmol) and sodium triacetoxyborohydride (0.19 g, 0.86 mmol). The crude product was purified by flash chromatography (0–15% MeOH gradient in AcOEt) to give 31 (0.18 g, 0.33 mmol) as a light yellow solid with 58% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.33 (s, 1H, NH), 8.30 (s, 1H), 7.65 (dd, J = 7.4, 0.8 Hz, 1H), 7.53 (d, J = 8.1 Hz, 1H), 7.47 (t, J = 2.8 Hz, 1H), 7.37–7.34 (m, 2H), 7.24–7.16 (m, 3H), 7.10–7.09 (m, 1H, Ar-H), 6.88–6.84 (m, 1H, Ar-H), 6.76 (s, 1H, Ar-H), 6.51 (s, 1H, Ar-H), 6.11 (d, J = 7.7 Hz, 1H), 3.86–3.84 (m, 4H, morph.), 3.79–3.78 (m, 4H, morph.), 3.67 (s, 2H, CH2), 3.49–3.48 (m, 1H), 2.83–2.80 (m, 2H, CH2), 2.22–2.17 (m, 2H, CH2), 1.98–1.80 (m, 2H, CH2), 1.47–1.38 (m, 2H, CH2). 13C NMR (101 MHz, DMSO-d6) δ 157.9, 154.5, 154.2, 151.0, 149.6, 140.5, 136.8, 130.0, 128.6, 126.5, 125.6, 120.9, 120.8, 119.6, 117.5, 113.2, 101.8, 94.7, 91.5, 65.6, 56.2, 51.7, 47.9, 46.0, 32.2. HRMS (ESI): m/z calcd for C31H34N8O2 [M+H]+: 551.2887; found 551.2880.
Compound 32 was prepared from aldehyde 22 (0.18 g, 0.52 mmol), 1-(4-methoxyphenyl)piperazine (0.12 g, 0.63 mmol), DCM (3.5 mL) and sodium triacetoxyborohydride (0.17 g, 0.79 mmol). The crude product was purified by flash chromatography (0–5% MeOH gradient in AcOEt) to give 32 (0.22 g, 0.42 mmol) as a light yellow solid with 81% yield. 1H NMR (300 MHz, CDCl3) δ: 8.52 (s; 1H, NH); 7.61 (d; J = 7.4 Hz; 1H, Ar-H); 7.49 (d; J = 8.1 Hz; 1H, Ar-H); 7.35–7.27 (m; 2H, Ar-H); 7.14–7.09 (m; 1H, Ar-H); 6.96–6.80 (m; 4H); 6.67 (s; 1H, Ar-H); 6.65 (s; 1H, Ar-H); 4.04–3.95 (m; 4H, morph.); 3.88 (s; 2H, CH2); 3.82–3.76 (m; 4H, morph.); 3.77 (s; 3H, CH3); 3.19–3.11 (m; 4H, piperaz.); 2.84–2.74 (m; 4H, piperaz.). 13C NMR (75 MHz, CDCl3) δ 158.8, 154.3, 153.8, 151.7, 150.3, 145.8, 136.8, 131.3, 126.1, 125.6, 122.0, 120.3, 118.3, 114.5, 112.9, 102.7, 96.1, 92.5, 66.4, 56.8, 55.7, 53.4, 50.7, 48.6. HRMS (ESI): m/z calcd for C30H33N7O2 [M+H]+: 524.2769; found 524.2770.
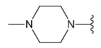
Compound 33 was prepared from aldehyde 22 (85 mg, 0.24 mmol), 1-methylpiperazine, (33 mL, 29 mg, 0.29 mmol), DCM (4.0 mL) and sodium triacetoxyborohydride (78 mg, 0.37 mmol). The crude product was purified by flash chromatography (0–20% MeOH gradient in AcOEt) to give 33 (91 mg, 0.21 mmol) as a light yellow solid with 86% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.41 (s, 1H, NH), 7.64 (d, J = 7.4 Hz, 1H, Ar-H), 7.53 (d, J = 8.1 Hz, 1H, Ar-H), 7.46 (t, J = 2.7 Hz, 1H, Ar-H), 7.21 (t, J = 7.7 Hz, 1H, Ar-H), 7.09 (t, J = 2.0 Hz, 1H, Ar-H), 6.76 (s, 1H, Ar-H), 6.48 (s, 1H, Ar-H), 3.85–3.83 (m, 4H, morph.), 3.78–3.76 (m, 4H, morph.), 3.65 (s, 2H, CH2), 2.50–2.45 (m, 4H, piperaz.), 2.32–2.32 (m, 4H, piperaz.), 2.14 (s, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ 157.9, 154.0, 151.0, 149.6, 136.8, 129.9, 126.5, 125.6, 120.7, 119.6, 113.2, 101.8, 94.7, 91.5, 65.6, 56.0, 54.7, 52.6, 47.9, 45.7. HRMS (ESI): m/z calcd for C24H29N7O [M+H]+: 432.2506; found 432.2511.
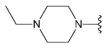
Compound 34 was prepared from aldehyde 22 (85 mg, 0.24 mmol), 1-ethylpiperazine (37 mL, 33 mg, 0.29 mmol), DCM (4.0 mL) and sodium triacetoxyborohydride (78 mg, 0.37 mmol). The crude product was purified by flash chromatography (0–20% MeOH gradient in AcOEt) to give 34 (85 mg, 0.19 mmol) as a light yellow solid with 78% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.37 (s, 1H, NH), 7.64 (d, J = 7.4 Hz, 1H, Ar-H), 7.53 (d, J = 8.1 Hz, 1H, Ar-H), 7.46 (t, J = 2.7 Hz, 1H, Ar-H), 7.21 (t, J = 7.7 Hz, 1H, Ar-H), 7.09–7.08 (m, 1H, Ar-H), 6.76 (s, 1H, Ar-H), 6.49 (s, 1H, Ar-H), 3.86–3.84 (m, 4H, morph.), 3.78–3.77 (m, 4H, morph.), 3.67 (s, 2H, CH2), 2.63–2.44 (m, 8H, piperaz.), 2.39 (q, J = 7.2 Hz, 2H, CH2), 1.00 (t, J = 7.2 Hz, 3H, CH3). 13C NMR (101 MHz, DMSO-d6) δ 172.0, 157.9, 153.8, 151.0, 149.6, 136.8, 129.9, 126.5, 125.6, 120.7, 119.6, 113.2, 101.8, 94.8, 91.5, 65.6, 55.9, 52.1, 51.4, 47.9, 21.1. HRMS (ESI): m/z calcd for C25H31N7O [M+H]+: 446.2663; found 446.2661.
Compound 35 was prepared from aldehyde 22 (85 mg, 0.24 mmol), methyl isonipecotate, (42 mg, 0.29 mmol), DCM (4.0 mL) and sodium triacetoxyborohydride (78 mg, 0.37 mmol). The crude product was purified by flash chromatography (0–10% MeOH gradient in CHCl3) to give 35 (76 mg, 0.16 mmol) as a light yellow solid with 65% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.35 (s, 1H, NH), 7.64 (dd, J = 7.4, 0.7 Hz, 1H, Ar-H), 7.53 (d, J = 8.1 Hz, 1H, Ar-H), 7.47 (t, J = 2.8 Hz, 1H, Ar-H), 7.21 (t, J = 7.7 Hz, 1H, Ar-H), 7.10–7.09 (m, 1H, Ar-H), 6.76 (s, 1H, Ar-H), 6.49 (s, 1H, Ar-H), 3.86–3.83 (m, 4H, morhp.), 3.78–3.77 (m, 4H, morph.), 3.65 (s, 2H, CH2), 3.58 (s, 3H, CH3), 2.87–2.84 (m, 2H, CH2), 2.32–2.27 (m, 1H, CH), 2.11–2.06 (m, 2H, CH2), 1.82–1.79 (m, 2H, CH2), 1.63–1.57 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 174.8, 157.9, 154.0, 151.0, 149.5, 136.8, 130.0, 126.5, 125.6, 120.7, 119.6, 113.2, 101.8, 94.7, 91.5, 65.6, 56.2, 52.2, 51.3, 47.9, 40.1, 28.0. HRMS (ESI): m/z calcd for C26H30N6O3 [M+H]+: 475.2452; found 475.2458.
Compound 36 was prepared from aldehyde 22 (0.18 g, 0.52 mmol), 2-(4-piperidyl)-2-propanol (93 mg, 0.62 mmol), DCM (3.5 mL) and sodium triacetoxyborohydride (0.17 g, 0.78 mmol). The crude product was purified by flash chromatography (0–5% MeOH gradient in AcOEt) to give 36 (0.183 g, 0.39 mmol) as an off-white solid with 74% yield. 1H NMR (300 MHz, CDCl3) δ: 8.57 (s; 1H, NH); 7.64–7.58 (m; 1H, Ar-H); 7.52–7.46 (m; 1H, Ar-H); 7.36–7.25 (m; 2H, Ar-H); 7.14–7.09 (m; 1H, Ar-H); 6.64 (s; 1H, Ar-H); 6.64 (s; 1H, Ar-H); 4.03–3.95 (m; 4H, morph.); 3.82 (s; 2H, CH2); 3.81–3.71 (m; 4H, morph.); 3.20–3.10 (m; 2H, CH2); 2.18–2.03 (m; 2H, CH2); 1.81–1.69 (m; 2H, CH2); 1.56–1.38 (m; 2H, CH2); 1.35–1.30 (m; 1H); 1.18 (s; 6H). 13C NMR (75 MHz, CDCl3) δ 158.7, 151.7, 150.2, 136.8, 131.3, 126.1, 125.6, 122.0, 120.3, 112.9, 102.7, 96.2, 92.4, 72.7, 66.4, 56.9, 54.2, 48.6, 47.3, 27.1, 27.0. HRMS (ESI): m/z calcd for C27H34N6O6 [M+H]+: 475.2816; found 475.2815.
Compound 37 was prepared from aldehyde 22 (0.12 g, 0.35 mmol), N-tert-butylpiperazine (59 mg, 0.42 mmol), DCM (2.0 mL) and sodium triacetoxyborohydride (0.11 g, 0.52 mmol). The crude product was purified by flash chromatography (0–20% MeOH gradient in AcOEt) to give 37 (0.15 g, 0.32 mmol) as a yellow solid with 93% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.33 (s, 1H, NH), 7.64 (dd, J = 7.4, 0.9 Hz, 1H, Ar-H), 7.53 (dt, J = 8.1, 0.8 Hz, 1H, Ar-H), 7.46 (t, J = 2.8 Hz, 1H, Ar-H), 7.21 (t, J = 7.7 Hz, 1H, Ar-H), 7.10–7.08 (m, 1H, Ar-H), 6.76 (s, 1H, Ar-H), 6.48 (s, 1H, Ar-H), 3.86–3.84 (m, 4H, morph.), 3.78–3.76 (m, 4H, morph.), 3.63 (s, 2H, CH2), 2.53–2.45 (m, 8H, piperaz.), 0.98 (s, 9H, t-Bu.). 13C NMR (101 MHz, DMSO-d6) δ 157.8, 154.0, 151.0, 149.5, 136.8, 130.0, 126.5, 125.6, 120.7, 119.6, 113.2, 101.8, 94.8, 91.5, 65.6, 56.0, 53.5, 53.1, 47.9, 45.2, 25.7. HRMS (ESI): m/z calcd for C27H35N7O [M+H]+: 474.2976; found 474.2976.
Compound 38 was prepared from aldehyde 22 (0.20 g, 0.58 mmol), 2-methyl-2-(piperazin-1-yl)propenamide dihydrochloride (0.18 g, 0.69 mmol), DCM (3.0 mL), triethylamine (0.194 mL, 1.38 mmol) and sodium triacetoxyborohydride (0.18 g, 0.86 mmol). The crude product was purified by flash chromatography (0–10% MeOH gradient in AcOEt) to give 38 (0.21 g, 0.42 mmol) as a light yellow solid with 73% yield. 1H NMR (500 MHz, DMSO-d6) δ 11.32 (s, 1H, NH), 7.65 (d, J = 7.3 Hz, 1H, Ar-H), 7.54 (d, J = 8.0 Hz, 1H, Ar-H), 7.49–7.45 (m, 1H, Ar-H), 7.22 (t, J = 7.7 Hz, 1H, Ar-H), 7.12–7.08 (m, 1H, Ar-H), 7.07–7.01 (m, 1H, Ar-H), 6.95–6.90 (m, 1H), 6.77 (s, 1H), 6.50 (s, 1H), 3.89–3.82 (m, 4H, morph.), 3.82–3.76 (m, 4H, morph.), 3.68 (s, 2H, CH2), 2.57–2.51 (m, 4H), 2.49–2.40 (m, 4H), 1.06 (s, 6H). 13C NMR (126 MHz, DMSO-d6) δ 178.1, 157.9, 153.9, 151.0, 149.5, 136.8, 130.0, 126.5, 125.6, 120.8, 119.6, 113.2, 101.8, 94.8, 91.5, 65.6, 62.4, 56.0, 53.2, 47.9, 46.1, 20.4. HRMS (ESI): m/z calcd for C27H34N8O2 [M+H]+: 503.2877; found 503.2901.
Compound 39 was prepared from aldehyde 22 (85 mg, 0.25 mmol), 1-cyclopropylpiperazine (35 mL, 37 mg, 0.29 mmol), DCM (3.0 mL) and sodium triacetoxyborohydride (78 mg, 0.37 mmol). The crude product was purified by flash chromatography (0–15% MeOH gradient in AcOEt) to give 39 (84 mg, 0.18 mmol) as a light yellow solid with 75% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.32 (s, 1H, NH), 7.64 (dd, J = 7.4, 0.7 Hz, 1H, Ar-H), 7.53 (d, J = 8.1 Hz, 1H, Ar-H), 7.47 (t, J = 2.8 Hz, 1H, Ar-H), 7.21 (t, J = 7.7 Hz, 1H, Ar-H), 7.09 (t, J = 2.1 Hz, 1H), 6.76 (s, 1H, Ar-H), 6.48 (s, 1H, Ar-H), 3.85–3.83 (m, 4H, morph.), 3.78–3.76 (m, 4H, morph.), 3.64 (s, 2H, CH2), 2.54 (s, 4H, piperaz.), 2.43 (s, 4H, piperaz.), 1.58 (s, 1H, CH), 0.38–0.36 (m, 2H, CH2), 0.26–0.24 (m, 2H, CH2). 13C NMR (101 MHz, DMSO-d6) δ 157.9, 154.0, 151.0, 149.5, 136.8, 130.0, 126.5, 125.6, 120.7, 119.6, 113.2, 101.8, 94.7, 91.5, 65.6, 56.0, 52.7, 52.7, 47.9, 38.0, 5.6. HRMS (ESI): m/z calcd for C26H31N7O [M+H]+: 458.2663; found 458.2666.
Compound 40 was prepared from aldehyde 22 (70 mg, 0.20 mmol), 1-cyclopentylpiperazine (39 mL, 38 mg, 0.24 mmol), DCM (4.0 mL) and sodium triacetoxyborohydride (64 mg, 0.30 mmol). The crude product was purified by flash chromatography (0–15% MeOH gradient in AcOEt) to give 40 (84 mg, 0.17 mmol) as a light yellow solid with 86% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.34 (s, 1H, NH), 7.64 (dd, J = 7.4, 0.7 Hz, 1H, Ar-H), 7.53 (d, J = 8.1 Hz, 1H, Ar-H), 7.46 (t, J = 2.8 Hz, 1H, Ar-H), 7.21 (t, J = 7.7 Hz, 1H, Ar-H), 7.09 (t, J = 2.1 Hz, 1H, Ar-H), 6.76 (s, 1H, Ar-H), 6.48 (s, 1H, Ar-H), 3.86–3.83 (m, 4H, morph.), 3.78–3.76 (m, 4H, morph.), 3.64 (s, 2H, CH2), 2.53–2.42 (m, 9H), 1.75–1.72 (m, 2H, CH2), 1.59–1.55 (m, 2H), 1.48–1.44 (m, 2H), 1.31–1.26 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 157.9, 153.9, 151.0, 149.5, 136.8, 130.0, 126.4, 125.6, 120.7, 119.6, 113.2, 101.8, 94.7, 91.5, 66.7, 65.6, 56.0, 52.7, 51.6, 47.9, 29.8, 23.6. HRMS (ESI): m/z calcd for C28H35N7O [M+H]+: 486.2976; found 486.2973.
Compound 41 was prepared from aldehyde 22 (0.10 g, 0.29 mmol), 4-(tert-butyl)piperidine hydrochloride (61 mg, 0.35 mmol), DCM (4.0 mL), triethylamine (0.097 mL, 0.69 mmol) and sodium triacetoxyborohydride (94 mg, 0.43 mmol). The crude product was purified by flash chromatography (0–20% MeOH gradient in CHCl3) to give 41 (0.10 g, 0.21 mmol) as a light yellow solid with 76% yield. 1H NMR (400 MHz, DMSO-d6) 11.32 (s, 1H, NH), 7.64 (dd, J = 7.4, 0.7 Hz, 1H, Ar-H), 7.53 (d, J = 8.1 Hz, 1H, Ar-H), 7.46 (t, J = 2.8 Hz, 1H, Ar-H), 7.21 (t, J = 7.7 Hz, 1H, Ar-H), 7.10–7.09 (m, 1H, Ar-H), 6.75 (s, 1H, Ar-H), 6.48 (s, 1H, Ar-H), 3.85–3.83 (m, 4H, morph.), 3.78–3.76 (m, 4H, morph.), 3.61 (s, 2H, CH2), 2.98–2.95 (m, 2H, CH2), 1.95–1.89 (m, 2H, CH2), 1.59–1.56 (m, 2H, CH2), 1.27–1.17 (m, 2H, CH2), 0.96–0.90 (m, 1H, CH), 0.81 (s, 9H, t-Bu.). 13C NMR (101 MHz, DMSO-d6) δ 157.8, 154.3, 151.0, 149.5, 136.8, 130.0, 126.4, 125.6, 120.7, 119.5, 113.2, 101.9, 94.7, 91.4, 65.6, 56.4, 54.0, 47.9, 45.8, 31.8, 27.2, 26.4. HRMS (ESI): m/z calcd for C28H36N6O [M+H]+: 473.3023; found 473.3028.
The multistep preparation of compound 42 started from 1-(oxetan-3-yl) piperazine.
Step 1.
To the solution of 3-oxetanone (0.23 mL, 0.28 g, 3.9 mmol) in dry DCM (39.0 mL), 1-Boc-piperazine (0.60 g, 3.2 mmol) was added, and then the mixture was stirred at room temperature. After four h, sodium triacetoxyborohydride (1.35 g, 6.4 mmol) was added, and stirring was continued at room temperature overnight. Then, water (30 mL) was added to the reaction mixture, and the phases were separated. The aqueous phase was extracted three times with chloroform (25 mL). Combined organic phases were dried over anhydrous sodium sulfate, filtrated the drying agent, and the solvent was evaporated under reduced pressure to obtain tert-butyl 4-(oxetan-3-yl)piperazin-1-carboxylate (0.61 g, 2.52 mmol) with 65% yield without purification. 1H NMR (300 MHz, CDC13) δ 4.68–4.52 (m; 4H, piperaz.); 3.50–3.32 (m; 5H); 2.31–2.09 (m; 4H); 1.43 (s; 9H, t-Bu.). MS-ESI: (m/z) calcd for C12H22N2O3 [M+H]+: 243.17; found 243.2.
Step 2.
To the solution of the product of Step 1 (0.55 g, 2.8 mmol) in DCM (28 mL), trifluoroacetic acid (16.8 mL) was added. The reaction was carried out at room temperature for two h. Then, the water was added (30 mL), and the reaction mixture was alkalized with saturated sodium carbonate solution (10 mL). Phases were separated, and the aqueous phase was extracted three times with chloroform (25 mL). Combined organic phases were dried over anhydrous sodium sulfate. The drying agent was filtered off and the solvent evaporated under reduced pressure to obtain 1-(oxetan-3-yl)piperazine (0.23 g, 1.61 mmol) with 57% yield without purification. 1H NMR (300 MHz, CDCl3) δ 4.66–4.56 (m; 4H); 3.66–3.56 (m; 1H); 3.30–3.12 (m; 4H); 2.68–2.51 (m; 4H). MS-ESI: (m/z) calcd for C7H14N2O [M+H]+: 143.12; found 143.1.
Compound 42 was prepared from aldehyde 22 (0.20 g, 0.58 mmol), 1-(oxetan-3-yl)piperazine (98 mg, 0.69 mmol), DCM (4.0 mL) and sodium triacetoxyborohydride (0.19 g, 0.86 mmol). The crude product was purified by flash chromatography (0–10% MeOH gradient in AcOEt) to give 42 (0.15 g, 0.32 mmol) as a light yellow solid with 54% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.33 (s, 1H, NH), 7.64 (dd, J = 7.5, 0.8 Hz, 1H, Ar-H), 7.54–7.52 (m, 1H, Ar-H), 7.47 (t, J = 2.8 Hz, 1H, Ar-H), 7.21 (t, J = 7.7 Hz, 1H, Ar-H), 7.10–7.08 (m, 1H, Ar-H), 6.76 (s, 1H, Ar-H), 6.49 (s, 1H, Ar-H), 4.50 (t, J = 6.5 Hz, 2H, CH2), 4.39 (t, J = 6.1 Hz, 2H, CH2), 3.86–3.84 (m, 4H, morph.), 3.78–3.76 (m, 4H, morph.), 3.67 (s, 2H, CH2), 3.40–3.33 (m, 1H, CH), 2.53–2.48 (m, 4H, piperaz.), 2.27–2.27 (m, 4H, piperaz.). 13C NMR (101 MHz, DMSO-d6) δ 157.9, 153.9, 151.0, 149.5, 136.8, 130.0, 126.5, 125.6, 120.7, 119.6, 113.2, 101.8, 94.8, 91.5, 74.4, 65.6, 58.5, 56.0, 52.3, 49.0, 47.9. HRMS (ESI): m/z calcd for C26H31N7O2 [M+H]+: 474.2612; found 474.2616.
5-(1H-indol-4-yl)-2-(((1S, 4S)-2-(oxetan-3-yl)-2,5-diaza-bicyclo [2.2.1]hept-2-yl)methyl)-7-(morpholin-4-yl)pyrazolo[1,5-a]pyrimidine (43)
The preparation of compound 43 started from (1S, 4S)-2-(oxetan-3-yl)-2,5-diazabicyclo [2.2.1]heptane, which was prepared analogously, as described for the synthesis of 1-(oxetan-3-yl) piperazine.
Compound 43 was prepared from aldehyde 22 (0.20 g, 0.58 mmol), (1S, 4S)-2-(oxetan-3-yl)-2,5-diazabicyclo [2.2.1]heptane (0.11 g, 0.69 mmol), DCM (4.0 mL) and sodium triacetoxyborohydride (0.19 g, 0.86 mmol). The crude product was purified by flash chromatography (0–10% MeOH gradient in AcOEt) to give 43 (0.18 g, 0.37 mmol) as a light yellow solid with 63% yield. 1H NMR (600 MHz, DMSO-d6) δ 11.34 (s, 1H, NH), 7.63 (dd, J = 7.4, 0.7 Hz, 1H, Ar-H), 7.53 (d, J = 8.0 Hz, 1H, Ar-H), 7.46 (t, J = 2.8 Hz, 1H, Ar-H), 7.21 (t, J = 7.7 Hz, 1H, Ar-H), 7.09–7.08 (m, 1H, Ar-H), 6.75 (s, 1H, Ar-H), 6.50 (s, 1H, Ar-H), 4.59–4.51 (m, 2H, CH2), 4.41–4.34 (m, 2H, CH2), 3.89–3.82 (m, 6H), 3.79–3.75 (m, 5H), 3.40 (s, 1H, CH), 3.22 (s, 1H), 2.84 (d, J = 9.4 Hz, 1H), 2.65–2.59 (m, 2H, CH2), 2.54–2.53 (m, 1H), 1.65–1.54 (m, 2H, CH2). 13C NMR (151 MHz, DMSO-d6) δ 157.8, 150.9, 149.5, 136.8, 130.0, 126.4, 125.6, 120.7, 119.5, 113.2, 101.8, 94.3, 91.4, 75.8, 75.3, 65.6, 61.6, 59.4, 57.3, 55.0, 52.7, 52.2, 47.8, 32.7. HRMS (ESI): m/z calcd for C27H31N7O2 [M+H]+: 486.2612; found 486.2614.
Compound 44 was prepared from aldehyde 22 (0.12 g, 0.36 mmol), 1-(cyclopropylcarbonyl)piperazine (64 µL, 70 mg, 0.43 mmol), DCM (4.0 mL) and sodium triacetoxyborohydride (0.11 g, 0.54 mmol). The crude product was purified by flash chromatography (0–10% MeOH gradient in AcOEt) to give 44 (0.14 g, 0.29 mmol) as a light yellow solid with 78% yield. 1H NMR (500 MHz, CDCl3) δ 8.67 (s, 1H, NH), 7.60 (d, J = 7.3 Hz, 1H, Ar-H), 7.48 (d, J = 8.1 Hz, 1H, Ar-H), 7.33–7.27 (m, 2H, Ar-H), 7.10 (s, 1H, Ar-H), 6.65 (s, 1H, Ar-H), 6.64 (s, 1H, Ar-H), 4.01–3.95 (m, 4H, morph.), 3.84 (s, 2H, CH2), 3.80–3.75 (m, 4H, morph.), 3.75–3.65 (m, 4H), 2.69–2.55 (m, 4H), 1.75–1.69 (m, 1H, CH), 1.01–0.95 (m, 2H, CH2), 0.77–0.72 (m, 2H, CH2). 13C NMR (126 MHz, CDCl3) δ 174.81, 153.02, 139.60, 128.89, 128.34, 124.80, 123.14, 115.62, 105.57, 98.80, 95.28, 69.17, 59.40, 51.37, 13.80, 10.22. HRMS (ESI): m/z calcd for C27H31N7O2 [M+H]+: 486.2612; found 486.2619.
Compound 45 was prepared from aldehyde 22 (85 mg, 0.25 mmol), 1-(cyclopropylmethyl)piperazine (44 µL, 41 mg, 0.29 mmol), DCM (3.0 mL) and sodium triacetoxyborohydride (78 mg, 0.37 mmol). The crude product was purified by flash chromatography (0–20% MeOH gradient in AcOEt) to give 45 (97 mg, 0.21 mmol) as a light yellow solid with 84% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.34 (s, 1H, NH), 7.65–7.63 (m, 1H, Ar-H), 7.53 (d, J = 8.1 Hz, 1H, Ar-H), 7.47–7.46 (m, 1H, Ar-H), 7.21 (t, J = 7.7 Hz, 1H, Ar-H), 7.10–7.09 (m, 1H, Ar-H), 6.76 (s, 1H, Ar-H), 6.48 (s, 1H, Ar-H), 3.86–3.84 (m, 4H, morph.), 3.78–3.77 (m, 4H, morph.), 3.65 (s, 2H, CH2), 2.53–2.45 (m, 8H, piperaz.), 2.15 (d, J = 6.6 Hz, 2H, CH2), 0.84–0.77 (m, 1H, CH), 0.45–0.40 (m, 2H, CH2), 0.06–0.02 (m, 2H, CH2). 13C NMR (101 MHz, DMSO-d6) δ 157.9, 154.0, 151.0, 149.5, 136.8, 130.0, 126.4, 125.6, 120.7, 119.5, 113.2, 101.8, 94.7, 91.5, 65.6, 62.8, 56.1, 52.7, 52.7, 47.9, 8.2, 3.7. HRMS (ESI): m/z calcd for C27H33N7O [M+H]+: 472.2819; found 472.2825.
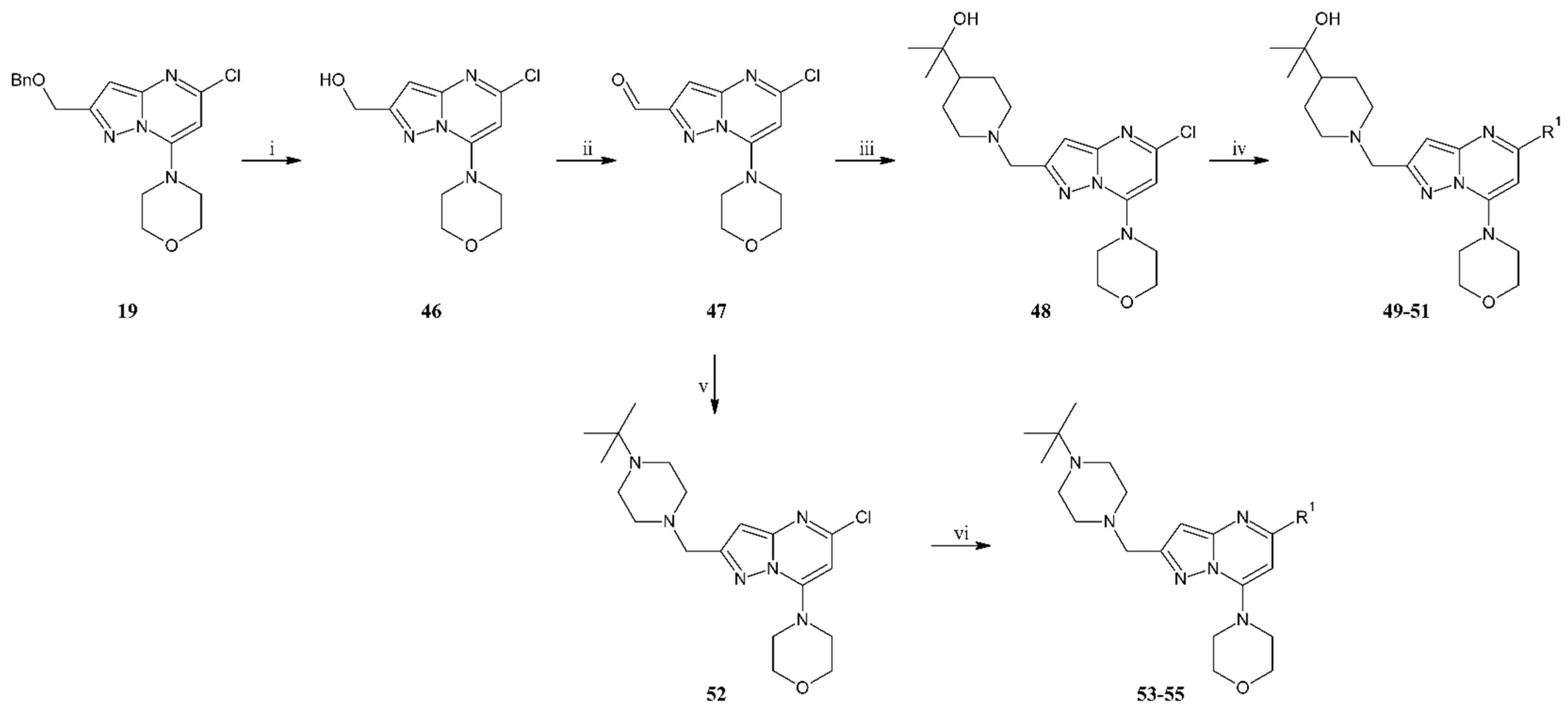
To the solution of compound 19 (16.6 g, 46.3 mmol) in CHCl3 (150 mL), methanesulfonic acid (61 mL, 925 mmol) was added, and then the reaction mixture was stirred at room temperature. After two h, the reaction mixture was poured onto the water containing ice and alkalized with 15% sodium hydroxide solution (25 mL). The aqueous phase was extracted with ethyl acetate (35 mL), and after separation, the organic phase was dried over anhydrous sodium sulfate. After filtration of the drying agent and evaporation of the solvent, the residue was purified by column chromatography (0–80% ethyl acetate gradient in heptane) to give 46 (12 g, 44.76 mmol) with 97% yield as an off-white solid. 1H NMR (300 MHz, CDCl3) δ: 6.49 (s, 1H, Ar-H), 6.07 (s, 1H, Ar-H), 4.87 (s, 2H, CH2), 4.00–3.90 (m, 4H, morph.), 3.83–3.73 (m, 4H, morph.). MS-ESI: m/z calcd for C11H13ClN4O2 [M+H]+: 269.08; found 269.0.
To a solution of compound 46 (3.00 g, 10.9 mmol) in DMF (30.0 mL) in argon atmosphere was added Dess–Martin periodinane (97%, 5.74 g, 13.1 mmol). The resulting mixture was stirred at room temperature for 2 h. The solvent was evaporated. The residue was washed with AcOEt and filtered. The filtrate was concentrated, and the crude product was purified by flash chromatography (0–100% AcOEt gradient in heptane) to give 47 (1.34 g, 5.02 mmol) with 46% yield. 1H NMR (500 MHz, DMSO-d6) δ 10.09 (s, 1H, CHO), 6.97 (s, 1H, Ar-H), 6.63 (s, 1H, Ar-H), 3.90–3.85 (m, 4H, morph.), 3.86–3.78 (m, 4H, morph.).
To the solution of compound 47 (3.4 g, 12.5 mmol) in dry DCM (30 mL), 2-(4-piperidyl)-2-propanol (2.24 g, 15.0 mmol) was added and then stirred at room temperature. After one hour, sodium triacetoxyborohydride (4.59 g, 21.2 mmol) was added, stirring the mixture at room temperature for a further 15 h. Then, water (45 mL) was added to the reaction mixture, and water-organic phases were separated. The aqueous phase was extracted three times with DCM (30 mL). Combined organic phases were dried over anhydrous sodium sulfate, filtered, and evaporated under reduced pressure. The residue was purified by flash chromatography (0–10% methanol gradient in ethyl acetate) to give 48 (3.1 g, 7.88 mmol) with a 63% yield as a slightly yellow solid. 1H NMR (500 MHz, CDCl3) δ 6.47 (s, 1H, Ar-H), 6.01 (s, 1H, Ar-H), 3.96–3.91 (m, 4H, morph.), 3.81–3.76 (m, 4H, morph.), 3.71 (s, 2H), 3.11–3.00 (m, 2H), 2.09–1.98 (m, 2H), 1.78–1.67 (m, 2H), 1.48–1.35 (m, 2H), 1.30–1.23 (m, 1H), 1.17 (s, 6H, 2xCH3). MS-ESI: m/z calcd for C19H28ClN5O2 [M+H]+: 394.20; found 394.1.
Compound 49 was prepared according to the general procedure for the Suzuki reaction. Synthesized from 48 (0.15 g, 0.381 mmol), 5-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole (0.16 g, 0.571 mmol), tetrakis(triphenylphosphine)palladium(0) (90 mg, 0.076 mmol), 2M aqueous sodium carbonate solution (0.38 mL, 0.762 mmol) and DME (6 mL). The crude product was purified by flash chromatography (50–100% ethyl acetate gradient in heptane) to give 49 (0.11 g, 0.22 mmol) with 60% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.34 (s, 1H, NH), 7.50–7.46 (m, 2H, Ar-H), 7.07–7.02 (m, 1H, Ar-H), 6.72–6.71 (m, 1H, Ar-H), 6.56 (d, J = 1.7 Hz, 1H, Ar-H), 6.49 (s, 1H), 4.02 (bs, 1H), 3.84–3.80 (m, 4H, morph.), 3.77–3.73 (m, 4H, morph.), 3.63 (s, 2H), 2.98–2.95 (m, 2H), 1.95–1.90 (m, 2H), 1.65–1.62 (m, 2H), 1.31–1.23 (m, 3H), 1.01 (s, 6H, 2xCH3). 13C NMR (101 MHz, DMSO-d6) δ 154.5, 154.2, 153.6, 150.8, 149.2, 132.8, 128.0, 126.9, 116.6, 113.5, 109.6, 101.8, 94.8, 93.9, 70.2, 65.6, 56.4, 53.9, 47.8, 46.9, 26.9, 26.6. HRMS (ESI): m/z calcd for C27H33FN6O2 [M+H]+: 493.2722; found 493.724.
Compound 50 was prepared according to the general procedure for the Suzuki reaction. Synthesized from 48 (0.15 g, 0.381 mmol), 6-azaindole-4-boronic acid pinacol ester (0.15 g, 0.571 mmol), tetrakis(triphenylphosphine)palladium(0) (88 mg, 0.076 mmol), 2M aqueous sodium carbonate solution (0.38 mL, 0.762 mmol) and DME (6 mL). The crude product was purified by flash chromatography (0–2% methanol gradient in ethyl acetate) to give 50 (0.13 g, 0.27 mmol) with 72% yield. 1H NMR(300 MHz, CDCl3) δ 10.43 (bs; 1H, NH); 8.80–8.78 (m; 1H, Ar-H); 8.72 (s; 1H, Ar-H); 7.51 (d; J = 3.1 Hz; 1H, Ar-H); 7.18 (d; J = 2.6 Hz; 1H, Ar-H); 6.65 (s; 1H, Ar-H); 6.61 (s; 1H, Ar-H); 4.02–3.90 (m; 4H, morph.); 3.84–3.72 (m; 6H); 3.20–3.09 (m; 2H, CH2); 2.16–2.03 (m; 2H, CH2); 1.80–1.70 (m; 2H); 1.53–1.31 (m; 3H); 1.18 (s; 6H, 2xCH3). 13C NMR (75 MHz, CDCl3) δ 156.4, 154.9, 151.7, 150.5, 138.2, 135.1, 133.8, 131.2, 130.0, 126.6, 102.7, 96.3, 91.5, 72.6, 66.4, 57.0, 54.3, 48.6, 47.4, 27.1, 27.1. HRMS (ESI): m/z calcd for C26H33N7O2 [M+H]+: 476.2769; found 476.2775.
Compound 51 was prepared according to the general procedure for the Suzuki reaction. Synthesized from 48 (0.15 g, 0.381 mmol), 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrrolo [2,3-b]pyridine (0.14 g, 0.571 mmol), tetrakis(triphenylphosphine) palladium(0) (88 mg, 0.076 mmol), 2M aqueous sodium carbonate solution (0.38 mL, 0.762 mmol) and DME (6 mL). The crude product was purified by flash chromatography (0–5% methanol gradient in ethyl acetate) to give 51 (0.12 g, 0.25 mmol) with 67% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.85 (s, 1H, NH), 8.34 (d, J = 5.0 Hz, 1H, Ar-H), 7.67 (d, J = 5.0 Hz, 1H, Ar-H), 7.61–7.59 (m, 1H, Ar-H), 7.10–7.09 (m, 1H, Ar-H), 6.84 (s, 1H, Ar-H), 6.55 (s, 1H, Ar-H), 4.02 (s, 1H), 3.84–3.83 (m, 8H), 3.63 (s, 2H), 2.97–2.94 (m, 2H), 1.95–1.89 (m, 2H), 1.65–1.62 (m, 2H), 1.27–1.20 (m, 3H), 1.01 (s, 6H, 2xCH3). 13C NMR (101 MHz, DMSO-d6) δ 155.5, 154.8, 150.9, 149.8, 149.7, 142.5, 137.0, 127.4, 117.1, 114.1, 100.8, 95.2, 91.1, 70.2, 65.6, 56.4, 53.9, 47.9, 46.8, 26.9, 26.6.HRMS (ESI): m/z calcd for C26H33N7O2 [M+H]+: 476.2769; found 476.2776.
To the solution of compound 47 (4.1 g, 15.4 mmol) in dry DCM (60 mL), N-t-butylpiperazine (2.62 g, 18.4 mmol) was added and then stirred at room temperature. After one h, sodium triacetoxyborohydride (5.54 g, 26.1 mmol) was added, and the mixture was stirred at room temperature for a further 15 h. Then, water (50 mL) was added to the reaction mixture, and the phases were separated. The aqueous phase was extracted three times with DCM (45 mL). Combined organic phases were dried over anhydrous sodium sulfate, filtered, and evaporated under reduced pressure. The residue was purified by flash chromatography (0–10% methanol gradient in ethyl acetate) to give 52 (3.2 g, 8.15 mmol) with 53% yield as a slightly yellow solid. 1H NMR (300 MHz, CDCl3) δ 6.47 (s, 1H, Ar-H), 6.03 (s, 1H, Ar-H), 3.99–3.89 (m, 4H, morph.), 3.84–3.76 (m, 4H, morph.), 3.74 (s, 2H, CH2), 2.63 (s, 8H, piperaz.), 1.08 (s, 9H, t-Bu.). MS-ESI: m/z calcd for C19H29ClN6O [M+H]+: 393.22; found 393.1.
Compound 53 was prepared according to the general procedure for the Suzuki reaction. Synthesized from 52 (0.12 g, 0.305 mmol), 5-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole (0.13 g, 0.458 mmol), tetrakis(triphenylphosphine)palladium(0) (72 mg, 0.061 mmol), 2M aqueous sodium carbonate solution (0.31 mL, 0.611 mmol) and DME (5 mL). The crude product was purified by flash chromatography (0–5% methanol gradient in ethyl acetate) to give 53 (0.10 g, 0.20 mmol) with 68% yield. 1H NMR (600 MHz, DMSO-d6) δ 11.36 (s, 1H, NH), 7.50–7.48 (m, 1H, Ar-H), 7.47–7.46 (m, 1H, Ar-H), 7.06–7.03 (m, 1H, Ar-H), 6.71–6.70 (m, 1H, Ar-H), 6.57–6.57 (m, 1H, Ar-H), 6.51 (s, 1H, Ar-H), 3.83–3.82 (m, 4H, morph.), 3.75–3.74 (m, 4H, morph.), 3.67 (s, 2H, CH2), 2.65–2.37 (m, 8H, piperaz.), 1.03 (s, 9H, t-Bu.). 13C NMR (151 MHz, DMSO-d6) δ 154.9, 153.7, 153.4, 150.8, 149.2, 132.8, 128.0, 126.9, 116.6, 113.6, 109.7, 101.8, 95.0, 94.0, 65.6, 55.8, 53.3, 47.8, 45.3, 40.0, 25.5. HRMS (ESI): m/z calcd for C27H34FN7O [M+H]+: 492.2881; found 492.2886.
Compound 54 was prepared according to the general procedure for the Suzuki reaction. Synthesized from 52 (0.12 g, 0.305 mmol), 6-azaindole-4-boronic acid pinacol ester (0.12 g, 0.458 mmol), tetrakis(triphenylphosphine)palladium(0) (71 mg, 0.061 mmol), 2M aqueous sodium carbonate solution (0.305 mL, 0.611 mmol) and DME (5 mL). The crude product was purified by flash chromatography (0–5% methanol gradient in ethyl acetate) to give 54 (0.11 g, 0.23 mmol) with 77% yield. 1H NMR (600 MHz, DMSO-d6) δ 11.84 (s, 1H, NH), 8.83–8.83 (m, 1H, Ar-H), 8.76–8.75 (m, 1H, Ar-H), 7.73–7.73 (m, 1H, Ar-H), 7.17–7.16 (m, 1H, Ar-H), 6.84 (s, 1H, Ar-H), 6.51 (s, 1H, Ar-H), 3.85–3.84 (m, 5H), 3.82–3.81 (m, 4H), 3.63 (s, 2H, CH2), 2.54–2.46 (m, 8H, piperaz.), 0.97 (s, 9H, t-Bu.). 13C NMR (151 MHz, DMSO-d6) δ 155.8, 154.1, 151.0, 149.6, 137.9, 133.6, 130.6, 129.8, 125.1, 101.7, 94.9, 90.9, 65.6, 56.0, 53.6, 52.9, 47.9, 45.2, 40.0, 25.7. HRMS (ESI): m/z calcd for C26H34N8O [M+H]+: 475.2928; found 472.2929.
Compound 55 prepared according to the general procedure for the Suzuki reaction. Synthesized from 52 (0.15 g, 0.382 mmol), 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrrolo [2,3-b]pyridine (0.14 g, 0.573 mmol), tetrakis(triphenylphosphine) palladium(0) (88 mg, 0.076 mmol), 2M aqueous sodium carbonate solution (0.38 mL, 0.763 mmol) and DME (5 mL). The crude product was purified by flash chromatography (0–5% methanol gradient in ethyl acetate) to give 55 (0.13 g, 0.27 mmol) with 72% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.85 (s, 1H, NH), 8.34 (d, J = 5.0 Hz, 1H, Ar-H), 7.67 (d, J = 5.1 Hz, 1H, Ar-H), 7.61–7.59 (m, 1H, Ar-H), 7.10–7.08 (m, 1H, Ar-H), 6.85 (s, 1H, Ar-H), 6.56 (s, 1H, Ar-H), 3.84–3.83 (m, 8H, morph.), 3.64 (s, 2H, CH2), 2.50–2.43 (m, 8H, piperaz.), 0.97 (s, 9H, t-Bu.). 13C NMR (101 MHz, DMSO-d6) δ 155.5, 154.4, 150.9, 149.8, 142.5, 137.0, 127.4, 117.1, 114.1, 100.8, 95.3, 91.2, 65.6, 55.9, 53.5, 53.1, 47.9, 45.2, 40.2, 25.7. HRMS (ESI): m/z calcd for C26H34N8O [M+H]+: 475.2928; found 472.2936.


 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}