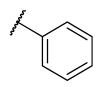
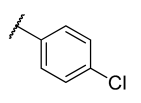
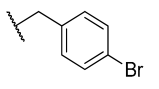
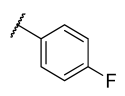
All chemicals and reagents were purchased from Merck, Johannesburg, South Africa and were used without any purification. The reaction progress was monitored with thin layer chromatography (TLC) plates. Purification of the final products was achieved using column chromatography with silica gel (0.063–0.200 mm) at different gradients of EtOAc-hexane eluents. Melting points were determined with open-end capillary tubes in a Stuart melting point instrument (SMP-3) and were uncorrected. The 1H, 13C, and 2D-NMR spectra of all synthesized compounds were recorded on 400 and 600 MHz Bruker AvanceIII spectrometers. The chemical shifts were recorded in parts per million (ppm) with deuterated dimethyl sulfoxide-d6 (δH 2.50 and δC 39.50 ppm) and chloroform-d (δH 7.26 and δC 77.00 ppm), wherein, tetramethylsilane (TMS) at δH = 0 was used as an internal standard. The splitting pattern is abbreviated as singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), pentet (p), doublet of triplet (dt), doublet of doublet (dd), triplet of doublet (td) and doublet of quartet (dq), while coupling constants J are reported in Hertz (Hz). High-resolution mass spectra were recorded using a Water Micromass LCT Premier TOF-MS spectrometer. The general synthetic procedure for azides is outlined in the supporting information.
4.2. General Synthetic Procedure for Quinoline-1,3,4-Oxadiazole Conjugates (4a–t)
To a solution of compound 3 (103.7 mg, 0.4 mmol) in DMF (3 mL), potassium carbonate (82.92 mg, 1.5 eq) was added and stirred until the reaction formed a paste in the round bottom flask. Then, 1.05 eq of appropriate benzyl bromides and aliphatic alkyl halides were added and stirring continued at r.t. for 1–2 h. Thereafter, the flask’s contents were poured in a slurry of ice and extracted with ethyl acetate. The crude product was purified with column chromatography using EtOAc-hexane eluents to obtain quinoline-1,3,4-oxadiazole conjugates in moderate to quantitative yields.
2-(Ethylthio)-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (4a)
Brown oil; Chemical formula: C14H13N3O2S, Yield; 53%, Mol wt: 287.34 gmol−1, M.p: 80.7–83.1 °C.
1H NMR (600 MHz, DMSO-d6) δ 8.88 (dd, J = 4.1, 1.8 Hz, 1H, H-2), 8.35 (dd, J = 8.3, 1.8 Hz, 1H, H-4), 7.62 (dd, J = 8.2, 1.2 Hz, 1H, H-5), 7.57 (dd, J = 8.3, 4.1 Hz, 1H, H-3), 7.54 (t, J = 7.9 Hz, 1H, H-6), 7.40 (dd, J = 7.7, 1.2 Hz, 1H, H-7), 5.63 (s, 2H H-6′), 3.26 (q, J = 7.3 Hz, 2H, H-7′), 1.37 (t, J = 7.3 Hz, 3H, H-8′).
13C NMR (151 MHz, DMSO-d6) δ 164.90, 163.58, 152.93, 149.39, 139.73, 135.93, 129.15, 126.53, 121.99, 121.49, 111.62, 60.56, 26.61, 14.70.
HRMS: (ESI+-MS, m/z) calcd for C14H13N3O2S (M + Na)+: 310.0626; found: 310.0631.
2-(Propylthio)-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (4b)
Brown oil; Chemical formula: C15H15N3O2S, Yield; 100%, Mol wt: 301.36 gmol−1.
1H NMR (600 MHz, DMSO-d6) δ 8.88 (dd, J = 4.1, 1.7 Hz, 1H, H-2), 8.35 (dd, J = 8.3, 1.7 Hz, 1H, H-4), 7.62 (dd, J = 8.3, 1.2 Hz, 1H, H-5), 7.57 (dd, J = 8.3, 4.1 Hz, 1H, H-3), 7.54 (t, J = 8.0 Hz, 1H, H-6), 7.39 (dd, J = 7.7, 1.2 Hz, 1H, H-7), 5.61 (s, 2H, H-6′), 3.23 (t, J = 7.1 Hz, 2H, H-7′), 1.73 (m, J = 7.3 Hz, 2H, H-8′), 0.96 (t, J = 7.4 Hz, 3H, H-9′).
13C NMR (151 MHz, DMSO-d6) δ 165.49 (C-2′), 164.08 (C-5′), 153.42 (C-8), 149.88 (C-2), 140.24 (C-8a), 136.43 (C-4), 129.65 (C-4a), 127.02 (C-6), 122.49 (C-3), 122.01 (C-5), 112.20 (C-7), 61.08 (C-6′), 34.38 (C-7′), 22.79 (C-8′), 13.19 (C-9′).
HRMS: (ESI+-MS, m/z) calcd for C15H15N3O2S (M + Na)+: 324.0783; found: 324.0786.
2-(Butylthio)-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (4c)
Yellow solid; Chemical formula: C16H17N3O2S, Yield; 53%, Mol wt: 315.39 gmol−1, M.p: 88.3–93.5 °C.
1H NMR (600 MHz, DMSO-d6) δ 8.88 (dd, J = 4.2, 1.8 Hz, 1H, H-2), 8.36 (dd, J = 8.3, 1.8 Hz, 1H, H-4), 7.63 (d, J = 8.4 Hz, 1H, H-5), 7.58 (dd, J = 8.3, 4.1 Hz, 1H, H-3), 7.54 (t, J = 8.0 Hz, 1H, H-6), 7.39 (dd, J = 7.8, 1.2 Hz, 1H, H-7), 5.62 (s, 2H H-6′), 3.25 (t, J = 7.3 Hz, 2H, H-7′), 1.74–1.65 (m, 2H, H-8′), 1.39 (hept, J = 7.7 Hz, 2H, H-9′), 0.87 (t, J = 7.4 Hz, 3H, H-10′).
13C NMR (151 MHz, DMSO-d6) δ 165.47 (C-2′), 164.10 (C-5′), 153.44 (C-8), 149.88 (C-2), 140.28 (C-8a), 136.43 (C-4), 129.66 (C-4a), 127.02 (C-6), 122.49 (C-3), 122.03 (C-5), 112.28 (C-7), 61.13 (C-6′), 32.23 (C-7′), 31.42 (C-8′), 21.44 (C-9′), 13.72 (C-10′).
HRMS: (ESI+-MS, m/z) calcd for C16H17N3O2S (M + Na)+: 338.0939; found: 338.0943.
2-(Pentylthio)-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (4d)
Brown solid; Chemical formula: C17H19N3O2S, Yield; 77%, Mol wt: 329.42 gmol−1, M.p: 84.6–90.1 °C.
1H NMR (600 MHz, DMSO-d6) δ 8.88 (dd, J = 4.1, 1.7 Hz, 1H, H-2′), 8.36 (dd, J = 8.3, 1.7 Hz, 1H, H-4′), 7.63 (dd, J = 8.2, 1.2 Hz, 1H, H-5), 7.58 (dd, J = 8.3, 4.1 Hz, 1H, H-3), 7.54 (t, J = 7.9 Hz, 1H, H-6), 7.38 (dd, J = 7.7, 1.2 Hz, 1H, H-7), 5.62 (s, 2H, H-6′), 3.24 (t, J = 7.3 Hz, 2H, H-7′), 1.71 (p, J = 7.3 Hz, 2H, H-8′), 1.38–1.30 (m, 2H, H-9′), 1.27 (m, J = 14.6, 7.0 Hz, 2H, H-10′), 0.85 (t, J = 7.2 Hz, 3H, H-11′).
13C NMR (151 MHz, DMSO-d6) δ 165.49 (C-2′), 164.09 (C-5′), 153.42 (C-8), 149.88 (C-2), 140.25 (C-8a), 136.43 (C-4), 129.66 (C-4a), 127.02 (C-6), 122.49 (C-3), 122.01 (C-5), 112.22 (C-7), 61.07 (C-6′), 32.49 (C-7′), 30.40 (C-8′), 29.04 (C-9′), 21.94 (C-10′),14.18 (C-11′).
HRMS: (ESI+-MS, m/z) calcd for C17H19N3O2S (M + Na)+: 352.1096; found: 352.1101.
2-(Hexylthio)-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (4e)
Brown oil; Chemical formula: C18H21N3O2S, Yield; 60%, Mol wt: 343.44 gmol−1.
1H NMR (600 MHz, DMSO-d6) δ 8.87 (dd, J = 4.1, 1.7 Hz, 1H, H-2), 8.36 (dd, J = 8.3, 1.7 Hz, 1H, H-4), 7.62 (dd, J = 8.3, 1.2 Hz, 1H, H-5), 7.57 (dd, J = 8.3, 4.1 Hz, 1H, H-3), 7.54 (t, J = 7.9 Hz, 1H, H-6), 7.38 (dd, J = 7.7, 1.2 Hz, 1H, H-7), 5.61 (s, 2H, H-6′), 3.24 (t, J = 7.3 Hz, 2H, H-7′), 1.70 (p, J = 7.5 Hz, 2H, H-9′), 1.36 (q, J = 7.1, 6.6 Hz, 2H, H-11′), 1.25 (m, J = 7.5, 4.5 Hz, 4H, H-8′,10′), 0.84 (t, J = 7.0 Hz, 3H, H-12′).
13C NMR (151 MHz, DMSO-d6) δ 165.49 (C-2′), 164.08 (C-5′), 153.41 (C-8), 149.87 (C-2), 140.23 (C-8a), 136.43 (C-4), 129.65 (C-4a), 127.02 (C-6), 122.49 (C-3), 121.99 (C-5), 112.14 (C-7), 61.05 (C-6′), 32.51 (C-7′), 31.01 (C-8′), 29.32 (C-9′), 27.90 (C-11′), 22.35 (C-10′), 14.26 (C-12′).
HRMS: (ESI+-MS, m/z) calcd for C18H21N3O2S (M + Na)+: 366.1252; found: 366.1252.
2-(Allylthio)-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (4f)
Yellow oil; Chemical formula: C15H13N3O2S, Yield; 52%, Mol wt: 299.35 gmol−1.
1H NMR (400 MHz, DMSO-d6) δ 8.88 (dd, J = 4.1, 1.8 Hz, 1H, H-2), 8.36 (dd, J = 8.3, 1.7 Hz, 1H, H-4), 7.63 (dd, J = 8.3, 1.2 Hz, 1H, H-5), 7.58 (dd, J = 8.3, 4.2 Hz, 1H, H-3), 7.54 (t, J = 7.9 Hz, 1H, H-6), 7.38 (dd, J = 7.7, 1.3 Hz, 1H, H-7), 5.95 (m, J = 17.0, 10.0, 7.0 Hz, 1H, H-8′), 5.63 (s, 2H, H-6′), 5.30 (m, J = 17.0, 1.4 Hz, 2H, H-9b), 5.12 (m, J = 10.0, 1.7, 0.9 Hz, 2H, H-9a), 3.92 (dd, J = 6.9, 1.1 Hz, 2H, H-7′).
13C NMR (101 MHz, DMSO-d6) δ 164.75 (C-2′), 164.31 (C-5′), 153.37 (C-8), 149.90 (C-2), 140.16 (C-8a), 136.45 (C-4), 132.87 (C-8′), 129.63 (C-4a), 127.05 (C-6), 122.53 (C-3), 121.95 (C-5), 119.93 (C-9), 111.99 (C-7), 60.94 (C-6′), 35.04 (C-7′).
HRMS: (ESI+-MS, m/z) calcd for C15H13N3O2S (M + Na)+: 322.0626; found: 322.0636.
2-[(3-Methylbut-2-en-1-yl)thio]-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (4g)
Yellow oil; Chemical formula: C17H17N3O2S, Yield; 30%, Mol wt: 327.40 gmol−1.
1H NMR (400 MHz, DMSO-d6) δ 8.88 (dd, J = 4.2, 1.8 Hz, 1H, H-2), 8.36 (dd, J = 8.3, 1.7 Hz, 1H, H-4), 7.63 (dd, J = 8.2, 1.2 Hz, 1H, H-5), 7.60–7.51 (m, 2H, H-3,6), 7.38 (dd, J = 7.7, 1.3 Hz, 1H, H-7), 5.61 (s, 2H, H-6′), 5.35 (dd, J = 7.8, 1.5 Hz, 1H, H-8′), 3.91 (d, J = 7.8 Hz, 2H, H-7′), 1.66 (s, 6H, H-10′,11′).
13C NMR (101 MHz, DMSO-d6) δ 164.60 (C-2′), 164.11 (C-5′), 153.29 (C-8), 149.87 (C-2), 140.04 (C-8a), 136.60 (C-4), 129.64 (C-4a), 127.11 (C-6), 122.54 (C-3), 122.03 (C-5), 112.21 (C-7), 79.26 (C-8′), 75.51 (C-9′), 61.05 (C-6′), 21.19 (C-7′).
HRMS: (ESI+-MS, m/z) calcd for C17H17N3O2S (M + Na)+: 350.0939; found: 350.0950.
2-[(2-Bromoethyl)thio]-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (4h)
Yellow oil; Chemical formula: C14H12BrN3O2S, Yield; 54%, Mol wt: 326.33 gmol−1.
1H NMR (600 MHz, DMSO-d6) δ 8.88 (dd, J = 4.2, 1.7 Hz, 1H, H-2), 8.36 (dd, J = 8.3, 1.7 Hz, 1H, H-4), 7.63 (dd, J = 8.3, 1.2 Hz, 1H, H-5), 7.58 (dd, J = 8.3, 4.1 Hz, 1H, H-3), 7.55 (t, J = 7.9 Hz, 1H, H-6), 7.40 (dd, J = 7.7, 1.2 Hz, 1H, H-7), 5.62 (s, 2H, H-6′), 3.83 (t, J = 7.3 Hz, 2H, H-7′), 3.72 (t, J = 7.1 Hz, 2H, H-8′).
13C NMR (151 MHz, DMSO-d6) δ 164.59 (C-2′), 164.38 (C-5′), 153.43 (C-8), 149.90 (C-2), 140.25 (C-8a), 136.45 (C-4), 129.65 (C-4a), 127.04 (C-6), 122.50 (C-3), 122.05 (C-5), 112.30 (C-7), 61.13 (C-6′), 34.36 (C-7′), 31.19 (C-8′).
HRMS: (ESI+-MS, m/z) calcd for C14H12BrN3O2S (M + Na)+: 387.9731; found: 387.9747.
2-[(5-Bromopentyl)thio]-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (4i)
Yellow oil; Chemical formula: C17H18BrN3O2S, Yield; 61%, Mol wt: 408.31 gmol−1.
1H NMR (600 MHz, DMSO- d6) δ 8.89 (dd, J = 4.2, 1.6 Hz, 1H, H-2), 8.42 (dd, J = 8.3, 1.7 Hz, 1H, H-4), 7.65 (d, J = 8.2 Hz, 1H, H-5), 7.62 (dd, J = 8.3, 4.2 Hz, 1H, H-3), 7.57 (t, J = 8.0 Hz, 1H, H-6), 7.42 (d, J = 7.8 Hz, 1H, H-7), 5.63 (s, 2H, H-6′), 3.51 (t, J = 6.7 Hz, 2H, H-11′), 3.26 (t, J = 7.3 Hz, 2H, H-7′), 1.81 (p, J = 6.9 Hz, 2H, H-9′), 1.74 (p, J = 7.4 Hz, 2H, H-8′), 1.49 (p, J = 7.7 Hz, 2H, H-10′).
13C NMR (151 MHz, DMSO-d6) δ 165.48 (C-2′), 164.02 (C-5′), 152.99 (C-8), 149.58 (C-2), 139.88 (C-8a), 137.27 (C-4), 129.68 (C-4a), 127.30 (C-6), 122.59 (C-3), 122.01 (C-5), 112.40 (C-7), 61.07 (C-6′), 35.26 (C-11′), 32.30 (C-7′), 31.97 (C-9′), 28.46 (C-8′), 26.82 (C-10′).
HRMS: (ESI+-MS, m/z) calcd for C17H18BrN3O2S (M + Na)+: 430.0201; found: 430.0212.
2-(Benzylthio)-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (4j)
Light brown solid; Chemical formula: C19H15N3O2S, Yield; 50%, Mol wt: 349.41 gmol−1, M.p: 75.3–79.8 °C.
1H NMR (600 MHz, DMSO-d6) δ 8.87 (dd, J = 4.1, 1.8 Hz, 1H, H-2), 8.36 (dd, J = 8.3, 1.8 Hz, 1H, H-4), 7.63 (dd, J = 8.2, 1.2 Hz, 1H, H-5), 7.58 (dd, J = 8.3, 4.1 Hz, 1H, H-3), 7.54 (t, J = 8.0 Hz, 1H, H-6), 7.42 (dd, J = 7.6, 1.7 Hz, 2H, H-9′, 13′), 7.37 (dd, J = 7.8, 1.2 Hz, 1H, H-7), 7.33–7.24 (m, 3H, H-10′, 11′,12′), 5.62 (s, 2H, H-6′), 4.52 (s, 2H, H-7′).
13C NMR (151 MHz, DMSO-d6) δ 164.87 (C-2′), 164.31 (C-5′), 153.38 (C-8), 149.90 (C-2), 140.21 (C-8a), 136.82 (C-8′), 136.44 (C-4), 129.65 (C-4a), 129.43 (C-9′,13′), 129.01 (C-10′,12′), 128.23 (C-11′), 127.04 (C-6), 122.52 (C-3), 121.98 (C-5), 112.05 (C-7), 60.98 (C-6′), 36.34 (C-7′).
HRMS: (ESI+-MS, m/z) calcd for C19H15N3O2S (M + Na)+: 372.0783; found: 372.0790.
2-[(4-Fluorobenzyl)thio]-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (4k)
Yellow crystals; Chemical formula: C19H16FN3O2S, Yield; 32%, Mol wt: 367.40 gmol−1, M.p: 106.9–109.4 °C.
1H NMR (600 MHz, DMSO-d6) δ 8.87 (dd, J = 4.1, 1.7 Hz, 1H, H-2), 8.36 (dd, J = 8.3, 1.7 Hz, 1H, H-4), 7.63 (dd, J = 8.3, 1.2 Hz, 1H, H-5), 7.58 (dd, J = 8.3, 4.1 Hz, 1H, H-3), 7.54 (t, J = 7.9 Hz, 1H, H-6), 7.47 (dd, J = 8.5, 5.6 Hz, 2H, H-9′,13′), 7.36 (dd, J = 7.7, 1.2 Hz, 1H, H-7), 7.09 (d, J = 8.9 Hz, 2H, H-10′,12′), 5.61 (s, 2H, H-6′), 4.51 (s, 2H, H-7′).
13C NMR (151 MHz, DMSO-d6) δ 164.82 (C-2′), 164.32 (C-5′), 162.05 (d, J = 244.2 Hz, C-11′), 153.33 (C-8), 149.90 (C-2), 140.12 (C-8a), 136.46 (C-4), 133.22 (d, J = 3.0 Hz, C-8′), 131.58 (d, J = 8.3 Hz, C-9′,13′), 129.63 (4a), 127.05 (C-6), 122.55 (C-3), 121.92 (C-5), 115.80 (d, J = 21.5 Hz, C-10′,12′), 111.82 (C-7), 60.85 (C-6′), 35.46 (C-7′).
HRMS: (ESI+-MS, m/z) calcd for C19H14FN3O2S (M + Na)+: 390.0688; found: 390.0696.
2-[(4-Chlorobenzyl)thio]-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (4l)
Cream crystals; Chemical formula: C19H14ClN3O2S, Yield; 85%, Mol wt: 383.85 gmol−1, M.p: 101.4–104.2 °C.
1H NMR (600 MHz, DMSO-d6) δ 8.87 (dd, J = 4.1, 1.7 Hz, 1H, H-2), 8.36 (dd, J = 8.3, 1.8 Hz, 1H, H-4), 7.63 (dd, J = 8.3, 1.2 Hz, 1H, H-5), 7.58 (dd, J = 8.3, 4.1 Hz, 1H, H-3), 7.53 (t, J = 8.0 Hz, 1H, H-6), 7.45 (d, J = 8.4 Hz, 2H, H-9′,13′), 7.36 (dd, J = 7.8, 1.2 Hz, 1H, H-7), 7.32 (d, J = 8.4 Hz, 2H, H-10′,12′), 5.61 (s, 2H, H-6′), 4.51 (s, 2H, H-7′).
13C NMR (151 MHz, DMSO-d6) δ 164.71 (C-2′), 164.37 (C-5′), 153.37 (C-8), 149.89 (C-2), 140.19 (C-8a), 136.45 (C-4), 136.13 (C-8′), 132.86 (C-11′), 131.32 (C-9′,13′), 129.65 (C-4a), 128.94 (C-10′,12′), 127.04 (C-6), 122.53 (C-3), 121.98 (C-5), 112.00 (C-7), 60.96 (C-6′), 35.52 (C-7′).
HRMS: (ESI+-MS, m/z) calcd for C19H14ClN3O2S (M + Na)+: 406.0393; found: 406.0407.
2-[(4-Bromobenzyl)thio]-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (4m)
Yellow solid; Chemical formula: C19H14BrN3O2S, Yield; 27%, Mol wt: 428.30 gmol−1, M.p: 120.9–124.1 °C.
1H NMR (600 MHz, DMSO-d6) δ 8.87 (dd, J = 4.1, 1.7 Hz, 1H, H-2), 8.36 (dd, J = 8.3, 1.7 Hz, 1H, H-4), 7.63 (dd, J = 8.3, 1.2 Hz, 1H, H-5), 7.58 (dd, J = 8.3, 4.1 Hz, 1H, H-3), 7.53 (t, J = 8.0 Hz, 1H, H-6), 7.46 (d, J = 8.4 Hz, 2H, H-10′,12′), 7.38 (d, J = 8.4 Hz, 2H, H-9′,13′), 7.36 (dd, J = 7.8, 1.2 Hz, 1H, H-7), 5.61 (s, 2H, H-6′), 4.49 (s, 2H, H-7′).
13C NMR (151 MHz, DMSO-d6) δ 164.69 (C-2′), 164.38 (C-5′), 153.38 (C-8), 149.89 (C-2), 140.20 (C-8a), 136.57 (C-8′), 136.45 (C-4), 131.87 (C-10′,12′), 131.64 (C-9′,13′), 129.65 (C-4a), 127.04 (C-6), 122.52 (C-3), 121.98 (C-5), 121.40 (C-11′), 112.02 (C-7), 60.97 (C-6′), 35.57 (C-7′).
HRMS: (ESI+-MS, m/z) calcd for C19H14BrN3O2S (M + Na)+: 449.9888; found: 449.9895.
2-[(4-Nitrobenzyl)thio]-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (4n)
Yellow crystals; Chemical formula: C19H14N4O4S, Yield; 104%, Mol wt: 394.40 gmol−1, M.p: 156.6–160.1 °C.
1H NMR (600 MHz, DMSO-d6) δ 8.88 (dd, J = 4.2, 1.8 Hz, 1H, H-2), 8.35 (dd, J = 8.3, 1.8 Hz, 1H, H-4), 8.13 (dd, J = 8.7, 6.8 Hz, 2H, H-10′,12′), 7.71 (dd, J = 8.7, 6.7 Hz, 2H, H-9′,13′), 7.63 (dd, J = 8.3, 1.2 Hz, 1H, H-5), 7.57 (dd, J = 8.3, 4.1 Hz, 1H, H-3), 7.53 (t, J = 8.0 Hz, 1H, H-6), 7.37 (dd, J = 7.8, 1.2 Hz, 1H, H-7), 5.62 (s, 2H, H-6′), 4.65 (s, 2H, H-7′).
13C NMR (151 MHz, DMSO-d6) δ 163.98 (C-2′), 163.97 (C-5′), 152.88 (C-8), 149.38 (C-2), 146.85 (C-11′), 144.68 (C-8′), 139.67 (C-8a), 135.95 (C-4), 130.25 (C-9′,13′), 129.14 (4a), 126.52 (C-6), 123.53 (C-10′,12′), 122.02 (C-3), 121.45 (C-5), 111.41 (C-7), 60.43 (C-6′), 34.86 (C-7′).
HRMS: (ESI+-MS, m/z) calcd for C19H14N4O4S (M + Na)+: 417.0633; found: 417.0645.
2-[(3-Fluorobenzyl)thio]-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (4o)
Yellow oil; Chemical formula: C19H14FN3O2S, Yield; 63%, Mol wt: 367.40 gmol−1.
1H NMR (600 MHz, DMSO-d6) δ 8.91 (d, J = 4.2 Hz, 1H, H-2), 8.48 (d, J = 8.2 Hz, 1H, H-4), 7.68 (d, J = 8.2 Hz, 1H, H-5), 7.65 (dd, J = 8.3, 4.3 Hz, 1H, H-3), 7.59 (t, J = 8.0 Hz, 1H, H-6), 7.43 (d, J = 7.7 Hz, 1H, H-7), 7.33 (q, J = 7.5 Hz, 1H, H-10′), 7.31–7.24 (m, 2H, H-9′,13′), 7.10 (td, J = 8.6, 2.6 Hz, 1H, H-11′), 5.64 (s, 2H, H-6′), 4.54 (s, 2H, H-7′).
13C NMR (151 MHz, DMSO-d6) δ 164.79 (d, J = 3.9 Hz, C-2′), 164.26 (d, J = 3.4 Hz, C-5′), 162.42 (d, J = 244.1 Hz, C-12′), 152.59 (C-8), 149.30 (C-2), 139.79 (d, J = 7.9 Hz, C-8a), 137.98 (C-4), 130.94 (d, J = 8.4 Hz, C-10′), 129.71 (d, J = 2.4 Hz, C-4a), 127.51 (d, J = 3.9 Hz, C-6), 125.57 (t, J = 3.2 Hz, C-9′), 122.65 (d, J = 3.5 Hz, C-3), 122.04 (d, J = 7.7 Hz, C-5), 116.21 (dd, J = 21.9, 2.9 Hz, C-13′), 115.09 (dd, J = 21.0, 1.4 Hz, C-11′), 122.65 (d, J = 3.5 Hz, C-7), 61.13 (C-6′), 35.62 (C-7′).
HRMS: (ESI+-MS, m/z) calcd for C19H14FN3O2S (M + Na)+: 390.0688; found: 390.0697.
2-[(3-Chlorobenzyl)thio]-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (4p)
Yellow oil; Chemical formula: C19H14ClN3O2S, Yield; 27%, Mol wt: 383.85 gmol−1.
1H NMR (600 MHz, DMSO-d6) δ 8.87 (dd, J = 4.2, 1.8 Hz, 1H, H-2), 8.36 (dd, J = 8.3, 1.8 Hz, 1H, H-4), 7.62 (d, J = 8.2 Hz, 1H, H-5), 7.57 (dd, J = 8.3, 4.1 Hz, 1H, H-3), 7.54 (s, 1H, H-13′), 7.52 (d, J = 8.0 Hz, 1H, H-6), 7.39 (dd, J = 7.1, 1.7 Hz, 1H, H-9′), 7.36 (dd, J = 7.7, 1.2 Hz, 1H, H-7), 7.34–7.26 (m, 2H, H-10′,11′), 5.60 (s, 2H, H-6′), 4.53 (s, 2H, H-7′).
13C NMR (151 MHz, DMSO-d6) δ 164.72 (C-2′), 164.40 (C-5′), 153.38 (C-8), 149.90 (C-2), 140.20 (C-8a), 139.59 (C-8′), 136.45 (C-4), 133.47 (C-12′), 130.82 (C-10′), 129.64 (C-4a), 129.29 (C-13′), 128.19 (C-9′), 128.14 (C-11′), 127.03 (C-6), 122.51 (C-3), 122.02 (C-5), 112.11 (C-7), 61.04 (C-6′), 35.51 (C-7′).
HRMS: (ESI+-MS, m/z) calcd for C19H14ClN3O2S (M + Na)+: 406.0393; found: 406.0405.
2-[(3-Bromobenzyl)thio]-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (4q)
Yellow oil; Chemical formula: C19H14BrN3O2S, Yield; 51%, Mol wt: 428.30 gmol−1.
1H NMR (600 MHz, DMSO-d6) δ 8.90 (d, J = 4.0 Hz, 1H, H-2), 8.45 (d, J = 8.2 Hz, 1H, H-4), 7.69 (s, 1H, H-13′), 7.67 (d, J = 8.2 Hz, 1H, H-5), 7.64 (dd, J = 8.3, 4.2 Hz, 1H, H-3), 7.58 (t, J = 8.0 Hz, 1H, H-6), 7.47 (dd, J = 8.0, 1.9 Hz, 1H, H-11′), 7.44 (d, J = 7.7 Hz, 1H, H-9′), 7.42 (d, J = 7.7 Hz, 1H, H-7), 7.25 (t, J = 7.8 Hz, 1H, H-10′), 5.63 (s, 2H, H-6′), 4.52 (s, 2H, H-7′).
13C NMR (151 MHz, DMSO-d6) δ 164.76 (C-2′), 164.37 (C-5′), 153.30 (C-8), 149.47 (C-2), 139.86 (C-8a), 139.82 (C-8′), 137.58 (C-4), 137.47 (C-10′), 132.15 (C-13′), 131.08 (C-11′), 129.73 (C-4a), 128.50 (C-9′), 127.36 (C-6), 122.58 (C-3), 122.07 (C-5), 122.02 (C-12′), 112.95 (C-7), 61.38 (C-6′), 35.61 (C-7′).
HRMS: (ESI+-MS, m/z) calcd for C19H14BrN3O2S (M + Na)+: 449.9888; found: 449.9903.
2-[(2,4-Difluorobenzyl)thio]-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (4r)
Cream solid; Chemical formula: C19H13F2N3O2S, Yield; 29%, Mol wt: 385.39 gmol−1, M.p: 98.7–105.2 °C.
1H NMR (600 MHz, DMSO-d6) δ 8.87 (dd, J = 4.1, 1.7 Hz, 1H, H-2), 8.36 (dd, J = 8.3, 1.8 Hz, 1H, H-4), 7.63 (dd, J = 8.2, 1.2 Hz, 1H, H-5), 7.61–7.51 (m, 3H, H-3.9′,6), 7.37 (dd, J = 7.7, 1.2 Hz, 1H, H-7), 7.24 (ddd, J = 10.6, 9.2, 2.6 Hz, 1H, H-12′), 6.99 (td, J = 8.5, 2.4 Hz, 1H, H-10′), 5.61 (s, 2H, H-6′), 4.53 (s, 2H, H-7′).
13C NMR (151 MHz, DMSO-d6) δ 164.58 (d, J = 7.5 Hz, C-5′), 164.31 (C-2′), 161.97 (d, J = 7.8 Hz, C-11′), 161.10 (d, J = 26.8 Hz, C-8′), 153.45 (d, J = 13.5 Hz, C-8), 149.89 (C-2), 140.32 (d, J = 25.4 Hz, C-8a), 136.43 (d, J = 3.8 Hz, C-4), 132.96 (dd, J = 9.9, 5.2 Hz, C-9′), 129.68 (d, J = 7.7 Hz, C-4a), 127.01 (d, J = 5.3 Hz, C-6), 122.48 (d, J = 8.6 Hz, C-3), 122.09 (d, J = 20.1 Hz, C-5), 120.44–120.17 (m, C-13′), 112.60 (C-7), 112.05 (dd, J = 21.5, 3.6 Hz, C-10′), 104.57 (dd, J = 26.1, 25.4 Hz, C-12′), 61.20 (d, J = 40.4 Hz, C-6′), 29.97–29.67 (m, C-7′).
HRMS: (ESI+-MS, m/z) calcd for C19H13F2N3O2S (M + Na)+: 408.0594; found: 408.0604.
2-[(3,4-Difluorobenzyl)thio]-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (4s)
Cream solid; Chemical formula: C19H13F2N3O2S, Yield; 62%, Mol wt: 385.39 gmol−1, M.p: 98.8–101.2 °C.
1H NMR (600 MHz, DMSO-d6) δ 8.88 (dd, J = 4.1, 1.8 Hz, 1H, H-2), 8.35 (dd, J = 8.3, 1.7 Hz, 1H, H-4), 7.63 (dd, J = 8.3, 1.2 Hz, 1H, H-5), 7.61–7.50 (m, 3H, H-3,13′,6), 7.37 (dd, J = 7.7, 1.2 Hz, 1H, H-7), 7.20 (td, J = 9.9, 2.5 Hz, 1H, H-12′), 6.98 (td, J = 8.5, 2.6 Hz, 1H, H-9′), 5.62 (s, 2H, H-6′), 4.52 (s, 2H, H-7′).
13C NMR (151 MHz, DMSO-d6) δ 164.56 (C-5′), 164.31 (C-2′), 162.55 (dd, J = 236.3, 11.0 Hz, C-10′), 160.90 (dd, J = 241.0, 12.0 Hz, C-11′), 153.42 (C-8), 149.88 (C-2), 140.25 (C-8a), 136.43 (C-4), 132.98 (dd, J = 10.0, 4.9 Hz, C-13′), 129.66 (C-4a), 127.01 (C-6), 122.49 (C-3), 122.03 (C-5), 120.29 (dd, J = 14.6, 3.6 Hz, C-8′), 112.42–112.16 (m, C-9), 112.04 (dd, J = 21.4, 3.6 Hz, C-7), 104.57 (t, J = 25.8 Hz, C-12′), 61.10 (C-6′), 29.81 (C-7′).
HRMS: (ESI+-MS, m/z) calcd for C19H13F2N3O2S (M + Na)+: 408.0594; found: 408.0606.
2-[(2,4-Dichlorobenzyl)thio]-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (4t)
Cream solid; Chemical formula: C19H13Cl2N3O2S, Yield; 40%, Mol wt: 418.30 gmol−1, M.p: 116.7–119.7 °C.
1H NMR (400 MHz, DMSO-d6) δ 8.86 (dd, J = 4.2, 1.7 Hz, 1H, H-2), 8.36 (dd, J = 8.3, 1.7 Hz, 1H, H-4), 7.64 (d, J = 2.2 Hz, 1H, H-12′), 7.62 (dd, J = 8.3, 1.2 Hz, 1H, H-5), 7.60–7.55 (m, 2H, H-9′,3), 7.53 (t, J = 8.0 Hz, 1H, H-6), 7.36 (dd, J = 7.7, 1.2 Hz, 1H, H-6), 7.32 (dd, J = 8.3, 2.2 Hz, 1H, H-10′), 5.61 (s, 2H, H-6′), 4.57 (s, 2H, H-7′).
13C NMR (101 MHz, DMSO-d6) δ 169.37 (C-5′), 168.94 (C-2′), 158.14 (C-8), 154.71 (C-2), 145.02 (C-8a), 141.19 (C-4), 139.51 (C-11′), 138.81 (C-13′), 138.15 (C-8′), 137.94 (C-9′), 134.40 (C-4a), 134.30 (C-12′), 132.75 (C-10′), 131.78 (C-6), 127.26 (C-3), 126.74 (C-5), 116.77 (C-7), 65.77 (C-6′), 38.86 (C-7′).
HRMS: (ESI+-MS, m/z) calcd for C19H13Cl2N3O2S (M + Na)+: 440.0003; found: 440.0016.
4.4. General Synthetic Procedure for Quinoline-1,3,4-Oxadiazole-1,2,3-Triazole Hybrids (12a–t)
To a solution of alkyne intermediate 11 (0.15 g) in DCM (10 mL) in a round bottom flask, sodium ascorbate (22 mol%) and copper(II) sulphate pentahydrate (10 mol%) in water (10 mL) were added and stirred at r.t for few minutes. Then, the appropriate azides (1.1 eq) in DCM were added and the mixture was stirred for 2–4 h. After this time, TLC analysis showed the alkyne was consumed, then the reaction was diluted with water and filtered to remove residual salts. The resulting filtrate was extracted with DCM, washed with brine and dried over anhydrous sodium sulphate, filtered, and concentrated under reduced pressure. The crude product was purified with column chromatography in EtOAc-hexane eluents to obtain pure quinoline-1,3,4-oxadiazole-1,2,3-triazole hybrids 12a–t in excellent yields (43–91%).
2-{[(1-Pentyl-1H-1,2,3-triazol-4-yl)methyl]thio-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (12a)
Yellow solid; Chemical Formula: C20H22N6O2S, Yield; 59%, Mol wt: 410.49 gmol−1, M.p: 90–94 °C.
1H NMR (400 MHz, DMSO-d6) δ 8.88 (dd, J = 4.2, 1.8 Hz, 1H), 8.37 (dd, J = 8.3, 1.8 Hz, 1H), 8.13 (s, 1H), 7.63 (dd, J = 8.3, 1.3 Hz, 1H), 7.60–7.57 (m, 1H), 7.54 (d, J = 8.1 Hz, 1H), 7.40 (dd, J = 7.7, 1.3 Hz, 1H), 5.62 (s, 2H), 4.60 (s, 2H), 4.28 (t, J = 7.1 Hz, 2H), 1.73 (p, J = 7.2 Hz, 2H), 1.31–1.05 (m, 4H), 0.80 (t, J = 7.2 Hz, 3H).
13C NMR (101 MHz, DMSO-d6) δ 164.70, 164.35, 153.42, 149.92, 142.24, 140.15, 136.49, 129.65, 127.08, 124.24, 122.55, 121.97, 111.98, 61.02, 49.82, 29.78, 28.37, 27.30, 21.91, 14.18.
HRMS: (ESI+-MS, m/z) calcd for C20H22N6O2S (M + Na)+: 433.1423; found: 433.1413.
2-{[(1-Hexyl-1H-1,2,3-triazol-4-yl)methyl]thio}-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (12b)
Yellow solid; Chemical Formula: C21H24N6O2S, Yield; 75%, Mol wt: 424.52 gmol−1, M.p: 72–78 °C.
1H NMR (400 MHz, DMSO-d6) δ 8.88 (dd, J = 4.1, 1.7 Hz, 1H), 8.37 (dd, J = 8.3, 1.7 Hz, 1H), 8.13 (s, 1H), 7.67–7.51 (m, 3H), 7.40 (dd, J = 7.7, 1.3 Hz, 1H), 5.62 (s, 2H), 4.60 (s, 2H), 4.28 (t, J = 7.1 Hz, 2H), 1.72 (p, J = 7.2 Hz, 2H), 1.41–1.04 (m, 6H), 0.84–0.76 (m, 3H).
13C NMR (101 MHz, DMSO-d6) δ 164.70, 164.34, 153.41, 149.92, 142.25, 140.14, 136.49, 129.64, 127.09, 124.25, 122.56, 121.96, 111.96, 61.01, 49.83, 30.96, 30.04, 27.29, 25.85, 22.32, 14.25.
HRMS: (ESI+-MS, m/z) calcd for C21H24N6O2S (M + Na)+: 447.1579; found: 447.1568.
2-{[(1-Benzyl-1H-1,2,3-triazol-4-yl)methyl]thio}-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (12c)
Yellow solid; Chemical Formula: C22H18N6O2S, Yield; 82%, Mol wt: 430.48 gmol−1, M.p: 68–71 °C.
1H NMR (400 MHz, Chloroform-d) δ 8.86 (dd, J = 4.2, 1.7 Hz, 1H), 8.08 (dd, J = 8.3, 1.7 Hz, 1H), 7.59 (s, 1H), 7.46–7.34 (m, 3H), 7.25 (dd, J = 5.1, 2.0 Hz, 3H), 7.23–7.16 (m, 1H), 7.20–7.10 (m, 2H), 5.45 (s, 2H), 5.37 (s, 2H), 4.44 (s, 2H).
13C NMR (101 MHz, Chloroform-d) δ 165.77, 163.48, 153.07, 149.59, 143.05, 139.95, 136.32, 134.43, 129.65, 129.11, 128.76, 128.01, 126.58, 123.27, 121.95, 121.73, 110.75, 60.75, 54.19, 26.91.
HRMS: (ESI+-MS, m/z) calcd for C20H22N6O2S (M + Na)+: 453.1110; found: 453.1098.
2-{[(1-(4-Chlorobenzyl)-1H-1,2,3-triazol-4-yl)methyl]thio}-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (12d)
Cream white solid; Chemical Formula: C22H17ClN6O2S, Yield; 63%, Mol wt: 464.93 gmol−1, M.p: 92–96 °C.
1H NMR (400 MHz, Chloroform-d) δ 8.87–8.81 (m, 1H), 8.08 (dd, J = 8.3, 1.7 Hz, 1H), 7.64 (s, 1H), 7.45–7.32 (m, 3H), 7.26–7.14 (m, 3H), 7.11–7.01 (m, 2H), 5.44 (s, 2H), 5.33 (s, 2H), 4.43 (s, 2H).
13C NMR (101 MHz, Chloroform-d) δ 165.60, 163.58, 153.19, 149.57, 143.27, 140.19, 136.15, 134.76, 133.00, 129.67, 129.27, 129.26, 126.51, 123.25, 121.87, 121.83, 111.10, 60.92, 53.37, 26.90.
HRMS: (ESI+-MS, m/z) calcd for C22H17ClN6O2S (M + Na)+: 487.0720; found: 487.0706.
2-{[(1-(4-Fluorobenzyl)-1H-1,2,3-triazol-4-yl)methyl]thio}-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (12e)
Yellow solid; Chemical Formula: C22H17FN6O2S, Yield; 62%, Mol wt: 448.47 gmol−1, M.p: 96–101 °C.
1H NMR (400 MHz, Chloroform-d) δ 8.95 (d, J = 3.7 Hz, 1H, H-2), 8.20 (dd, J = 8.2, 1.7 Hz, 1H, H-4), 7.70 (s, 1H, H-5″), 7.56–7.42 (m, 3H, H-3,H-5, H-6), 7.28 (dd, J = 7.3, 1.6 Hz, 1H, H-7), 7.21 (dd, J = 8.5, 5.3 Hz, 2H, H-2′″&H-6′″), 7.07–6.96 (m, 2H, H-3′″&H-5′″), 5.54 (s, 2H, H-6′), 5.42 (s, 2H, H-7″), 4.51 (s, 2H, H-6″).
13C NMR (101 MHz, Chloroform-d) δ 165.81, C-5′, 164.08, C-4′″, 163.45, C-2′, 161.61, C-4′″, 152.92, C-8, 149.42, C-2, 143.25,C-4″, 139.60, C-8a, 136.66, C-4, 130.34 (d, J = 3.4 Hz, C-1′″), 129.92 (d, J = 8.4 Hz, C-2′″&6′″), 129.68, C-4a, 126.72, C-6, 123.24. C-5″, 121.96, C-3, 121.73, C-5, 116.11 (d, J = 21.9 Hz, C-3′″&5′″), 110.84, C-7, 60.74, C-6′, 53.43, C-7″, 26.87, C-6″.
HRMS: (ESI+-MS, m/z) calcd for C22H17ClN6O2S (M + Na)+: 471.1004; found: 471.0999.
2-{[(1-(4-Bromobenzyl)-1H-1,2,3-triazol-4-yl)methyl]thio}-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (12f)
Cream solid; Chemical Formula: C22H17BrN6O2S, Yield; 53%, Mol wt: 509.38 gmol−1, M.p: 78–82 °C.
1H NMR (400 MHz, Chloroform-d) δ 8.89–8.83 (m, 1H), 8.10 (dd, J = 8.3, 1.6 Hz, 1H), 7.65 (s, 1H), 7.47–7.35 (m, 5H), 7.20 (dd, J = 7.4, 1.5 Hz, 1H), 7.05–6.97 (m, 2H), 5.46 (s, 2H), 5.33 (s, 2H), 4.45 (s, 2H).
13C NMR (101 MHz, Chloroform-d) δ 165.75, 163.50, 153.07, 149.59, 143.33, 139.94, 136.34, 133.48, 132.26, 129.66, 129.60, 126.59, 123.36, 122.91, 121.96, 121.75, 110.69, 60.73, 53.46, 26.85.
HRMS: (ESI+-MS, m/z) calcd for C22H17BrN6O2S (M + Na)+: 517.0058; found: 517.0046.
2-{[(1-(4-Nitrobenzyl)-1H-1,2,3-triazol-4-yl)methyl]thio}-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (12g)
Bright yellow solid; Chemical Formula: C22H17N7O4S, Yield; 74%, Mol wt: 475.48 gmol−1, M.p: 100–106 °C.
1H NMR (400 MHz, Chloroform-d) δ 8.83 (dd, J = 4.2, 1.7 Hz, 1H), 8.12–8.04 (m, 3H), 7.77 (s, 1H), 7.46–7.34 (m, 3H), 7.28–7.14 (m, 2H), 5.49 (s, 2H), 5.44 (s, 2H), 4.45 (s, 2H).
13C NMR (101 MHz, Chloroform-d) δ 165.55, 163.66, 149.53, 136.23, 129.72, 128.54, 126.52, 124.21, 123.70, 121.89, 111.17, 60.98, 53.06, 26.87.
HRMS: (ESI+-MS, m/z) calcd for C22H17N7O4S (M + Na)+: 498.0960; found: 498.0951.
2-{[(1-(3-Chlorobenzyl)-1H-1,2,3-triazol-4-yl)methyl]thio}-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (12h)
Cream white solid; Chemical Formula: C22H17ClN6O2S, Yield; 76%, Mol wt: 464.93 gmol−1, M.p: 78–84 °C.
1H NMR (600 MHz, Chloroform-d) δ 8.87 (dd, J = 4.3, 1.7 Hz, 1H), 8.11 (dd, J = 8.2, 1.7 Hz, 1H), 7.67 (s, 1H), 7.46–7.37 (m, 3H), 7.25–7.16 (m, 3H), 7.13 (d, J = 1.9 Hz, 1H), 7.01 (dt, J = 7.4, 1.5 Hz, 1H), 5.47 (s, 2H), 5.35 (s, 2H), 4.46 (s, 2H).
13C NMR (101 MHz, Chloroform-d) δ 165.69, 163.56, 153.17, 149.70, 143.33, 140.19, 136.38, 136.11, 134.95, 130.41, 129.65, 128.97, 128.03, 126.48, 126.02, 123.44, 121.95, 121.77, 110.63, 60.75, 53.43, 26.85.
HRMS: (ESI+-MS, m/z) calcd for C22H17ClN6O2S (M + Na)+: 487.0720; found: 487.0730.
2-{[(1-(2,4-Dichlorobenzyl)-1H-1,2,3-triazol-4-yl)methyl]thio}-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (12i)
Cream white solid; Chemical Formula: C22H16Cl2N6O2S, Yield; 57%, Mol wt: 499.37 gmol−1, M.p: 124–130 °C.
1H NMR (400 MHz, Chloroform-d) δ 8.97 (dd, J = 4.3, 1.7 Hz, 1H), 8.22 (dd, J = 8.4, 1.7 Hz, 1H), 7.82 (s, 1H), 7.59–7.46 (m, 3H), 7.42 (d, J = 2.1 Hz, 1H), 7.31 (dd, J = 7.4, 1.6 Hz, 1H), 7.22 (dd, J = 8.3, 2.1 Hz, 1H), 7.04 (d, J = 8.3 Hz, 1H), 5.57 (d, J = 3.4 Hz, 4H), 4.56 (s, 2H).
13C NMR (101 MHz, Chloroform-d) δ 165.73, 163.47, 152.89, 149.41, 143.17, 139.57, 136.67, 134.04, 130.95, 130.91, 129.74, 129.67, 127.93, 126.72, 123.76, 121.96, 121.72, 110.83, 60.73, 50.84.
HRMS: (ESI+-MS, m/z) calcd for C22H16Cl2N6O2S (M + Na)+: 521.0411; found: 521.0421.
2-{[(1-(3,4-Difluorobenzyl)-1H-1,2,3-triazol-4-yl)methyl]thio}-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (12j)
Cream white solid; Chemical Formula: C22H16F2N6O2S, Yield; 61%, Mol wt: 466.46 gmol−1, M.p: 70–75 °C.
1H NMR (400 MHz, Chloroform-d) δ 8.97 (dd, J = 4.2, 1.7 Hz, 1H), 8.20 (dd, J = 8.4, 1.7 Hz, 1H), 7.78 (s, 1H), 7.56–7.45 (m, 3H), 7.34–7.19 (m, 2H), 6.92–6.80 (m, 2H), 5.56 (s, 2H), 5.49 (s, 2H), 4.54 (s, 2H).
13C NMR (101 MHz, Chloroform-d) δ 165.76, 164.55 (dd, J = 250.5, 12.1 Hz), 163.49, 160.65 (dd, J = 251.3, 27.0 Hz), 153.00, 149.52, 143.18, 139.79, 136.47, 131.51 (dd, J = 9.9, 4.7 Hz), 129.67, 126.64, 123.41, 121.95, 121.73, 117.78 (dt, J = 14.6, 3.7 Hz), 112.15 (dd, J = 21.5, 3.8 Hz), 110.77, 104.40 (t, J = 25.4 Hz), 60.74, 47.17 (d, J = 3.8 Hz), 26.84.
HRMS: (ESI+-MS, m/z) calcd for C22H16Cl2N6O2S (M + Na)+: 521.0411; found: 521.0421.
2-{[(1-Phenyl-1H-1,2,3-triazol-4-yl)methyl]thio}-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (12k)
Light brown solid; Chemical Formula: C21H16N6O2S, Yield; 91%, Mol wt: 416.46 gmol−1, M.p: 122–128 °C.
1H NMR (400 MHz, Chloroform-d) δ 8.83 (dd, J = 4.3, 1.7 Hz, 1H), 8.11 (s, 1H), 8.05 (dd, J = 8.3, 1.8 Hz, 1H), 7.60 (dd, J = 7.6, 1.8 Hz, 2H), 7.45–7.28 (m, 6H), 7.18 (dd, J = 7.3, 1.7 Hz, 1H), 5.47 (s, 2H), 4.54 (s, 2H).
13C NMR (101 MHz, Chloroform-d) δ 165.75, 163.64, 153.18, 149.71, 143.46, 140.23, 136.82, 136.06, 129.70, 129.63, 128.85, 126.44, 121.91, 121.79, 121.59, 120.60, 110.77, 60.83, 26.83.
HRMS: (ESI+-MS, m/z) calcd for C21H16N6O2S (M + Na)+: 439.0953; found: 439.0959.
2-{[(1-(4-Chlorophenyl)-1H-1,2,3-triazol-4-yl)methyl]thio}-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (12l)
Yellow solid; Chemical Formula: C21H15ClN6O2S, Yield; 79%, Mol wt: 450.90 gmol−1, M.p: 132–136 °C.
1H NMR (400 MHz, Chloroform-d) δ 8.85 (dd, J = 4.3, 1.7 Hz, 1H), 8.15–8.05 (m, 2H), 7.61–7.52 (m, 2H), 7.48–7.33 (m, 5H), 7.20 (dd, J = 7.3, 1.6 Hz, 1H), 5.48 (s, 2H), 4.54 (s, 2H).
13C NMR (101 MHz, Chloroform-d) δ 165.73, 163.64, 153.06, 149.59, 143.79, 139.94, 136.34, 135.30, 134.62, 129.87, 129.66, 126.56, 121.96, 121.79, 121.76, 121.57, 110.77, 60.80, 26.74.
HRMS: (ESI+-MS, m/z) calcd for C21H16N6O2S (M + Na)+: 473.0563; found: 473.0560.
2-{[(1-(4-Fluorophenyl)-1H-1,2,3-triazol-4-yl)methyl]thio}-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (12m)
Brown crystalline; Chemical Formula: C21H15FN6O2S, Yield; 84%, Mol wt: 434.45 gmol−1, M.p: 134–140 °C.
1H NMR (400 MHz, Chloroform-d) δ 8.83 (dd, J = 4.3, 1.8 Hz, 1H), 8.10–8.02 (m, 2H), 7.64–7.53 (m, 2H), 7.44–7.31 (m, 3H), 7.19 (d, J = 7.4 Hz, 1H), 7.13–7.03 (m, 2H), 5.47 (s, 2H), 4.53 (s, 2H).
13C NMR (101 MHz, Chloroform-d) δ 165.72, 161.21, 153.20, 149.72, 143.64, 140.23, 136.05, 133.09, 129.64, 126.44, 122.65, 122.56, 121.93, 121.80, 116.76, 116.53, 110.71, 60.81, 26.76.
HRMS: (ESI+-MS, m/z) calcd for C21H16N6O2S (M + Na)+: 457.0859; found: 457.0851.
2-{[(1-(4-Bromophenyl)-1H-1,2,3-triazol-4-yl)methyl]thio}-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (12n)
Brown solid; Chemical Formula: C21H15BrN6O2S, Yield; 77%, Mol wt: 495.35 gmol−1, M.p: 126–132 °C.
1H NMR (400 MHz, Chloroform-d) δ 8.84 (dd, J = 4.2, 1.7 Hz, 1H), 8.13–8.03 (m, 2H), 7.56–7.46 (m, 4H), 7.45–7.32 (m, 3H), 7.19 (dd, J = 6.8, 2.2 Hz, 1H), 5.48 (s, 2H), 4.54 (s, 2H).
13C NMR (101 MHz, Chloroform-d) δ 165.71, 163.68, 153.20, 149.73, 143.83, 140.24, 136.07, 135.79, 132.84, 129.65, 126.45, 122.51, 121.98, 121.94, 121.82, 121.47, 110.73, 60.82, 26.74.
HRMS: (ESI+-MS, m/z) calcd for C21H15BrN6O2S (M + Na)+: 517.0058; found: 517.0046.
2-{[(1-(4-Nitrophenyl)-1H-1,2,3-triazol-4-yl)methyl]thio}-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (12o)
Bright yellow crystals; Chemical Formula: C21H15N7O4S, Yield; 83%, Mol wt: 461.45 gmol−1, M.p: 150–156 °C.
1H NMR (400 MHz, Chloroform-d) δ 8.94 (dd, J = 4.3, 1.7 Hz, 1H), 8.42 (s, 1H), 8.40–8.32 (m, 2H), 8.20 (dd, J = 8.3, 1.7 Hz, 1H), 8.00–7.92 (m, 2H), 7.56–7.44 (m, 3H), 7.30 (dd, J = 7.3, 1.6 Hz, 1H), 5.57 (s, 2H), 4.65 (s, 2H).
13C NMR (101 MHz, Chloroform-d) δ 165.62, 163.67, 152.99, 149.52, 147.24, 144.57, 140.96, 139.75, 136.53, 129.68, 126.64, 125.44, 122.00, 121.83, 121.67, 120.61, 110.76, 60.78, 26.59.
HRMS: (ESI+-MS, m/z) calcd for C21H15N7O4S (M + Na)+: 484.0804; found: 484.0799.
2-[(Quinolin-8-yloxy)methyl]-5-{[(1-(p-tolyl)-1h-1,2,3-triazol-4-yl)methyl]thio}-1,3,4-oxadiazole (12p)
Light brown solid; Chemical Formula: C22H18N6O2S, Yield; 73%, Mol wt: 430.48 gmol−1, M.p: 78–80 °C.
1H NMR (400 MHz, Chloroform-d) δ 8.87 (dd, J = 4.2, 1.8 Hz, 1H), 8.10 (dd, J = 8.3, 1.7 Hz, 1H), 8.07 (s, 1H), 7.52–7.45 (m, 2H), 7.40 (dddd, J = 9.7, 8.3, 5.9, 1.5 Hz, 3H), 7.21 (dd, J = 8.7, 7.3 Hz, 3H), 5.50 (d, J = 1.8 Hz, 2H), 4.55 (s, 2H), 2.33 (s, 3H).
13C NMR (101 MHz, Chloroform-d) δ 165.83, 163.58, 152.96, 149.43, 143.29, 139.71, 138.99, 136.58, 134.58, 130.19, 129.68, 126.68, 121.92, 121.77, 121.55, 111.09, 60.88, 26.89, 21.08.
HRMS: (ESI+-MS, m/z) calcd for C21H15N7O4S (M + Na)+: 453.1110; found: 453.1097.
2-{[(1-(4-Methoxyphenyl)-1H-1,2,3-triazol-4-yl)methyl]thio}-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (12q)
Black solid; Chemical Formula: C22H18N6O3S, Yield; 76%, Mol wt: 446.48 gmol−1, M.p: 122–128 °C.
1H NMR (400 MHz, Chloroform-d) δ 8.84 (dd, J = 4.3, 1.7 Hz, 1H), 8.07 (dd, J = 8.3, 1.7 Hz, 1H), 8.02 (s, 1H), 7.53–7.45 (m, 2H), 7.44–7.32 (m, 3H), 7.20 (dd, J = 7.3, 1.6 Hz, 1H), 6.93–6.84 (m, 2H), 5.48 (s, 2H), 4.53 (s, 2H), 3.76 (s, 3H).
13C NMR (101 MHz, Chloroform-d) δ 165.80, 163.60, 159.87, 153.12, 149.62, 143.21, 140.08, 136.20, 130.28, 129.64, 126.52, 122.23, 121.91, 121.77, 121.71, 114.71, 110.84, 60.83, 55.62, 26.88.
HRMS: (ESI+-MS, m/z) calcd for C22H18N6O3S (M + Na)+: 469.1059; found: 469.1049.
2-{[(1-(2,4-Dichlorophenyl)-1H-1,2,3-triazol-4-yl)methyl]thio}-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (12r)
Light brown solid; Chemical Formula: C21H14Cl2N6O2S, Yield; 73%, Mol wt: 485.35 gmol−1, M.p: 112–116 °C.
1H NMR (400 MHz, Chloroform-d) δ 8.85 (dd, J = 4.3, 1.7 Hz, 1H), 8.14 (s, 1H), 8.08 (dd, J = 8.3, 1.7 Hz, 1H), 7.55 (d, J = 2.4 Hz, 1H), 7.45–7.30 (m, 5H), 7.20 (dd, J = 7.2, 1.7 Hz, 1H), 5.48 (s, 2H), 4.56 (s, 2H).
13C NMR (101 MHz, Chloroform-d) δ 165.59, 163.61, 153.04, 149.57, 142.75, 139.91, 136.31, 135.29, 133.67, 131.59, 130.81, 129.64, 127.79, 126.74, 126.55, 125.39, 121.93, 121.74, 110.69, 60.73, 26.68.
HRMS: (ESI+-MS, m/z) calcd for C21H14Cl2N6O2S (M + Na)+: 521.0411; found: 521.0421.
2-{[(1-(2,6-Dichlorophenyl)-1H-1,2,3-triazol-4-yl)methyl]thio}-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (12s)
Light brown solid; Chemical Formula: C21H14Cl2N6O2S, Yield; 77 %, Mol wt: 485.35 gmol−1, M.p: 113–118 °C.
1H NMR (400 MHz, Chloroform-d) δ 8.84 (dd, J = 4.3, 1.7 Hz, 1H), 8.07 (dd, J = 8.3, 1.7 Hz, 1H), 7.89 (s, 1H), 7.44–7.26 (m, 6H), 7.23–7.16 (m, 1H), 5.49 (s, 2H), 4.58 (s, 2H).
13C NMR (101 MHz, Chloroform-d) δ 163.63, 153.09, 149.53, 142.67, 140.02, 136.23, 131.72, 129.63, 128.76, 126.54, 125.80, 121.89, 121.74, 110.92, 60.88, 26.91.
HRMS: (ESI+-MS, m/z) calcd for C21H14Cl2N6O2S (M + Na)+: 508.01; found: 508.35.
2-{[(1-(2,6-Dimethylphenyl)-1H-1,2,3-triazol-4-yl)methyl]thio}-5-[(quinolin-8-yloxy)methyl]-1,3,4-oxadiazole (12t)
Brown solid; Chemical Formula: C23H20N6O2S, Yield; 43%, Mol wt: 444.51 gmol−1, M.p: 110–116 °C.
1H NMR (400 MHz, Chloroform-d) δ 8.82 (dd, J = 4.2, 1.7 Hz, 1H), 8.05 (dd, J = 8.4, 1.7 Hz, 1H), 7.71 (s, 1H), 7.43–7.31 (m, 2H), 7.25–7.15 (m, 1H), 7.05 (d, J = 7.6 Hz, 2H), 5.49 (s, 2H), 4.57 (s, 2H), 1.83 (s, 6H).
13C NMR (101 MHz, Chloroform-d) δ 165.71, 163.61, 153.09, 149.58, 142.63, 140.05, 136.19, 135.69, 135.34, 130.03, 129.63, 128.41, 126.48, 125.09, 121.90, 121.77, 110.82, 60.81, 27.03, 17.31.
HRMS: (ESI+-MS, m/z) calcd for C23H20N6O2S, (M + Na)+: 467.1266; found: 467.1267.


 ,
, 






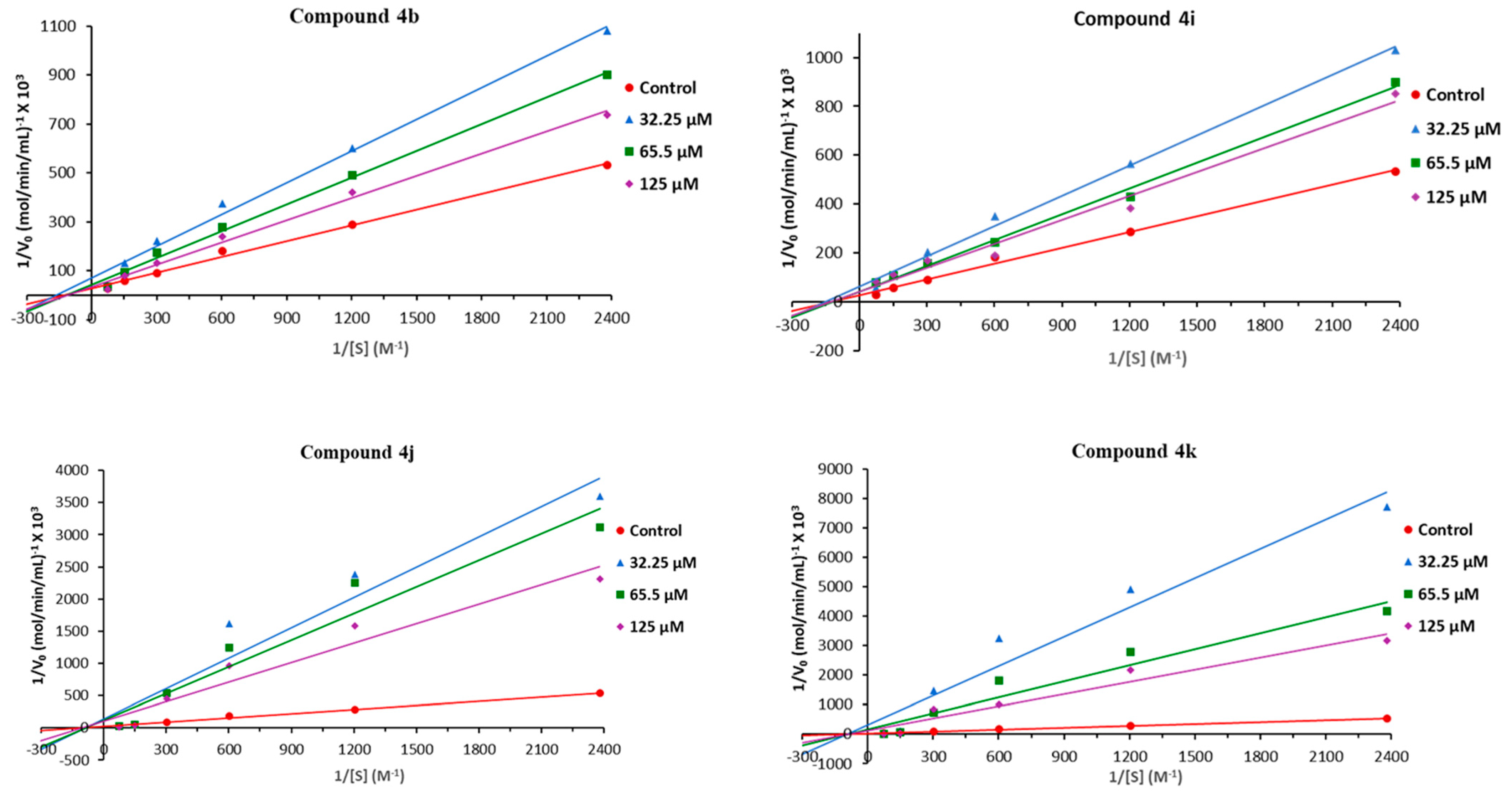



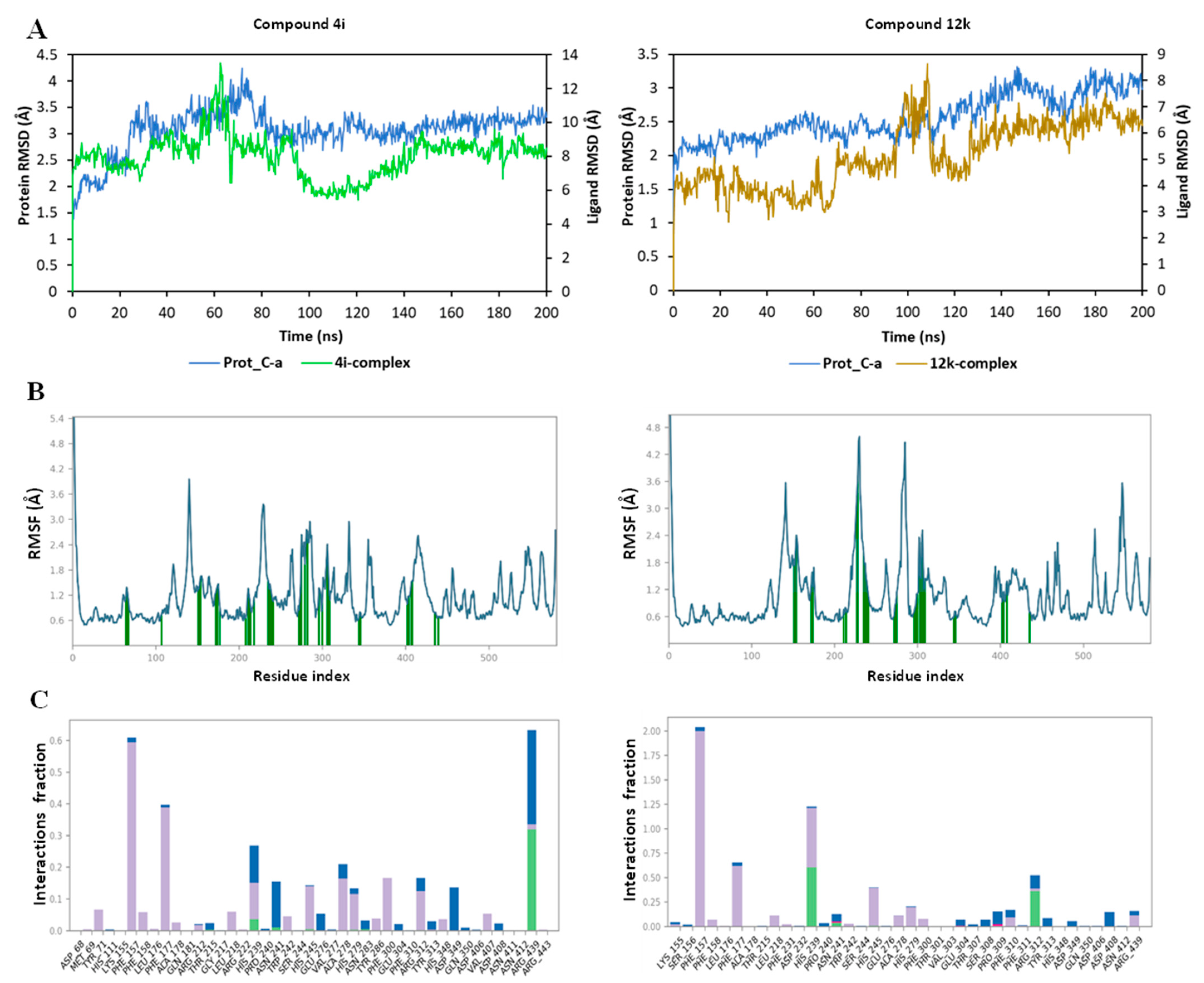

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
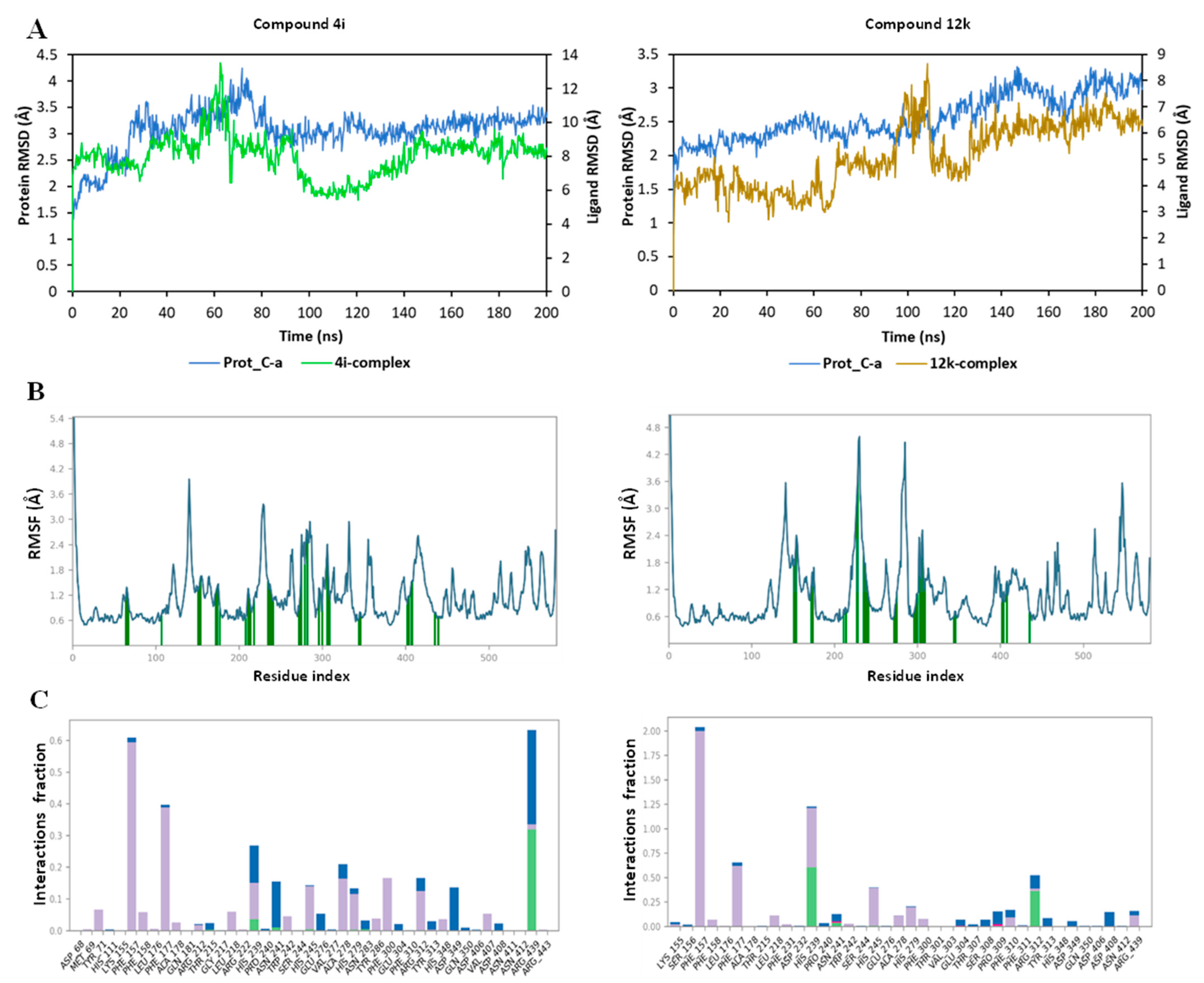{kind=link}
{kind=link}
{kind=link}
{kind=link}